Advances in Biological Chemistry
Vol. 2 No. 4 (2012) , Article ID: 24812 , 7 pages DOI:10.4236/abc.2012.24049
Determination of bacterial viability by selective capture using surface-bound siderophores
![]()
1Biosciences Division, Los Alamos National Laboratory, Los Alamos, USA
2Chemistry Division, Los Alamos National Laboratory, Los Alamos, USA
3ISR3, Space Data Systems, Los Alamos National Laboratory, Los Alamos, USA
Email: *harshini@lanl.gov
Received 18 August 2012; revised 24 September 2012; accepted 30 September 2012
Keywords: Siderophores; Viability Detection; Desferrioxamine; Fluorescence Microscopy; Self-Assembled Monolayers
ABSTRACT
A significant challenge in bacterial detection is the identification of viable bacteria over debris, specifically post decontamination. Of increasing concern are antibiotic resistant strains that require accurate and rapid post decontamination analysis. Current strategies are fraught with disadvantages and most of them are not selective for viable bacteria. However, bacteria are critically dependent upon iron sequestration, synthesizing and releasing siderophores (SDPs) to tightly bind iron, with the subsequent uptake of iron bound SDPs. This is a highly conserved process that occurs only in intact bacteria. Herein we report a facile method to use bacterial SDPs to selectively and rapidly identify only viable bacteria in complex matrices, and discriminate them from their dead counterparts. Desferrioxamine B (Desf B) tethered to a glass slide is used to specifically capture viable bacteria from a mixture of viable and dead Escherichia coli, as demonstrated by fluorescence microscopy. We report both direct conjugation of Desf B on thin-filmcoated glass slides as well as biotin-streptavidin conjugation strategies, both of which are successful in the said goal. We have analyzed the density of images obtained upon fluorescence staining using edge detection with a Canny edge detector. This novel application of a software analysis tool originally developed for satellite imaging to biological staining allows for accurate quantitation of observed data.
1. INTRODUCTION
Recent Escherichia coli (E. coli) outbreaks in Europe have resulted in over 4300 cases of infection leading to 50 deaths [1]. Erroneous identification led to blanket bans on vegetables from the European Union, an action that had devastating economic consequences (billions of US dollars) [2]. These outbreaks and their associated costs have emphasized the need for improved detection methods to rapidly assess viable pathogens for food safety. Current diagnostic techniques are obviously inadequate. Real-time PCR and culture confirmation for viability are laboratory intensive processes requiring at least 18 hours to complete [3]. Antibody-based methods can offer greater speed but are associated with a high rate of false positives, [4,5] possibly due to the rapid mutation rates of pathogenic strains, or the fact that antibodies cannot always distinguish between debris and viable pathogens. Furthermore, antibodies suffer from instability upon prolonged storage and use in ambient environments. Rapid and selective interrogation of bacterial viability is critical to many fields besides food safety, namely rapid determination of the efficiency of a decontamination process, efficacy assessment after initial medical intervention to infection, and detection of exposure to a biological threat agent (e.g., in enclosed buildings).
Using siderophores (SDPs)—molecules employed by the bacteria to sequester vital iron within a host—show promise as a way to selectively interrogate for viable bacteria [6]. Iron is a key nutrient for survival and free iron is rare in nature [7]. Bacteria synthesize and release SDPs to bind iron with a remarkably high affinity, up to 1052 M–1 [8]. Iron-bound SDPs are recognized by specific cell surface receptors and internalized to the periplasm, where processing and degradation transfers the iron to the bacterial cell [9]. This nutrient sequestration route is absolutely necessary for bacterial survival.
