American Journal of Molecular Biology
Vol.2 No.3(2012), Article ID:20988,11 pages DOI:10.4236/ajmb.2012.23028
Suppression of Sup35 amyloid fibril formation by group II chaperonin from Thermoplasma acidophilum
![]()
1Department of Biomedical Science, Institute of Regenerative Medicine and Biofunction, Graduate School of Medical Science, Tottori University, Tottori, Japan
2Department of Chemistry and Biotechnology, Graduate School of Engineering, Tottori University, Tottori, Japan
Email: *kawata@bio.tottori-u.ac.jp
Received 11 April 2012; revised 8 May 2012; accepted 23 May 2012
Keywords: Group II Chaperonin Monomer; Thermoplasma acidophilum; Structure and Function; Suppression of Amyloid Fibril; Sup35NM Amyloid
ABSTRACT
The Group II chaperonin from Thermoplasma acidophilum was added to the in vitro amyloid fibrillation reaction of yeast Sup35NM protein to assess its effects. By measuring the formation of Sup35NM fibrils in real time using the fluorescent dye Thioflavin T, we found that the addition of T. acidophilum-cpn α16, α1, and β1 proteins suppressed fibril formation. Addition of a 0.1 molar-equivalent T. acidophilum-cpn α16 relative to Sup35NM prolonged the initial lagtime of fibril formation and decreased the rate of fibril extension. Addition of 1 or 3 molar-equivalents of T. acidophilum-cpn monomers also produced a similar effect. Delayed addition of these chaperonins after the initial lag phase did not suppress fibril formation. Interestingly, these effects were also observed upon adding only the apical domain segments of α and β-subunits, and we also found that deletion of the helical protrusion in the apical domain of these segments led to an abolishment of the suppression effects. A synthetic peptide whose sequence corresponded to the helical protrusion also displayed a suppression effect, which indicated that archaeal group II chaperonin binds to Sup35NM through the helical protrusion of the apical domain. These findings suggest that group II chaperonin might be actively involved in suppressing amyloid fibril formation, in addition to acting as a protein folding assistant.
1. INTRODUCTION
The irreversible aggregation and fibrillation of various proteins in the cell, implicated in various neurological diseases, is a phenomenon that is currently being researched extensively, with a view to developing an eventual treatment or method of prevention for such diseases. In the cellular context, the molecular chaperones, an endogenous group of proteins that are postulated to specifically interact with and prevent the association of denatured proteins, are likely and obvious candidates for studies related to protein fibrillation. Various members of the molecular chaperone family, such as Hsp104, Hsp70, and Hsp40, have been shown to interact with proteins that form amyloid fibrils and in certain cases prevent their fibrillation [1-3] . Notably, studies involving the effects of the chaperonins (Hsp60) on various fibrilforming proteins have been relatively scarce to date, considering the broad and wide effects that this subgroup of the molecular chaperones have on the overall maintenance of cellular protein solubility and stability in general. It was reported that Escherichia coli GroEL chaperonin binds prion protein and induces a protease-resistant aggregation, rather than suppression [4] of fibrillation. Also, TCP-1 ring complex (called TRiC or CCT) from eukaryotic cytosols suppress the huntingtin protein aggregation by binding to a short sequence element of the N-terminal poly-Q tract [5,6] of this protein. Although the structural and functional features between GroEL and group II chaperonins are closely related [7- 12] , there seem to be differences in their interactions with other proteins as well as their effects in suppressing protein aggregation.
Chaperonins form large oligomeric complexes, and assist protein synthesis by folding nascent polypeptides in an ATP-dependent manner [13-18] . Group I chaperonins such as GroEL from E. coli are generally homooligomeric, and assemble heptameric rings stacked back to back. Group I chaperonins require association with a co-chaperonin such as GroES to isolate and fold substrate protein [19,20] . In the eukaryotic cytosol and various archaeal organisms, group II chaperonins that form homo-oligomeric or hetero-oligomeric molecules may be found. The group II chaperonins share the same overall subunit architecture with group I chaperonins, such as an equatorial domain that is responsible for ATP-binding and signal transduction between rings, an apical domain that binds substrate proteins and the co-chaperonins such as GroES, and an intermediate domain that links these two domains together [12] . However, when compared in more detail with the group I chaperonins such as E. coli GroEL, many aspects of archaeal group II chaperonins are still unclear. A most prominent structural difference between group I and group II chaperonins is the presence of a long helical protrusion in the apical domain of group II chaperonins, which is used for encapsulation of substrate protein [21]. This protrusion is said to substitute for the co-chaperonin (GroES) of the group I system.
