Advances in Biological Chemistry
Vol.3 No.1(2013), Article ID:28107,10 pages DOI:10.4236/abc.2013.31010
Topological conformational changes of human papillomavirus (HPV) DNA bound to an insoluble aluminum salt—A study by low temperature PCR
![]()
Milford Hospital and Milford Molecular Laboratory, Milford, Connecticut, USA
Email: shlee01@snet.net
Received 5 October 2012; revised 9 November 2012; accepted 28 November 2012
Keywords: Low Temperature PCR; HPV; DNA Conformation; Gardasil®; Postmortem; AAHS Adjuvant; Aluminum
ABSTRACT
A low temperature (LoTemp®) polymerase chain reaction (PCR), conducted at cycling temperatures not to exceed 85˚C and catalyzed by a novel highly processive HiFi® DNA polymerase with proofreading function, was used to study the topological conformational changes of the human papillomavirus (HPV) L1 gene DNA fragments bound to the insoluble amorphous aluminum hydroxyphosphate sulfate (AAHS) adjuvant in the quadrivalent HPV vaccine, Gardasil®. L1 gene DNA fragments of HPV-11, HPV-18 and HPV-16 were detected in the AAHS particles by nested PCR, but all were lacking a region that was amplifiable by an MY09 degenerate primer. In addition, a pair of degenerate consensus GP6/MY11 primers was able to amplify a target segment of the HPV-11 L1 gene DNA and the HPV-18 L1 gene DNA bound to the AAHS particles as expected for any HPV DNA in the B-conformation. However, there was no co-amplification of the HPV-16 L1 gene DNA known to coexist in the same samples. The lack of coamplification was verified by direct DNA sequencing of the PCR amplicons. The companion HPV-16 L1 gene DNA in the same sample required repeated PCRs with a pair of modified non-degenerate GP6/ MY11 primers for detection. This melting profile of the HPV-16 L1 gene DNA was similar to that of the HPV-16 L1 gene DNA recently discovered in the postmortem blood of a young woman who suffered a sudden unexpected death 6 months after Gardasil® vaccination. The findings suggest that the topological conformational changes in the HPV L1 gene DNA residues bound to the AAHS adjuvant may be genotype-related. The special non-B-conformation may prevent the HPV-16 L1 gene DNA from being degraded in the body of the vaccine recipients after intramuscular injection.
1. INTRODUCTION
Molecular characterization of human papillomavirus (HPV) DNA in clinical specimens begins with primerdirected enzymatic polymerase chain reaction (PCR) amplification of the target DNA with a thermostable DNA polymerase. In practice, pre-PCR purification of DNA samples is required [1-4], and the DNA template must be soluble in an aqueous solution. When the viral load is low, attempts to purify a minute quantity of DNA from a complex sample may risk losing the target template all together.
A low temperature (LoTemp®) PCR system with a highly processive DNA polymerase which also has proof-reading function has been used to increase the sensitivity of PCR detection of HPV DNA in proteinase-K digested unpurified clinical samples without sacrificing specificity [5,6]. This novel PCR system depends on chemical denaturation of the dsDNA template at 85˚C so that the highly stable but less heat-resistant HiFi® DNA polymerase of high processivity can perform its function of cyclic primer extension at 65˚C. In this system, the annealing temperature is set at 50˚C for high stringency PCR, and at 40˚C for low stringency PCR amplification. Using LoTemp® PCR, residual HPV L1 gene DNA fragments have been detected in the proteinase K-digested insoluble fraction of the HPV vaccine Gardasil® (Merck and Co., Inc.) [7], and in the postmortem blood and spleen of a healthy young woman who suffered a sudden unexpected death 6 months following Gardasil® vaccination without a recognized cause of death after a full autopsy analysis [8]. The US Food and Drug Administration (FDA) has confirmed that the quadrivalent HPV vaccine Gardasil® does contain HPV L1 gene DNA fragments [9]. In theory, residual L1 gene DNA fragments of HPH-16, HPV-18, HPV-11 and HPV-6 may be present in this quadrivalent vaccine. These naked HPV DNA fragments in the vaccine are expected to be bound to the particulate amorphous aluminum hydroxy phosphate sulfate (ASHS) adjuvant [7]. Aluminum-based adjuvant nanoparticles are generally designed to be phagocytized by macrophages in the vaccine recipients [10-13]. This communication reports using low temperature PCR supported by direct automated DNA sequencing to study the topological changes of conformation of the HPV DNA bound to AAHS particles. DNA fragments in a non-Bconformation may be protected from degradation by nucleases after intramuscular injection into the host.
2. MATERIALS AND METHODS
2.1. Gardasil® Vaccine Samples
A total of 16 Gardasil® vials or manufacturer-prefilled vaccine syringes with intact original packages were purchased by licensed practitioners in nine countries and submitted to the author’s laboratory to be tested for residual HPV L1 gene DNA fragments. The first part of the test results based on degenerate consensus primer nested PCR amplification was previously reported [7].
2.2. Preparation of Samples for PCR
An aliquot of 100 μL of the vaccine suspension was centrifuged at ~16,000 × g for 10 min in a 1.5 mL microcentrifuge tube. The pellet was washed twice with 1 mL of 70% ethanol each and the final ethanol suspension was centrifuged at ~16,000 × g for 5 min. The washed pellet was air-dried. The dried pellet was re-suspended in 100 μL of 0.1 mg/mL proteinase K (Sigma Chemical Co., St. Louis, MO) in a buffer consisting of 50 mM Tris-HCl, 1 mM EDTA, 0.5% Tween 20, pH 8.1. The mixture was digested at 45˚C - 55˚C overnight and was exhaustively washed with the same Tween 20 buffer pH 8.1, 4 times, 1 mL each time. At this pH, both the AAHS particles and the DNA molecules carry a negative electrostatic charge. The final washed pellet, presumably consisting of protein-free AAHS particles, was resuspended in 100 μL of buffer. After heating at 95˚C for 10 min, 1 μL of the washed and heated insoluble particle suspension was used for each primary PCR followed by nested PCR.