Recent studies have utilized immobilized SDPs to identify pathogens and the receptors that bind the iron transporting species [10,11], highlighting the effective use of surface bound SDP’s. Furthermore, use of SDPsupported particles has been reported for aqueous iron detection at low concentrations [12], exhibiting SDP utility in solid support detection platforms. Doorneweerd et al. [10] reported selective capture of Pseudomonas aeruginosa using the SDP pyoverdine, attached to bovine serum albumin (BSA) scaffold non covalently printed on a polydimethylsiloxane lithographed surface. More recently a Desferrioxamine B (Desf B)/BSA scaffold has been used for effective detection and viability determination of Yersinia, impressively detecting down to 103 CFU/ml [13]. However, to use SDPs for the detection of viable organisms, effective surface functionalization strategies are required such that a tethered SDP can retain its structural integrity and ability to bind the intact pathogen, and non-specific interactions associated with complex samples are effectively minimized through more rigorous washing. We have shown that the selfassembled monolayers used herein for surface functionalization are intrinsically resistant to non-specific interactions associated with complex biological samples such as culture filtrate, serum and urine, and are compatible to washing with detergents and chemicals to minimize irrelevant surface interactions [14]. Previous methods for assessment of pathogen viability have involved evaluation of binding of the bacterium to immobilized antibodies [15,16], a method that suffers from intrinsic issues associated with antibody-dependent assays (specificity, sensitivity).
The method reported herein differentiates from that of Kim et al. [13] by employing three distinct innovations:
1) Covalent SDP attachment to the glass surface, which leads to greater structural integrity, and allows significantly simpler technology; 2) An innovative method to quantify fluorescence imaging, namely by using a software package initially developed for interpreting satellite images, and 3) Use of surface functionalization chemistry that facilitates direct tethering of a variety of SDPs while minimizing non-specific binding.
We elected to assess this method with Desf B, a bacterial SDP characteristic of Streptomyces species, although it is recognized by various bacteria including E. coli [17]. It is a well studied linear SDP with three hydroxamate iron binding epitopes to coordinate in a hexadentate sphere [18]. The binding of Desf B to the E. coli SDP receptor, FhuD, is well documented, including the reported crystal structure of Desf B bound to FhuD [19]. Desf B is commercially available as the mesylate salt and is used for iron chelation to treat iron poisoning and hemochromatosis [20]. Synthesis of Desf B: antibiotic conjugates to deliver therapeutics in a “Trojan horse” manner has also been accomplished [21].
We herein report a robust, facile and rapid method for the detection of viable bacteria using a tethered SDP capture (Figure 1). Demonstrated for the detection of E. coli, the method can theoretically be adapted to all bacterial pathogens by choosing the appropriate SDP molecules.
2. MATERIALS AND METHODS
2.1. Materials
Glass microscope slides (“Fisher’s Finest”) were purchased from Thermo-Fisher (NJ, USA). Mesylated salt of Desf B and Diglycolic anhydride was obtained from Sigma Aldrich (MO, USA). Ethanol was obtained from Pharmco/Aaper (CT, USA). All other solvents and buffers were obtained from Sigma-Aldrich or Thermo-Fisher. 3-Aminopropylmethyldiethoxysilane (APMDES) was purchased from Gelest (PA, USA), while polyethylene glycol (PEG) reagents were obtained from Quanta Biodesign® (OH, USA); these materials were used as received without further purification. Live/Dead BacLight-TM bacterial viability kit was purchased from Invitrogen. Luria-Bertani Agar media and all other cell culture requirements were obtained from Invitrogen Photosciences, Inc. (CA, USA). Fluorescence microscopy was used to characterize the live/dead staining (Olympus IX81 microscope with FITC band-pass, (DIFCO, NY,
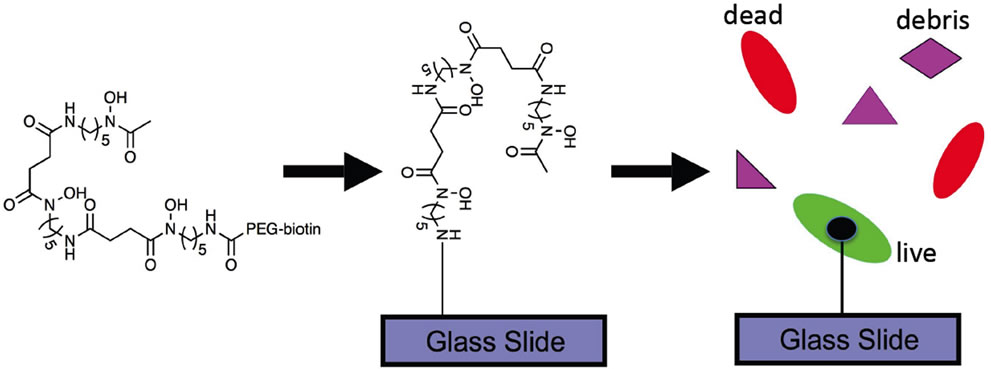
Figure 1. Schematic representation of the proposed approach. SDPs (Desf) are tethered to functionalized glass slides and then exposed to a mixture of viable and dead E. coli. Only living E. coli bind to the SDPs, allowing for determination of bacterial viability.