The group II chaperonin from Thermoplasma acidophilum (Ta-cpn) is composed of two stacked eightmembered rings of alternating α- and β-subunits [22] whose amino acid sequences are approximately 60% identical [12,23] (Figure 1). In both crystal structural studies [24] and NMR studies [25], a difference in secondary structure has been found between the α- and β-subunits in the vicinity of the helical protrusion of the
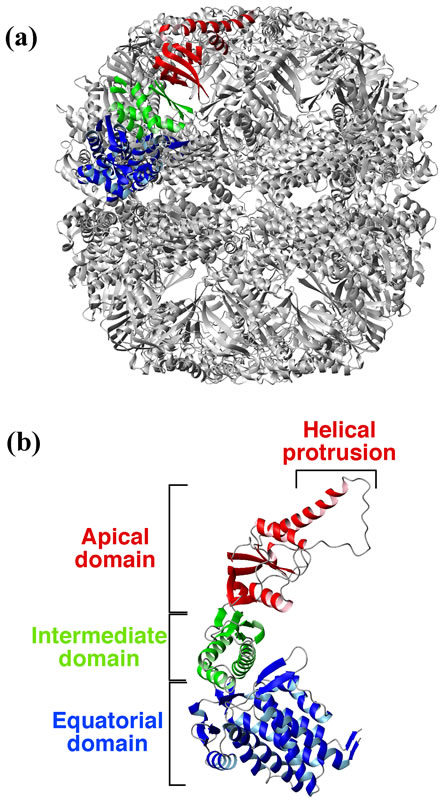
Figure 1. Structure of group II chaperonin from Thermoplasma acidophilum. The figures were generated using MOLMOL [26] and coordinates from PDB file 1A6D ADDIN EN.CITE ADDIN EN.CITE.DATA [12] , and are shown as ribbon models. The apical domain is indicated in red, the equatorial domain in blue, and the intermediate domain in green. (a) Hexadecamer (α8β8) of chaperonin from T. acidophilum. One α subunit is colored; (b) α subunit structure of the chaperonin.
apical domain. The recombinant α-subunit from T. acidophilum forms a hexadecameric oligomer that displays not only ATPase activity but also GTPase, CTPase, UTPase activities in the presence of Mg2+, Co2+, or Mn2+ ion, along with a corresponding chaperonin activity [27,28] . Also, the recombinant α- and β-subunits from T. acidophilum may be isolated in monomeric form, and these monomers displayed a strong nucleotide hydrolysis activity in the presence of divalent ions, especially Mn2+ or Co2+ ion [29]. In the original T. acidophilum cells, we found that a significant fraction (~50%) of the chaperonin was present as a monomer [29], and the biological function of these monomers, if any, is not known. Recently, it has been reported that the group II chaperonin from an antarctic methanogen, Methanococcoides burtonii, is also predominantly monomeric, with only 23% - 33% found in oligomeric form, both in vitro and in vivo [30] .
Here, in order to probe the involvement of Ta-cpn in preventing protein fibril formation, we studied the effects of Ta-cpn on the amyloid fibril formation of yeast Sup35NM protein in vitro. Yeast Sup35 is a translation termination factor which, after binding with eRF1 (Sup45p in yeast), catalyzes the release of the nascent polypeptide chain from the ribosome, and often used in studies of in vitro fibril formation. Sup35 is a 685-residue protein composed of three domains, N, M, and C. The N domain (residues 1 - 123), which is enriched in particular uncharged polar amino acid (Gln, Asn, Tyr), has five imperfect nine-residue repeats with consensus YQQYNPQGG, and forms the major part of the amyloid fibril core region. The M domain (residues 124 - 253) is highly charged and confers the soluble non-prion state in vitro and in vivo, and the C domain (residues 254 - 685) is a GTPase essential for translation termination [31] . In this study, we used the NM domains of Sup35 as a model protein for amyloid fibril formation. We found that both recombinant oligomeric and monomeric Ta-cpn were capable of suppressing Sup35NM amyloid fibril formation. Interestingly, isolated apical domain polypeptides were successful in suppressing fibril formation. When the helical protrusion segment was deleted from these apical domain polypeptides, the ability to suppress fibril formation was lost, which indicated that the helical protrusion segment was responsible for this effect. We concluded that the helical protrusion segment of the apical domain recognized and interacted with monomers or soluble oligomers of Sup35NM at an early stage of fibril formation. These results may suggest that helical protrusion segment of group II chaperonin not only acts as a lid of the central cavity during protein folding assistance, but also actively assists in recognizing and binding to proteins that may proceed to aggregates or fibrils. Our results may also suggest a possible novel role for the significant amounts of monomeric chaperonin that may be found in T. acidophilum cells.