2.3. Nested PCR Amplification of HPV L1 Gene DNA
Initially, a Taq DNA polymerase was used for the PCR amplification. In this Taq PCR protocol, 1 mL of the sample suspension, 2.5 units of GE rTaq DNA polymerase in 0.25 mL (GE Healthcare, Piscataway, NJ, USA), 2.5 mL of 10× PCR buffer, 2 mL of 25 mM MgCl2, 1 mL of 5 mM dNTPs, 1 mL of MY09 primer (10 mM), 1 mL of MY11 primer (10 mM), and water were added up to a final volume of 25 µL to initiate a primary PCR. The nested PCR mixture contained the same ingredients except that the primers were GP6/MY11 or GP5/MY09 and the 1 mL of the suspension was replaced with water. The primary PCR products were transferred to the nested PCR tubes by micro-glass rods to avoid micropipetting aerosol [6]. Thermocycling steps were set as preheating for 2 min at 94˚C, followed by 30 cycles, each set at 94˚C for 30 sec, 54˚C for 2 min and 72˚C for 1 min; the final extension was 72˚C for 10 min for both primary and nested PCR.
For the low temperature PCR, 20 µL of the LoTemp® master mix containing manufacturer-optimized HiFi® DNA polymerase, magnesium ions, denaturing agents, and dNTPs with stabilizing additives was used to replace the GE rTaq DNA polymerase, the buffer, the MgCl2 solution, and dNTPs described for the Taq PCR in a final volume of 25 µL. The amounts of template materials and the primers were identical to those described for the Taq PCR protocol. The LoTemp® thermocycling steps were set for an initial heating at 85˚C for 10 min, followed by 30 cycles, each set at 85˚C for 30 sec, 40˚C for 30 sec, and 65˚C for 1 min. The final extension was 65˚C for 10 min.
After completion of the primary and the nested PCR, a 5 µL aliquot of the PCR products was pipetted out from each tube and mixed with 2 µL loading fluid for electrophoresis in a 2% agarose gel containing ethidium bromide. The gel was examined under UV light for various PCR product bands in the agarose gel.
2.4. Direct Automated DNA Sequencing of the Nested PCR Amplicons
For DNA sequencing, a trace of the positive nested PCR product was transferred directly with a micro-glass rod from the positive nested PCR tube into a 20 µL volume of a cycle sequencing reaction mixture consisting of 14.5 µL water, 3.5 µL of 5 × buffer, 1 µL of BigDye Terminator 1.1 (Applied Biosystems) and 1 µL of 10 µM sequencing primer. After thermal cycling according to the manufacturer’s recommendation, the reaction mixture was loaded in an automated ABI 3130 four-capillary Genetic Analyzer for sequence analysis. Alignment analysis of a 45 - 60 base sequence in the hypervariable region of the L1 gene excised from the computer-generated basecalling electropherogram was performed against various standard HPV genotype sequences stored in the GenBank, using the on-line BLAST (Basic Local Alignment Search Tool) system to validate the specific HPV genotyping and for visual sequence analyses.
2.5. Primers
The sequences of the well characterized degenerate [14] and non-degenerate [15] primers with their modifications which were used in this study are listed as follows.
MY09 = 5’-CGTCCMARRGGAWACTGATC-3’
MY11 = 5’-GCMCAGGGWCATAAYAATGG-3’
Key to degenerate nucleotides: M = (A + C), R = (A + G), W = (A + T), Y = (C + T)
GP5 = 5’-TTTGTTACTGTGGTAGATAC-3’
GP6 = 5’-GAAAAATAAACTGTAAATCA-3’
GP6+ = 5’-GAAAAATAAACTGTAAATCATATTC- 3’
MY11(1) = 5’-GCACAGGGACATAACAATGG-3’
HPV16MY11+ = 5’-GCACAGGGCCACAATAATGGCAT-3’
The binding sites for the four principal primers in the HPV L1 gene ORF are positioned in following order:
5’---MY11----GP5-----GP6----MY09---3’
In this report, nested PCR refers to heminested PCR with the GP6/MY11 or the GP5/MY09 primer pair, except the “same-nested PCR” defined below.
2.6. Same-Nested PCR for Amplification of HPV-16 L1 Gene DNA in Vaccine and in Postmortem Blood
A “same-nested” PCR was designed for amplification of a 184 bp hypervariable region of an HPV-16 L1 gene DNA in the vaccine and in the postmortem blood of a young woman who suffered a sudden unexpected death 6 months after Gardasil® vaccination. For the same-nested PCR protocol, the primary PCR and the subsequent same-nested PCR(s) were conducted with an identical pair of non-degenerate PCR primers, or the subsequent same-nested PCR was conducted with a pair of the same primers having a few new bases added to the 3’-end of one or to both of the primers which had been used in the prior PCR. As a result, all same-nested PCR products were terminated by the first pair of PCR primers used to initiate the primary PCR. The postmortem blood DNA was prepared by Dr. Donald Love at Auckland Hospital Molecular Genetics Laboratory in New Zealand and sent to the author’s laboratory for HPV DNA testing by the order of the Wellington coronial court in connection of a public inquest hearing on August 8-9, 2012.
3. RESULTS
3.1. Comparing Taq PCR and Low Temperature PCR Amplifications
Both conventional Taq polymerase PCR and the low temperature PCR with HiFi® DNA polymerase amplified the control HPV-16 plasmid DNA to generate an amplicon of ~450 bp in the primary PCR with the degenerate MY09/MY11 primers and an amplicon of ~190 bp in the nested PCR with the degenerate consensus GP6/MY11 primers, respectively (Figure 1). These positive control GP6/MY11 nested PCR amplicons in both PCR systems were confirmed to be those of HPV-16 L1 gene DNA by direct automated DNA sequencing, using GP6 as the sequencing primer. Neither PCR system generated a primary PCR amplicon band from any of the Gardasil® vaccine lot samples tested. In all low temperature nested PCRs initiated with 1 µL of proteinase K-digested vaccine particles, a single robust HPV L1 gene DNA amplicon of ~190 bp in size was obtained as visualized at agarose gel electrophoresis (Figure 1). DNA sequencing with GP6 as the sequencing primer validated these ampliconsto be those of HPV-11 L1 gene DNA, HPV-18 L1 gene DNA or a mixture of both, as previously reported [7]. The Taq PCR protocol also generated a ~190 bp PCR amplicon in all nested PCRs, but in addition numerous non-target PCR products in the nested PCR. None of the PCR products generated by the Taq protocol was usable for direct DNA sequencing. In general, Taq DNA polymerase is effective in amplification of purified HPV DNA to generate a template for direct DNA sequencing, but frequently fails to selectively amplify the target HPV DNA in complex samples in a nested PCR setting without pre-PCR and inter-step sample purifications [6]. As a result, all experiments described in this report were conducted in low temperature PCR unless otherwise specified.