USA). The UV photomask was designed at the Los Alamos National Laboratory and fabricated by Photosciences, Inc. (CA, USA). Fluorescence microscopy was used to characterize the live/dead staining (Olympus IX81 microscope with FITC band-pass, TRITC bandpass, and FITC long-pass filter sets, and a DP71 camera) (Olympus America Inc. (PA, USA)).
2.2. Slide Preparation
Due to the commercial abundance of and facile functionalization of Desf B, we chose to evaluate it as a model SDP for the selective capture of viable E. coli, on a robust, chemically modified surface. Two methods were tested (Figures 2 and 3). First, the SDP was covalently tethered to the glass surface (direct method) and used to capture viable bacteria (Figure 2). The second (indirect) method (Figure 3) relied on the familiar biotin-streptavidin conjugation, where the biotinylated SDP is captured on a streptavidin-coated glass surface.
In both the methods, patterned, PEG-modified thin films were prepared as described previously [14]. Briefly, microscope slides were rinsed with ethanol, blown dry, and exposed to UV-ozone for 30 min. APMDES was vapor deposited onto the cleaned microscope slides, followed by rinsing with ethanol, drying under a stream of argon, and analysis by contact angle. The resulting amines were modified with a mixture of two discrete PEG chains, one with a methoxy group for non-specific binding resistance, and one with an fluorenylmethoxycarbonyl (Fmoc) protected amine for functionality.
The attachment was carried out using standard peptide coupling methods (benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP), N,N-diisopropylethylamine (DIEA), and N-methylpyrrolidinone (NMP)) at room temperature, overnight. After rinsing with NMP, acetone, and ethanol; drying under a stream of argon; and contact angle measurement; the Fmocgroups were removed (20% piperidine/NMP, 2 × 15 min). The slides were rinsed thoroughly with NMP, and the resulting amines were capped with either a carboxylic acid using diglycolic anhydride (for direct binding) or biotin using N-hydroxysuccinimidylbiotin (for indirect biotin/streptavidin mediated capture). Both reactions utilized DIEA in NMP. The slides were rinsed thoroughly (NMP, acetone, ethanol) and dried under a stream of argon. Irradiation of the slides with UV/O3 through a chrome photomask (500 μm feature size) removed the thin film in certain areas while leaving the carboxylic acidor biotin-terminated film in the masked areas. The cleaned areas were “backfilled,” i.e., reacted with another portion of APMDES and the methoxy-terminated PEG chain, to create patterned surfaces. This procedure allowed for the incorporation of an intrinsic negative control (in the regions that only contained methoxy groups) while controlling the number of reactive groups present on the surfaces (by the percentage of Fmocamine-protected PEG reagent included in the PEG deposition solution). The subsequent reactants could only conjugate to the portions of the film that contain the carboxylic acid terminus/biotin functionality (circular areas in Figures 2 and 3), but not in the areas with only methoxy-terminated SAM (remaining area of the glass slide in Figures 2 and 3). In previous studies using a waveguide-based method, we determined that methoxy-ter-

Figure 2. Schematic description of direct conjugation approach for the detection of viable E. coli. Microscopic images at 10× magnification, one square of the patterned area is shown. Green staining indicates binding of viable E. coli (106 CFU/ml) to the functionalized glass slides, and red staining shows binding (or lack thereof) of dead bacteria.