2. EXPERIMENTAL AND METHODS
2.1. Cloning and Plasmids
The structural genes (T. acidophilum Ta0980 and Ta1276) of T. acidophilum chaperonin α-subunit and β-subunit were amplified from genomic DNA by PCR. A C-terminal histidine tag was added to each amplified fragment and inserted into pET23a(+) vector (Novagen) to produce pET-Ta-cpn α and pET-Ta-cpn β respectively. The apical domains of Ta-cpn α-subunit (Ser218-Asn369) and β-subunit (Asn215-Asn366) were amplified from pET-Ta-cpn α and pET-Ta-cpn β using PCR and inserted into pET23a(+) vector to produce pET-Api-Ta-cpn α-His6 and pET-Api-Ta-cpn β-His6. ΔlidTa-cpnΔlidApi-Ta-cpn α and ΔlidApi-Ta-cpn β were constructed by replacing the helical protrusion segment of the apical domains of Ta-cpn α (Ile249-Asn275) and Ta-cpn β (Ile246-Asn272) with a short linker (Glu-Thr-Ala-Ser-Glu) [32], using site-directed mutagenesis by the QuikChange kit (Stratagene) on pET-Ta-cpn α, pET-Api-Ta-cpn α-His6 and pETApi-Ta-cpn β-His6, respectively. All mutants were confirmed by DNA sequence analysis.
The structural gene of Saccharomyces cerevisiae S288c Sup35(1-254) was amplified from genomic DNA by PCR, after which a C-terminal histidine tag was added. The resultant fragment was inserted into pET23a(+) vector (Novagen) to produce pET-Sup35NM-His6.
2.2. Purification of Chaperonin
For expression of Ta-cpn α, Ta-cpn β, and mutant proteins, E. coli BL21 (DE3) was transformed with the constructed plasmid and transformants were grown at 37˚C in LB medium in the presence of 50 μg/ml ampicillin over night. Purification of Ta-cpn α, Ta-cpn β proteins were described previously [29]. For purification of ApiTa-cpn α and ΔlidApi-Ta-cpn α, harvested cells were resuspended in 50 mM phosphate-buffer, pH 7.0, containing 300 mM KCl and 1 mg/ml Pefabloc, and lysed by sonication on ice and centrifuged. Soluble proteins were then loaded onto a column of 25 ml TALON® Resin (Clontech), pre-equilibrated with 50 mM phosphatebuffer, pH 7.0, containing 300 mM KCl. The column was washed with three volumes of 50 mM phosphate-buffer, pH 7.0, containing 300 mM KCl, and then further washed with three volumes of 50 mM phosphate-buffer, pH 7.0, containing 300 mM KCl and 20 mM imidazole. Api-Tacpn α and ΔlidApi-Ta-cpn α were eluted with 50 mM phosphate-buffer, pH 7.0, containing 300 mM KCl and 250 mM imidazole. Purification of Api-Ta-cpn β and ΔlidApi-Ta-cpn β were performed using the protocol described above, with the exception that the pH of the buffer used was changed to 8.0. The fractions containing cpn proteins were pooled and dialyzed against 50 mM Hepes-KOH, pH 7.4, containing 150 mM KCl at 4˚C. The proteins were concentrated using Amicon-10 (Mw 10,000 cut off filter). Next, the preparations were loaded onto a Superdex75 10/300 GL column (GE Healthcare) equilibrated with 50 mM Hepes-KOH, pH 7.4, containing 150 mM KCl using an ÄKTA-FPLC system at 4˚C. The purity of the purified proteins was checked by 15% SDS-PAGE. Protein concentrations of purified proteins were determined by using the commercial Protein Assay Kit (Bio-Rad) with BSA as a standard.