3.2. Lack of DNA Denaturation at MY09-Binding Site in HPV DNA
When the degenerate MY09/MY11 primary PCR products were blindly transferred to the PCR mixture containing the GP5/MY09 primer pair for nested PCR amplification, no visible nested PCR product band was obtained at agarose gel electrophoresis (Figure 2). In comparison, the GP5/MY09 primer pair invariably generated a ~400 bp nested PCR product band from the positive control HPV-16 plasmid DNA (Figure 2). This result suggested that DNA conformation changes occurred in the HPV L1 gene DNA fragments when the latter were bound to the particulate AAHS and the changes in conformation prevented a region of DNA in the L1 gene at the MY09-binding site from being effectively amplified even with a highly processive HiFi® DNA polymerase. The DNA segment at the GP5-binding site was not affected. The latter segment was properly denatured for GP6/MY11 amplification (Figure 1) and demonstrated in direct DNA sequencing as -GTATCTACCACAGTAACAAA- (Figure 3).
3.3. DNA Conformational Changes at the MY11-Binding Site of HPV-16 DNA
The HPV-16 L1 gene DNA bound to AAHS particles
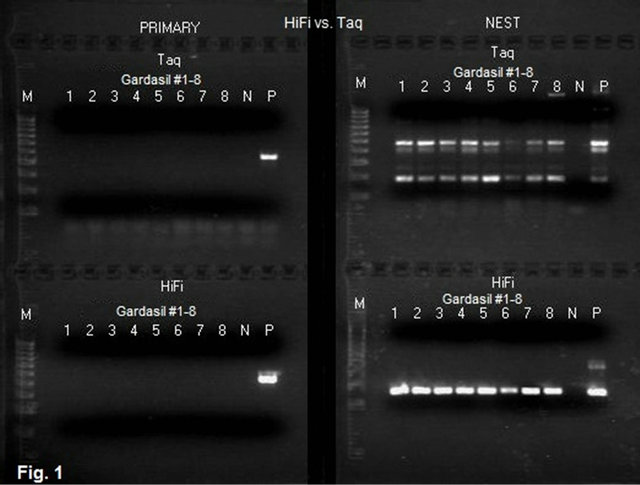
Figure 1. Agarose gel electrophoresis of the products of MY09/ MY11 primary PCR and GP6/MY11 nested PCR of HPV L1 gene DNA in Gardasil® with Taq and HiFi® DNA polymerases. Description: The suspensions of the pellets derived from 8 Gardasil® samples were used as the template materials to initiate the primary PCR. For both Taq and LoTemp® protocols, 1 mL of the suspension was used to start the MY09/MY11 primary PCR. The primers used in the nested PCR were GP6 and MY11. The primary PCR products were transferred to the nested PCR tubes by micro-glass rods. The ingredients and the thermocycling programs for Taq PCR and for LoTemp® PCR with HiFi® polymerase were described in the Materials and Methods section. Note: Neither Taq PCR nor LoTemp® HiFi® PCR generated a visible HPV amplicon in the primary PCR while the expected positive HPV MY09/MY11 control amplicon of ~450 bp is visualized in both primary PCRs. The LoTemp® HiFi® nested PCR protocol generated a single ~190 bp product band for all 8 Gardasil® samples and for the positive control HPV DNA, ready for direct DNA sequencing. All ~190 bp amplicons were confirmed by DNA sequencing to be composed of HPV-11 L1 gene DNA or HPV-18 L1 gene DNA, or a mixture of both. The Taq nested PCR generated several PCR product bands for all 8 Gardasil® samples and for the positive control HPV DNA. None of the Taq amplicons were useful as the template for direct DNA sequencing. PRIMARY = Primary PCR with MY09/ MY11 degenerate primers; Nest = Nested PCR with GP6/ MY11 primers; Taq = Taq PCR protocol; HiFi = LoTemp® PCR protocol with HiFi® DNA polymerase; M = molecular ruler 100 - 1000 bp; N = water control; P = HPV-16 plasmid DNA adjusted to 5 - 10 copies per mL dissolved in TE buffer.
appeared to acquire a non-B-conformation different from that of the HPV-11 and HPV-18 DNA. This phenomenon was further illustrated by PCR and DNA sequencing on Gardasil® vaccine lot #NL01490 which proved to contain both HPV-18 L1 gene DNA fragments and HPV-16 L1 gene DNA fragments. The degenerate consensus GP6/MY11 primer nested PCR product of Gardasil® lot #NL01490 was illustrated in lane 1, HiFi nest, Figure 1, showing a single amplicon band, and its corresponding DNA sequencing base-calling electropherogram was recorded in Figure 3, showing a 100% match with an HPV- 18 L1 gene sequence (GenBank Locus #EF202155.1).

Figure 2. Agarose gel electrophoresis of GP5/ MY09 nested PCR products of HPV L1 gene DNA in Gardasil®. Description: Each of the HiFi primary PCR products #1-5 described in Figure 1 was transferred by a micro-glass rod into a LoTemp® PCR mixture containing the GP5/MY09 primers instead of the GP6/MY11 primers. Note: A nested PCR amplicon of ~400 bp is visualized for the positive control HPV DNA, but not for any of the Gardasil® samples. The result of this experiment indicates that the melting profile prevented the MY09 primer-binding site of the HPV L1 gene DNA in Gardasil® from being amplified during PCR. M = molecular ruler 100 - 1000 bp; N = water control; P = HPV-16 plasmid DNA.
There was no evidence of co-amplification of any other HPV DNA by the 20-base degenerate MY11 primer, which was underlined on the right end with the three degenerate bases marked by a yellow dot on the top of each degenerate base.
In the course of this study, Gardasil® lot sample #NL01490 was found to also contain HPV-16 L1 gene DNA fragments which required two same-nested PCRs to amplify, namely one primary PCR and one samenested PCR with a pair of non-degenerate HPV16- MY11+/GP6 primers and a second nested PCR with a pair of non-degenerate HPV16MY11+/GP6+ primers to obtain a template (Figure 4) for DNA sequencing. The base-calling result had a 100% match with an HPV-16 L1 gene sequence (GenBank Locus #HQ644299.1), as illustrated in Figure 5. There was no mutation at the MY11 primer-binding site in the last 20 underlined bases to account for a failure of the degenerate consensus GP6/ MY11primer amplification.