Figure 3. Description of the indirect conjugation approach (using biotin-streptavidin chemistry) and results. Glass slides were patterned with biotinylated silane-based thin film chemistry. Biotin-streptavidin conjugation was used to attach biotinylated Desf to the glass slides, which were exposed to a mixture of differentially stained viable (green) and dead (red) E. coli (107 CFU/ml). Fluorescence microscopy clearly indicates selective binding of only viable bacteria to the patterned areas of the slide (containing Desf), Microscopic images at 4× magnification, demonstrating functionalized areas, and control background on the patterned glass slides.
minated SAMs as functional surfaces minimized nonspecific interactions in complex biological samples [14].
Both attachment strategies utilized the terminal amine of Desf B, which is not critical to iron binding. For direct tethering (Figure 2), patterned glass slides with carboxylic acid-terminated regions were incubated in an aqueous solution of ethyl dimethyl aminopropyl carbodiimide (EDC) and N-hydroxysuccinimide (NHS) (4:1) for 30 min, and were thoroughly washed with water. The slides were immersed in a solution containing Desf B, sodium carbonate, and FeCl3 (1 mM each) for 2 - 3 h at room temperature and then subsequently rinsed with water, which resulted in direct tethering of Desf B on the patterned areas of the slides.
For indirect tethering (biotin-streptavidin chemistry), the glass slides with biotinterminated regions were incubated in streptavidin solution (2 mM) for 10 min, and were thoroughly washed. The biotinylated Desf B, or E. coli incubated with biotinylated Desf B was then added on to the functionalized glass slides to bind the SDP on the patterned glass slide.
2.3. Binding Evaluation
For the evaluation of specific binding, E. coli was grown overnight in Luria Bertani media at 37˚C. The bacterial cells were harvested, re-suspended in phosphate buffered saline (PBS), and split into two portions. To one portion of the bacterial suspension, isopropanol (80%) was added to kill the bacterial cells. After incubation for 2 hrs, the cell suspension was washed thoroughly with PBS. This method is expected to kill 100% of bacterial cells. The dead cell lysates were not clarified and contain SDP receptor residues and other cell components.
To confirm this expectation, assessment of viability was confirmed by incubation (48 hr) in Luria Bertani agar media at 37˚C for both live and dead bacterial suspensions. Live and dead bacterial suspensions were mixed in equal proportion, and labeled using a Live/ Dead Bac-LightTM bacterial viability kit that differentially stains living and dead bacterial cells. Bac-LightTM kit consists of two nucleic acid stains, SYTO 9 and propidium iodide. SYTO 9 (green) penetrates most of the membranes freely whereas propidium iodide (red) only penetrates the damaged membranes and also reduces SYTO 9 fluorescence when both the dyes are present. Using this method, viable cells are dyed green, and dead cells, red. Live/dead E. coli labeled mixture (at different dilutions (CFU/ml)) was added to functionalized glass slides, and incubated for 45 min in a hydration chamber at room temperature. The glass slides were then washed with PBS or PBS with 0.01% Tween-20 to remove the unbound bacteria (1 ml wash volume, 2 washes), and imaged using fluorescence microscopy (Figures 2-4). This allows for the removal of non-specifically associated components, while preserving any covalent interactions of relevance to the assay. To test the sensitivity of the method, Live/dead E. coli labeled mixture was serially diluted in PBS and was added on to the glass slides. The bacterial suspensions were plated on the agar plate to obtain colony-forming units per ml (CFU/ml), which indicates the limit of detection of the assay.
2.4. Fluorescence Microscopy Quantification
The fluorescence images collected upon SDP capture of E. coli were subjected to edge detection using a Canny edge detector for identification of edge pixels around each site. A Delaunay triangulation of the edge points was computed to obtain proximity graph. Triangle edges of length greater than three pixel units were rejected to obtain polygons that tightly bound the sites (Figure 4(b)). As a novel adaptation of the software, an interactive input was used to provide known mask areas for enclosure of site polygons as well as sampled background areas, based on the patterning lattice. The ratio of the sum of the areas of the sites contained within a mask to the area of the mask was taken as the density of the sites in the mask, and used to develop the data represented in Figure 4(c).
3. RESULTS AND DISCUSSION
3.1. Direct Conjugation of Desf B to Glass Slides
Direct conjugation of Desf B was highly effective in the capture of viable bacteria (Figure 2). This image shows a single patterned area of the glass slide taken through a 10x objective. Since uptake of the SDP-iron complex is an active process, only viable pathogens bind to the functionalized glass slides. Furthermore, viable bacteria (green) were bound predominantly in the patterned areas of the glass slides, with little binding to the control areas. There was also significantly reduced binding of dead bacteria (red) to the patterned areas, indicating a selective determination of viability using this approach. Minimal non-specific interaction was observed owing to complex media (culture media without bacteria, bovine growth serum, data not shown), further demonstrating the specificity of this approach. The current limit of detection of binding to live cells was determined to be 105 CFU/ml, however efforts are underway to improve this sensitivity. The sensitivity of the assay can possibly be improved by increasing the reactive portion on the glass surface, which is currently 1%. A major advantage of this technique is the rapid result time (minutes, compared to days by culture). Any technique that occurs this quickly could be used at the point of detection, and could alleviate undue panic in the case of an outbreak or bio-weapon release. This is also true of evaluation of the efficacy of a decontamination protocol.
We used fluorescence microscopy for the feasibility demonstration shown here, but the approach can conceptually be integrated into other readout formats. The approach is directly compatible with reagent-free methods like interferometry and impedance-based detection. With the use of fluorescently labeled reporter ligands (antibody or aptamers), the binding of viable bacteria to surface SDPs can be read in any fluorescence or absorbance reader with appropriately functionalized plates.
3.2. Indirect Conjugation of Desf B to Glass Slides
With the indirect tethering strategy (Figure 3) using biotin-streptavidin conjugation, two methodologies were evaluated: first, biotinyl-Desf B was attached to streptavidin-coated thin films, then labeled E. coli was added

Figure 4. Representative processing of a fluorescence microscopy image using Canny edge detection. Panel A shows the fluorescence microscope image (4× magnification) of specific staining for live bacteria using direct Desf B capture. Panel B demonstrates edge detection of the image using a Canny edge detector, resulting in the identification of edge pixels around each site. Panel C demonstrates the conversion of the measured pixel intensity to spot density. Black bars indicate the quantitative spot density for the patterned areas when the slides were stained for viable (green) and dead (red) bacteria (107 CFU/ml). Clear bars indicate background staining under the same conditions.
and incubated. Second, biotinyl-Desf B was pre-incubated with labeled E. coli, and the mixture was added to the streptavidin-coated thin films. Both methods were able to selectively capture viable E. coli. However, the pre-incubation method resulted in less non-specific binding (Figure 3) which may be due to decreased distribution of surface SDPs. In all cases, binding of dead cells was significantly lower, and was further reduced by robust washing with detergent-containing buffers (e.g., 0.01% Tween-20). The limit of detection for the indirect method was determined to be 106 CFU/ml. One of the reasons for exploring this strategy was to create universal chips that can be used for detection of a variety of bacteria simply by varying the biotinylated-SDP used. However, this method resulted in greater non-specific interaction of E. coli with the biotinylated surface, indicating direct, binding of E. coli to the biotinylated surface, even in the absence of Desf B. Because of this observation, coupled with the fact that carboxylate-based attachment improved sensitivity, the direct conjugation method is preferred.