3.4. Detection of HPV-16 L1 Gene DNA Fragments in Postmortem Blood by Same-Nested PCR
Numerous attempts to amplify an HPV L1 gene DNA in the postmortem blood with a pair of degenerate consensus GP6/MY11 primers under high stringency and low stringency PCR condition failed. After several experiments, 0.5 µg of the extracted genomic DNA was chosen to start a same-nested PCR under high stringency
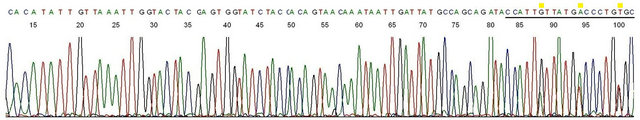
Figure 3. Sequencing validation of HPV L1 gene DNA in Gardasil®. Description: The nested PCR amplicon depicted in lane 1, Figure 1 (lot # NL01490)was used as the template and GP6 the sequencing primer. This base-calling electropherogram confirmed an HPV-18 L1 gene DNA in the amplicon. Note the 20-bases of degenerate MY11 primer (underlined) on the right end of the sequence.
condition with a pair of non-degenerate MY11(1) and GP6 primers. Only with a second same-nested PCR when the elongated 23-base non-degenerate HPV16- MY11+ was paired with a GP6 primer, a faint HPV DNA amplicon of ~190 bp was generated in 3 of 10 samples (Figure 6). An additional 3rd same-nested PCR with a pair of non-degenerate HPV16MY11+/GP6+ primers was needed to generate a clean template (Figure 7) for direct DNA sequencing. The 5 extra bases added to the 3’ end in the GP6+ primer increased the specificity of the HPV-16 DNA amplification in the same-nested PCR and overcame the primer-competing inhibitors of the human genomic DNA. The two-directional sequencing electropherogram showed a 184-base HPV-16 L1 gene DNA sequence matching 100% of the standard sequence of GenBank Locus #HQ644299.1 (Figures 8 and 9). There was no base mutation at the MY11 primer-binding site (underlined bases in Figure 8) or at the GP6 primerbinding site (underlined bases in Figure 9) that might have contributed to a failure of the degenerate consensus GP6/MY11 primer amplification.
4. DISCUSSION
Residual HPV L1 gene DNA in the vaccine Gardasil® [8] has provided the materials for this PCR-based study on aluminum-induced topological changes in DNA conformation. AAHS is Merck’s proprietary mineral-based particulate adjuvant which consists of amorphous precipitates prepared by mixing solutions of NaH2PO4, KAl(SO4)2 and ammonium hydroxide under special controlled conditions [16-18]. AAHS is the only certified insoluble non-protein ingredient in the Gardasil® formulation [19,20]. This adjuvant was originally created for binding phospholipid-containing antigens by ligand exchange of a phosphate group for a hydroxyl group on the adjuvant surface [21]. Since its point of zero charge (PZC) is pH 7 [18], antigens bound to the AAHS surface by electrostatic attraction will elute when exposed to interstitial fluid [21]. In the current study, the proteinase K-digested precipitates of the vaccine prepared for PCR have been exhaustively washed in a nonionic detergent solution at pH 8.1. Therefore, it is reasonable to assume that any HPV DNA residues left in the precipitates must be firmly attached to the AAHS particles via a chemical bond between the insoluble aluminum (Al3+) and the DNA molecule [22].
The DNA sequences of the type-specific HPV L1 genes used to manufacture the Gardasil® vaccine are well known. The highly conserved region of the HPV L1 gene, about 450 bp in size and terminated by the binding sites for the degenerate MY09 and MY11 primers, is well characterized. Heminested PCR based on degenerate consensus GP6/MY11 and GP5/MY09 amplification has been used for routine detection and genotyping of various HPV genotypes in clinical specimens [5,6,23, 24]. The hypervariable 181 - 187 bp L1 gene segment of HPV-11, HPV-16 and HPV-18 in the B-conformation is invariably amplified by the degenerate consensus GP6/ MY11 primer pair individually or co-amplified if more than one HPV genotype is present in a sample [6,23,24]. The lack of GP5/MY09 PCR primer amplification (Figure 2) for the HPV-11 and HPV-18 L1 gene DNA fragments in all Gardasil® vaccine lot samples tested indicates a topological change in DNA conformation at the MY09 primer-binding site that affects the melting profile of the HPV DNA molecules attached to AAHS. In heminested PCR on the same vaccine samples, the degenerate consensus GP6/MY11 primers are capable of amplifying the L1 gene DNA of at least one of these two genotypes from the template bound to insoluble AAHS particles in all vaccine samples tested, as for any HPV DNA in the B-conformation (Figure 1). Aluminum, unlike other metals, is known to stabilize and destabilize portions of a dsDNA molecule at different pHs, cause intrastrand cross-links, and create a “noncooperative melting profile” for the bound DNA molecule [25]. The data presented in this work indicate that DNA bound to insoluble Al3+ may also acquire a non-B-conformation.