3.3. Quantification of E. coli on the Glass Slides
Our quantification strategy (Figure 4, and described above) sets our method apart from others. Using software initially designed for satellite imaging, fluorescent areas are counted [22,23]. Fitting the data to the model indicated a significantly greater attachment of live cells than dead cells to the patterned surface. Viewing these images by the naked eye suggest almost exclusive binding of live (green) bacteria; upon quantification the dead (red) bacteria are definitely present, even though their concentration is much lower than their live counterparts. This is partly due to the human eye having greater sensitivity to the color green. Efforts are currently ongoing for the optimization and correction for threshold intensity, time of exposure and other parameters, which will allow for the better quantitation of such results.
4. CONCLUSION
In summary, we have developed a universal, simple yet robust platform that can distinguish between live and dead bacteria using the SDP-mediated bacterial iron uptake mechanism. The novel aspects of this approach are 1) use of bacterial SDPs for determination of viability; 2) use of self-assembled monolayer chemistry for surface functionalization that intrinsically resists non-specific interactions in complex biological samples; 3) flexibility of functionalization chemistry to multiple tethering strategies; 4) incorporation of an intrinsic control with patterned surfaces for accurate data interpretation; and 5) unique adaptation of a software application for the rapid and accurate translation of the observed images to quantitative data, with statistical relevance. Fluorescence microscopy is used as the readout in the current study, but the approach is not limited to any single transduction strategy. Although the method is universal in that all bacterial pathogens secrete SDPs, the widespread application of this approach relies on the availability of SDPs, either from commercial sources or using synthetic strategies. It is likely that a wider variety of SDPs will become available as SDPs become an essential part of biological detection; by sharing our strategy, we hope to help drive this innovation.
A simple method for viable bacterial capture can have far reaching application for pathogen detection, especially for identifying medical and food borne pathogens, and in assessing effectiveness of decontamination methods. Without the “live or dead” ambiguity, use of antibody recognition should be thoroughly revitalized, as after SDP capture, recognition can take place with the knowledge of observing a known live pathogen. Efforts are currently underway to apply this technology to further pathogens, develop multiplex readout assays, and provide a useable platform for application of a rapid assay to assess bacterial viability.
5. ACKNOWLEDGEMENTS
We thank the Los Alamos LDRD Exploratory Research (HM and JS) program for funding. The authors thank Drs. Basil I. Swanson and Jennifer S. Martinez, Los Alamos National Laboratory, for technical suggestions and discussions.
![]()
![]()
REFERENCES
- Robert, K. (2011) EHEC/HUS O104:H4: The outbreak is considered to be over. http://www.rki.de/EN/Home/PM_EHEC.html
- Flynn, D. (2011) Germany’s E. coli outbreak most costly in history. Food Safety News. http://www.foodsafetynews.com/2011/06/europes-o104-outbreak---most-costly-in-history/
- Bustin, S.A., Benes, V., Garson, J.A., Hellemans, J., Huggett, J., Kubista, M., Mueller, R., Nolan, T., Pfaffl, M.W., Shipley, G.L., Vandesompele, J. and Wittwer, C.T. (2009) The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clinical Chemistry, 55, 611-622. doi:10.1373/clinchem.2008.112797
- Drake, J.W. (1993) Rates of spontaneous mutation among RNA viruses. Proceedings of the National Academy of Sciences USA, 90, 4171-4175. doi:10.1073/pnas.90.9.4171
- Hensley, S.E., Das, S.R., Bailey, A.L., Schmidt, L.M., Hickman, H.D., Jayaraman, A., Viswanathan, K., Raman, R., Sasisekharan, R., Bennink, J.R. and Yewdell, J.W. (2009) Hemagglutinin receptor binding avidity drives influenza a virus antigenic drift. Science, 326, 734-736. doi:10.1126/science.1178258
- Ratledge, C. and Dover, L.G. (2000) Iron metabolism in pathogenic bacteria. Annual Review of Microbiology, 54, 881-941. doi:10.1146/annurev.micro.54.1.881
- Neilands, J.B. (1995) Siderophores: Structure and function of microbial iron transport compounds. The Journal of Biological Chemistry, 270, 26723-26726.