The degenerate consensus MY09/MY11, GP5/MY09 and GP6/MY11 PCR primer pairs all fail to amplify the HPV-16 L1 gene DNA fragments present in the Gardasil® vaccine lot samples tested [Manuscript under peer review]. In the current study, experiments were designed to use the degenerate consensus GP6/MY11 primer pair
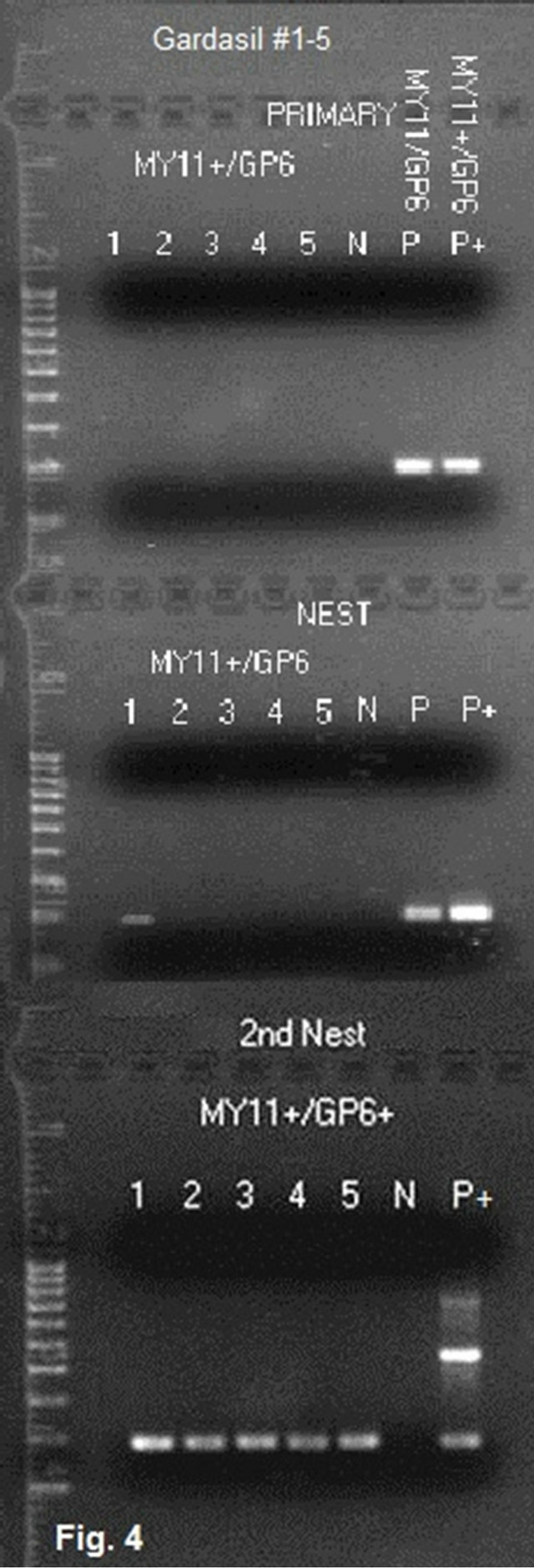
Figure 4. Same-nested PCR amplification for detection of HPV-16 L1 gene DNA in Gardasil®. Description: A pair of non-degenerate 23-base HPV-16 MY11+ and GP6 primers were used to amplify the HPV-16 L1 gene fragments in the proteinase Kdigested, exhaustively washed precipitates in 5 Gardasil® lot samples with a same-nested PCR protocol in which an identical pair of primers were used in the primary and nested PCR in tandem. Then the same-nested PCR products were transferred to a second nested PCR with a pair of HPV- 16 MY11+ and GP6+ primers for final amplification and preparation of the templates for DNA sequencing. Note: Only the products in lane 1 of the first same-nested PCR gel (lot #NL01490) show a weak ~190 bp HPV DNA amplicon, and all 5 samples show a specific HPV DNA amplicon in the second nested PCR. All 2nd nested PCR amplicons were confirmed to be of HPV-16 L1 gene DNA sequence (see Figure 5). 2nd Nest = 2nd nested PCR; MY11+ = HPV-16 MY11+; N = water control; P = HPV-16 plasmid DNA control amplified with degenerate consensus GP6/MY11 primers; P+ = HPV- 16 plasmid DNA control amplified with nondegenerate HPV-16 MY11+ and GP6 primers.
to amplify the HPV DNA in a vaccine sample known to contain both HPV-18 L1 gene DNA and HPV-16 L1 gene DNA in order to confirm that variable topological conformational changes in DNA of different genotypes may take place when bound to an insoluble aluminum salt. The results showed that only an HPV-18 amplicon was generated (Figure 3) without co-amplification of the HPV-16 DNA which was also present in the same sample. The companion HPV-16 L1 gene DNA was demonstrated by a same-nested PCR with the GP6 primer and a non-degenerate MY11 primer with a 3’-end extension referred to as HPV16MY11+ (Figure 4). No mutations were detected in this segment of HPV-16 L1 gene DNA (Figure 5). The failure of amplification of the HPV-16 DNA by the degenerate consensus GP6/MY11 primer PCR appears to be caused by a topological nonB-conformation of the target DNA template induced by the insoluble Al3+ which binds the phosphate backbone of the dsDNA [22]. The HPV-16 L1 gene DNA with this topological change apparently requires a non-degenerate primer with fully matched bases to initiate an enzymatic primer extension at the duplex.
The presence of HPV DNA fragments of vaccine origin in the body of vaccine recipients might be anticipated after intramuscular injections of Gardasil® since the vaccine is known to contain these DNA fragments [9]. But the finding of HPV-16 L1 gene DNA fragments only without the companion HPV-11 and/or HPV-18 DNA in a postmortem blood sample of a teenager who died 6 months after completion of 3 injections of a quadrivalent HPV vaccine was a surprise. As mentioned above, the HPV-11 and HPV-18 L1 gene DNA fragments in the vaccine are easily amplified with a pair of degenerate consensus GP6/MY11 primers in a heminested PCR setting. Similar to the HPV-16 L1 gene DNA fragments in the Gardasil® vaccine (Figure 4), the HPV-16 gene DNA fragments in the postmortem blood also require a same-nested PCR with specially designed non-degenerate primers for amplification (Figures 6 and 7). They also lack a PCR-amplifiable MY09 primer-binding site. A two-directional DNA sequencing (Figures 8 and 9) of the PCR amplicon proves that there is no base mutation in the entire length of the 184 bp amplicon which might have accounted for the failure of a degenerate consensus GP6/MY11 primer PCR amplification. The finding of only HPV-16 DNA in the postmortem blood following Gardasil® vaccination implies that the companion HPV- 11 and the HPV-18 L1 gene DNA fragments in the vaccine may be more accessible to the intracellular nucleases for degradation than those of HPV-16 L1 gene DNA fragments. The HPV-16 L1 gene DNA fragments in the postmortem blood might have been stabilized by aluminum in a non-B-conformation and protected from nuclease degradation. However, an aluminum-induced Zconformation of DNA has been reported to be stable even after the total removal of aluminum by a chelating agent [26]. Therefore, it is also possible that the nonB-conformation in the DNA fragments induced by aluminum in the Gardasil® vaccine might have persisted in some host cells without physically being bound to the aluminum salt.