- Carrano, C.J. and Raymond, K.N. (1979) Ferric Ion Sequestering Agents. 2. Kinetics and mechanism of iron removal from transferrin by enterobactin and synthetic tricatechols. Journal of the American Chemical Society, 101, 5401-5404. doi:10.1021/ja00512a047
- Braun, V. and Hantke, K. (2011) Recent insights into iron import by bacteria. Current Opinion in Chemical Biology, 15, 328-334. doi:10.1016/j.cbpa.2011.01.005
- Doorneweerd, D.D., Henne, W.A., Reifenberger, R.G. and Low, P.S. (2010) Selective capture and identification of pathogenic bacteria using an immobilized siderophore. Langmuir, 26, 15424-15429. doi:10.1021/la101962w
- Bugdahn, N., Peuckert, F., Albrecht, A.G., Miethke, M., Marahiel, M.A. and Oberthur, M. (2010) Direct identification of a siderophore import protein using synthetic petrobactin ligands. Angewandte Chemie International Edition, 49, 10210-10213. doi:10.1002/anie.201005527
- Roy, E.G., Jiang, C.H., Wells, M.L. and Tripp, C. (2008) Determining subnanomolar iron concentrations in oceanic seawater using a siderophore-modified film analyzed by infrared spectroscopy. Analytical Biochemistry, 80, 4689- 4695.
- Kim, Y., Lyvers, D.P., Wei, A., Reifenberger, R.G. and Low, P.S. (2012) Label-free detection of a bacterial pathogen using an immobilized siderophore, deferoxamine. Lab on a Chip, 12, 971-976. doi:10.1039/c2lc20904g
- Anderson, A.S., Dattelbaum, A.M., Montano, G.A., Price, D.N., Schmidt, J.G., Martinez, J.S., Grace, W.K., Grace, K.M. and Swanson, B.I. (2008) Functional PEG-modified thin films for biological detection. Langmuir, 24, 2240- 2247. doi:10.1021/la7033438
- Suo, Z.Y., Avci, R., Yang, X.H. and Pascual, D.W. (2008) Efficient immobilization and patterning of live bacterial cells. Langmuir, 24, 4161-4167. doi:10.1021/la7038653
- Suo, Z.Y., Yang, X.H., Avci, R., Deliorman, M., Rugheimer, P., Pascual, D.W. and Idzerda, Y. (2009) Antibody Selection for Immobilizing Living Bacteria. Analytical Biochemistry, 81, 7571-7578.
- Challis, G.L. (2005) A widely distributed bacterial pathway for siderophore biosynthesis independent of nonribosomal peptide synthetases. ChemBioChem, 6, 601-611. doi:10.1002/cbic.200400283
- Keberle, H. (1964) The biochemistry of desferrioxamine and its relation to iron metabolism. Annals of the New York Academy of Sciences, 119, 758-768. doi:10.1111/j.1749-6632.1965.tb54077.x
- Clarke, T.E., Braun, V., Winkelmann, G., Tari, L.W. and Vogel, H.J. (2002) X-ray crystallographic structures of the Escherichia coli periplasmic protein FhuD bound to hydroxamate-type siderophores and the antibiotic albomycin. The Journal of Biological Chemistry, 277, 13966- 13972. doi:10.1074/jbc.M109385200
- Miller, M.J. (1989) Syntheses and therapeutic potential of hydroxamic acid based siderophores and analogs. Chemical Reviews, 89, 1563-1579. doi:10.1021/cr00097a011
- Roosenberg, J.M., Lin, Y.M., Lu, Y. and Miller, M.J. (2000) Studies and syntheses of siderophores, microbial iron chelators, and analogs as potential drug delivery agents. Current Medicinal Chemistry, 7, 159-197. doi:10.2174/0929867003375353
- Prasad, L. and Skourikhine, A.N. (2006) Vectorized image segmentation via trixel agglomeration. Pattern Recognition, 39, 501-514. doi:10.1016/j.patcog.2005.10.014
- Prasad, L. (2007) Rectification of the chordal axis transform skeleton and criteria for shape decomposition. Image and Vision Computing, 25, 1557-1571.
NOTES
*Corresponding author.