This is the first time that PCR amplification followed by direct DNA sequencing has been used to study topological conformational changes in DNA fragments
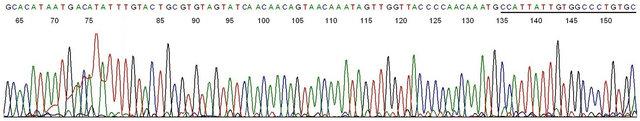
Figure 5. Sequencing validation of HPV-16 L1 gene DNA in Gardasil®. Description: The amplicon depicted in lane 1 of 2nd samenested PCR in Figure 4 (lot # NL01490)was used as the template and GP6 the sequencing primer. This base-calling electropherogram confirmed an HPV-16 L1 gene DNA in the amplicon. Note the 23-bases of non-degenerate HPV-16 MY11+ primer in the right end of the sequence. The last 20 bases (underlined) are substantially matched with those in a degenerate MY11 primer which is routinely used for PCR amplification of HPV-16 DNA in solution. However, a degenerate MY11 primer successfully amplified an HPV-18 L1 gene DNA (Figure 3), but not an HPV-16 L1 gene DNA bound to the same insoluble nanoparticles in the Gardasil® vaccine.

Figure 6. Same-nested PCR amplification in detection of HPV-16 L1 gene DNA fragments in postmortem blood after Gardasil® vaccination. Description: The whole blood DNA was extracted and prepared by using a Gentra® Puregene® Blood Kit (Qiagen) and finally dissolved in TE buffer at the concentration of 0.5 µg of DNA per µL to be used as the starting sample. Each LoTemp® HiFi® primary PCR tube contained 1 µL of the DNA sample, 1 µL of 10 µM GP6 primer and 1 µL of 10 µM of MY11(1), 20 µL of LoTemp® PCR master mix with HiFi® DNA polymerase, and water added up to 25 µL. A total of 10 parallel primary PCRs were performed. The ingredients in the 1st nested PCR were identical to those for the primary PCR except that 1 µL of water was used instead of the DNA sample. The ingredients for the 2nd nested PCR were identical to those for the 1st nested PCR except that the PCR primers were HPV16MY11+ and GP6. The results show that the MY11(1)/GP6 primer pair generated no visible amplicons in the primary PCRs, and nonspecific products at the 400 - 500 bp range in the 1st nested PCR. The HPV16MY11+/GP6 2nd nested PCR generated 3 weak HPV DNA amplicon bands of ~190 bp among non-specific products as shown in lanes 5, 7 and 8. Note: YV1824/1 = DNA Batch No. assigned by Auckland Hospital. Molecular ruler 100 - 1000 bp on the left; N = negative water control; P = HPV-16 plasmid DNA control amplified in parallel.

Figure 7. Third same-nested PCR in preparation of an HPV-16 gene DNA template for DNA sequencing. Description: The 2nd nested PCR products depicted in lane 8, Figure 6 was further amplified in a 3rd same-nested PCR with a pair of HPV16MY11+/ GP6+ primers. 8 = 3rd same-nested PCR amplicon using the 2nd nested PCR products as the template; N = negative water control; P = HPV-16 plasmid DNA control amplified in parallel.
bound to an insoluble aluminum salt. The study has been conducted with a low temperature PCR system in nested PCR settings. It may require more than one nested PCR to generate a useful template for direct DNA sequencing analysis when the initial template DNA is insoluble and in a non-B-conformation. In addition to using a highly processive DNA polymerase with proofreading function, the advantage of a low temperature PCR conducted at cycling temperatures not to exceed 85˚C reduces the rate of heat-induced mutations [27], namely depurination [28] and deamination [29] of the nitrogenous bases in the PCR products. Elimination of base mutations induced in PCR facilitates subsequent sequence analyses when minute quantities of PCR amplicons are re-amplified repeatedly to generate a final product to be used as the sequencing template.
The materials used for the current study were AAHS precipitates of a quadrivalent HPV vaccine in which the antigenic proteins were manufactured by a DNA recombinant technology. The viral genes encoding the L1 major capsid protein of HPV-16, HPV-18, HPV-11 and
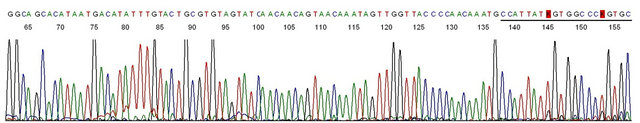
Figure 8. Base-calling electropherogram of DNA sequencing of the PCR amplicon shown in #8 of Figure 7, using GP6+ as the sequencing primer. Description: This is a typical HPV-16 L1 gene DNA sequence with the 20 bases of MY11 primer in the right end.
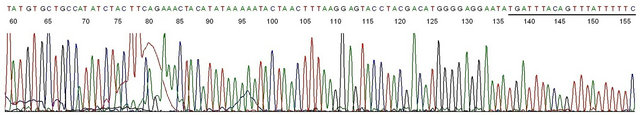
Figure 9. Base-calling electropherogram of DNA sequencing of the PCR amplicon shown in #8 of Figure 7, using HPV16MY11+ as the sequencing primer. Description: This is one of the two-directional DNA sequencing result showing a typical HPV-16 L1 gene DNA sequence with the 20 bases of GP6 primer in the right end.
HPV-6 were inserted into yeast cells for production of the four type-specific L1 capsid proteins used as antigens [30,31]. Based on the data presented in this report, the topological changes of the residual HPV L1 gene DNA when bound to the insoluble AAHS adjuvant seem to be genotype-related. For example, only the L1 gene DNA of the HPV-11, HPV-18 or HPV-16 has been detected, and the HPV-16 DNA needs a special method of amplification for its detection. Failure to detect HPV-6 DNA fragments may be due to a more effective removal of the HPV-6 L1 gene DNA residues from the antigen in the vaccine-manufacturing process; or the failure of detection may be caused by a special format of binding between the HPV-6 L1 gene DNA and the AAHS particles, a topological change that has turned both of its MY09 and MY11 binding sites into non-B-conformations. Since the physicochemical properties of AAHS depend on the conditions of precipitation [16-18], the dsDNA bound to the AAHS particles may vary in topology and may require different protocols for PCR amplification of different types of DNA in various vaccine batches.
Clinical trials have consistently demonstrated a higher anti-HPV-16 antibody response than an anti-HPV-18 antibody response following the use of the quadrivalent Gardasil® vaccine [32-34]. The mechanism for such a difference is poorly understood. The special conformational changes which can render the HPV-16 L1 gene DNA bound to the AAHS adjuvant less degradable in the human body than its HPV-18 or HPV-11 counterparts may provide an explanation for this difference. Codelivery of a DNA component in a protein vaccine with aluminum salts is known to stimulate a potent and multivalent immune response [35,36]. Intramuscular injection of free HPV-16 L1 plasmid DNA into BALB/C mice invariably induces a strong CD8 T cell response [18]. Persistence of the HPV-16 L1 gene DNA in a vaccinated person may play a role in boosting the typespecific immunogenicity of the HPV-16 capsid protein antigen via a not yet defined mechanism which warrants further investigation for vaccination efficacy and safety.
5. CONCLUSION
Naked HPV L1 gene DNA fragments bound to Al3+ in solid phase by ligand exchange have acquired a non-Bconformation. The resulting topological conformational changes may affect a region shared by many HPV genotypes, for example at the primer-binding site for the degenerate MY09 primer, or may affect a region of a specific genotype, for example at the degenerate MY11 primer-binding site of the HPV-16 L1 gene DNA. DNA conformational changes induced by a particulate aluminum-based adjuvant may have stabilized the residual HPV-16 L1 gene DNA fragments in an injectable vaccine and prevented their enzymatic degradation in a vaccine recipient.
6. ACKNOWLEDGEMENTS
This study was commissioned and sponsored by SANE-VAX, Inc. for a future payment not to exceed one US dollar. The author thanks Ms. Veronica S. Vigliotti and Ms. Jessica S. Vigliotti for donating their extremely valuable technical and professional time to assist completion of this study.
REFERENCES
- Vernon, S.D., Unger, E.R. and Williams, D. (2000) Comparison of human papillomavirus detection and typing by cycle sequencing, line blotting, and hybrid capture. Journal of Clinical Microbiology, 38, 651-655.
- Johnson, T., Bryder, K., Corbet, S. and Fomsgaard, A. (2003) Routine genotyping of human papillomavirus samples in Denmark. Acta Pathologica, Microbiologica et Immunologica Scandinavica, 111, 398-404. doi:10.1034/j.1600-0463.2003.t01-1-1110204.x
- Speich, N., Schmitt, C., Bollmann, R. and Bollmann, M. (2004) Human papillomavirus (HPV) study of 2916 cytological samples by PCR and DNA sequencing: Genotype spectrum of patients from the west German area. Journal of Clinical Microbiology, 53,125-128.
- Carvalho, N.O., del Castillo, D.M., Perone, C., Januário, J.N., de Melo, V.H. and Brasileiro, F.G. (2010) Comparison of HPV genotyping by type-specific PCR and sequencing. Memórias do InstitutoOswaldo Cruz, 105, 73- 78. doi:10.1590/S0074-02762010000100011
- Lee, S.H., Vigliotti, V.S., Vigliotti, J.S. and Pappu, S. (2007) Routine human papillomavirus genotyping by DNA sequencing in community hospital laboratories. Infectious Agents and Cancer, 2, 11.
- Lee, S.H. (2012) Guidelines for the use of molecular tests for the detection and genotyping of human papilloma virus from clinical specimens. Methods in Molecular Biology, 903, 65-101. doi:10.1007/978-1-61779-937-2_5
- Lee, S.H. (2012) Detection of human papillomavirus (HPV) L1 gene DNA possibly bound to particulate aluminum adjuvant in the HPV vaccine Gardasil®. Journal of Inorganic Biochemistry, 117, 85-92. doi:10.1016/j.jinorgbio.2012.08.015
- Lee, S.H. (2012) Detection of human papillomavirus L1 gene DNA fragments in postmortem blood and spleen after Gardasil® vaccination—A case report. Advances in Bioscience and Biotechnology, 3, 1214-1224.
- FDA Information on Gardasil (2011) Presence of DNA fragments expected, no safety risk. http://www.fda.gov/BiologicsBloodVaccines/Vaccines/ApprovedProducts/ucm276859.htm
- Lindblad, E.B. (2004) Aluminium compounds for use in vaccines. Immunology and Cell Biology, 82, 497-505. doi:10.1111/j.0818-9641.2004.01286.x
- Gherardi, R.K., Coquet, M., Cherin, P., Belec, L., Moretto, P., Dreyfus, P.A., Pellissier, J.F., Chariot, P. and Authier, F.J. (2001) Macrophagic myofasciitis lesions assess long-term persistence of vaccine-derived aluminium hydroxide in muscle. Brain, 124, 1821-1831. doi:10.1093/brain/124.9.1821
- Exley, C., Swarbrick, L., Gherardi, R.K. and Authier, F.J. (2009) A role for the body burden of aluminium in vaccine-associated macrophagicmyofasciitis and chronic fatigue syndrome. Medical Hypotheses, 72, 135-139. doi:10.1016/j.mehy.2008.09.040
- Gherardi, R.K. and Authier, F.J. (2012) Macrophagic myofasciitis: Characterization and pathophysiology. Lupus, 21,184-189. doi:10.1177/0961203311429557
- Manos, M.M., Ting, Y., Wright, D.K., Lewis, A.J., Broker, T.R. and Wolinsky, S.M. (1989) Use of polymerase chain reaction amplification for the detection of genital human papillomaviruses. Cancer Cells, 7, 209-214.
- Snijders, P.J., van den Brule, A.J., Schrijnemakers, H.F., Snow, G., Meijer, C.J. and Walboomers, J.M. (1990) The use of general primers in the polymerase chain reaction permits the detection of a broad spectrum of human papillomavirus genotypes. Journal of General Virology, 71, 173-181. doi:10.1099/0022-1317-71-1-173
- Burrell, L.S., Johnston, C.T., Schulze, D., Klein, J., White, J.L. and Hem, S.L. (2001) Aluminium phosphate adjuvants prepared by precipitation at constant pH, Part I: Composition and structure. Vaccine, 19, 275-281. doi:10.1016/S0264-410X(00)00160-2
- Burrell, L.S., Johnston, C.T., Schulze, D., Klein, J., White, J.L. and Hem, S. L. (2001) Aluminium phosphate adjuvants prepared by precipitation at constant pH. Part II: Physicochemical properties. Vaccine, 19, 282-287. doi:10.1016/S0264-410X(00)00162-6
- Caulfield, M.J. , Shi, L., Wang, S., Wang, B., Tobery, T. W., Mach, H., Ahl, P.L., Cannon, J.L., Cook, J.C., Heinrichs, J.H. and Sitrin, R.D. (2007) Effect of alternative aluminum adjuvants on the absorption and immunogenicity of HPV16 L1 VLPs in mice. Human Vaccines, 3, 139-146. doi:10.4161/hv.3.4.4309
- McCormack, P.L. and Joura, E.A. (2010) Quadrivalent human papillomavirus (types 6, 11, 16, 18) recombinant vaccine (Gardasil®): A review of its use in the prevention of premalignant genital lesions, genital cancer and genital warts in women. Drugs, 70, 2449-2474. doi:10.2165/11204920-000000000-00000
- Health Canada (2007) Summary basis of decision (SBD) GardasilTM. http://www.hc-sc.gc.ca/dhp-mps/alt_formats/hpfb-dgpsa/pdf/prodpharma/sbd_smd_2007_gardasil_102682-eng.pdf
- Egan, P.M., Belfast, M.T., Giménez, J.A., Sitrin, R.D. and Mancinelli, R.J. (2009) Relationship between tightness of binding and immunogenicity in an aluminumcontaining adjuvant-adsorbed hepatitis B vaccine. Vaccine, 27, 3175-3180. doi:10.1016/j.vaccine.2009.03.054
- Zhang, R.-Y., Liu, Y., Pang, D.-W., Cai, R.-X. and Qi, Y.-P. (2002) Spectroscopic and voltammetric study on the binding of aluminium(III) to DNA. Analytical Sciences, 18, 761-766. doi:10.2116/analsci.18.761
- Ge, S., Gong, B., Cai, X., Yang, X., Gan, X., Tong, X., Li, H., Zhu, M., Yang, F., Zhou, H. and Hong, G. (2012) Prevent cervical cancer by screening with reliable human papillomavirus detection and genotyping. Cancer Medicine, 1, 59-67. doi:10.1002/cam4.9
- Lee, S.H., Vigliotti, V.S., Vigliotti, J.S. and Pappu, S. (2009) Validation of human papillomavirus genotyping by signature DNA sequence analysis. BMC Clinical Pathology, 9, 3. doi:10.1186/1472-6890-9-3
- Karlik, S.J., Eichhorn, G.L., Lewis, P.N. and Crapper, D.R. (1980) Interaction of aluminum species with deoxyribonucleic acid. Biochemistry, 19, 5991-5998. doi:10.1021/bi00567a008
- Latha, K.S., Anitha, S., Rao, K.S. and Viswamitra, M.A. (2002) Molecular understanding of aluminum-induced topological changes in (CCG)12 triplet repeats: Relevance to neurological disorders, Biochimica et Biophysica Acta, 1588, 56-64. doi:10.1016/S0925-4439(02)00133-3
- Andre, P., Kim, A., Khrapko, K. and Thilly, W.G. (1997) Fidelity and mutational spectrum of Pfu DNA polymerase on a human mitochondrial DNA sequence. Genome Research, 7, 843-852.
- Lindahl, T. and Nyberg, B. (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry, 11, 3610- 3618. doi:10.1021/bi00769a018
- Lindahl, T. and Nyberg, B. (1974) Heat-induced deamination of cytosine residues in deoxyribonucleic acid. Biochemistry, 13, 3405-3410. doi:10.1021/bi00713a035
- Mach, H., Volkin, D.B., Troutman, R.D., Wang, B., Luo, Z., Jansen, K.U. and Shi, L. (2006) Disassembly and reassembly of yeast-derived recombinant human papillomavirus virus-like particles (HPV VLPs). Journal of Pharmaceutical Sciences, 95, 2195-2206. doi:10.1002/jps.20696
- Bryan, J.T. (2007) Developing an HPV vaccine to prevent cervical cancer and genital warts. Vaccine, 25, 3001- 3006. doi:10.1016/j.vaccine.2007.01.013
- Einstein, M.H., Baron, M., Levin, M.J., Chatterjee, A., Edwards, R.P., Zepp, F., Carletti, I., Dessy, F.J., Trofa, A.F., Schuind, A. and Dubin, G. and HPV-010 Study Group (2009) Comparison of the immunogenicity and safety of Cervarix and Gardasil human papillomavirus (HPV) cervical cancer vaccines in healthy women aged 18 - 45 years. Human Vaccines, 5, 705-719. doi:10.4161/hv.5.10.9518
- Giuliano, A.R., Lazcano-Ponce, E., Villa, L., Nolan, T., Marchant, C., Radley, D., Golm, G., McCarroll, K., Yu, J., Esser, M.T., Vuocolo, S.C. and Barr, E. (2007) Impact of baseline covariates on the immunogenicity of a quadrivalent (types 6, 11, 16, and 18) human papillomavirus virus-like-particle vaccine. Journal of Infectious Diseases, 196, 1153-1162. doi:10.1086/521679
- Villa, L.L., Ault, K.A., Giuliano, A.R., Costa, R.L., Petta, C.A., Andrade, R.P., Brown, D.R., Ferenczy, A., Harper, D.M., Koutsky, L.A., Kurman, R.J., Lehtinen, M., Malm, C., Olsson, S.E., Ronnett, B.M., Skjeldestad, F.E., Steinwall, M., Stoler, M.H., Wheeler, C.M., Taddeo, F.J., Yu, J., Lupinacci, L., Railkar, R., Marchese, R., Esser , M.T., Bryan, J., Jansen, K.U., Sings, H.L., Tamms, G.M., Saah, A.J. and Barr, E. (2006) Immunologic responses following administration of a vaccine targeting human papillomavirus types 6, 11, 16, and 18. Vaccine, 24, 5571-5583. doi:10.1016/j.vaccine.2006.04.068
- Kwissa, M., Lindblad, E.B., Schirmbeck, R. and Reimann, J. (2003) Codelivery of a DNA vaccine and a protein vaccine with aluminum phosphate stimulates a potent and multivalent immune response. Journal of Molecular Medicine, 81, 502-510. doi:10.1007/s00109-003-0452-9
- Davis, H.L., Weeratna, R., Waldschmidt, T.J., Tygrett, L., Schorr, J. and Krieg, A.M. (1998) CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. Journal of Immunology, 160, 870-876.
LIST OF ABBREVIATIONS
AAHS: Amorphous Aluminum Hydroxyphosphate Sulfate;
HPV: Human Papillomavirus;
BLAST: Basic Local Alignment Search Tool;
dNTP: Deoxynucleotide Triphosphates;
dsDNA: Double-Stranded DNA;
PCR: Polymerase Chain Reaction.