2.3. Preparation of Sup35NM
For expression of Sup35NM protein, E. coli BL21 (DE3) cells were transformed with the constructed plasmid (pET-Sup35NM-His6) and transformants were grown at 37˚C in LB medium in the presence of 50 μg/ml ampicillin. After induction with 1 mM IPTG at an OD600 of 0.6 - 0.8, cell were harvested after 2 hr of induction. Cells were lysed in 20 mM Tris-HCl, pH 8.0, containing 8 M urea at room temperature and by sonication, and centrifuged. Soluble proteins were then loaded onto a 25 ml Ni-NTA Superflow column (QIAGEN), pre-equilibrated with 20 mM Tris-HCl, pH 8.0, containing 8 M urea. The column was washed with five volumes of 20 mM Tris-HCl, pH 8.0, containing 8 M urea and 30 mM imidazole. Sup35NM was eluted using 20 mM Tris-HCl, pH 8.0, containing 8 M urea and 400 mM imidazole and the fractions containing Sup35NM were pooled. The pooled fractions were loaded onto a 25 ml Q-Sepharose Fast Flow column (GE Healthcare) pre-equilibrated with the elution buffer used in Ni-NTA chromatography. The column was washed with five volumes of 20 mM Tris-HCl, pH 8.0 containing 8 M urea and 100 mM NaCl. Sup35NM was eluted with 20 mM Tris-HCl, pH 8.0, containing 8 M urea and 300 mM NaCl and the fractions containing Sup35NM were pooled and dialyzed against 20 mM Tris-HCl, pH 7.4, containing 8 M urea. Anhydrous methanol (100%) was added on ice at a ratio of 5:1 to precipitate Sup35NM. The pellet was washed with 100% methanol using 1/2 of the volume of the supernatant. After centrifugation, the supernatant was removed and the pellet was stored in 70% (v/v) methanol (1/2 volume of supernatant) at –80˚C until use.
2.4. Peptides
Custom syntheses of the helical protrusion peptide of Ta-cpn α (249IKKTEIEAKVQISDPSKIQDFLNQET N275) was performed by PEPTIDE 2.0, Inc. (USA). Peptide purity was approximately >97% as determined by HPLC and electrospray mass spectrometry.
2.5. CD Spectra Measurements
CD measurements of all mutants of Ta-cpn used in this study were checked routinely to confirm their folded structures. Far-UV CD spectra from 190-250 nm were recorded with a Jasco J-720 spectropolarimeter equipped with a constant temperature cell holder at 25˚C. Sixteen independent scans were accumulated and recorded using a 1-mm light path length cell to obtain the display spectra. Protein concentrations were 0.05 mg/ml in 5 mM Tris-HCl, pH 7.4.
2.6. Amyloid Fibril Formation Kinetics
Recombinant protein Sup35NM was precipitated with methanol and resuspended in 50 mM Hepes-KOH, pH 7.4, containing 8 M urea. Sup35NM concentration was determined by measuring the OD at 280 nm. Sup35NM (250 μM) was diluted 100-fold in 50 mM Hepes-KOH, pH 7.4, containing 150 mM KCl, 20 mM MgCl2, 1 mM DTT, and 1.25 μM thioflavin T (Thio T). The concentration of urea was 80 mM during fibril formation. The reaction was started by stirring with a Teflon-coated stirring chip in a quartz cell at 25˚C using a Jasco FP-6300 spectrofluorometer. Amyloid fibril formation was followed by monitoring ThioT binding. All data were normalized, and the resulting curves were used to determine the lag times and apparent extension rates of fibril formation. The lag time was obtained by fitting a straight line to the slope of the growth phase between approximately 30% and 70% of the maximum relative amplitude, and the time at which this line intersected with the baseline was designated as the lag time. Apparent extension rates (RFU/hr) were determined by linear approximation of the growth curve. All fibril formation experiments were performed at least in triplicate and data are displayed as averages ± standard error in Table 1.
2.7. AFM Measurements
Atomic force microscopy measurements were performed using a Digital Instruments Nanoscope IV scanning microscope (model MMAFM-2) at 25˚C. Measurements were performed using air tapping mode. For the air tapping mode measurements, 15 μl of 10-fold diluted fibril solution was put onto freshly cleaved mica, incubated for 30 min, and then washed with 150 μl of water and dried.
Atomic force microscopy measurements of fibrils treated by gold-conjugated antibody were performed as follows. First, the mixed amyloid fibrils prepared according to the protocol described above in the presence of Api-Ta-cpn were incubated with 1500-fold diluted anti-Api-Ta-cpn antibody (from rabbit) at 25˚C overnight. After centrifugation at 20,000 g for 60 min, the pellet samples were resuspended in 300 μl of PBS buffer, pH 8.2, containing secondary antibody (anti-rabbit IgG antibody (from goat) conjugated with 40-nm colloidal gold (British BioCell International, Inc.)). After incubation at 25˚C for 1 hr, the
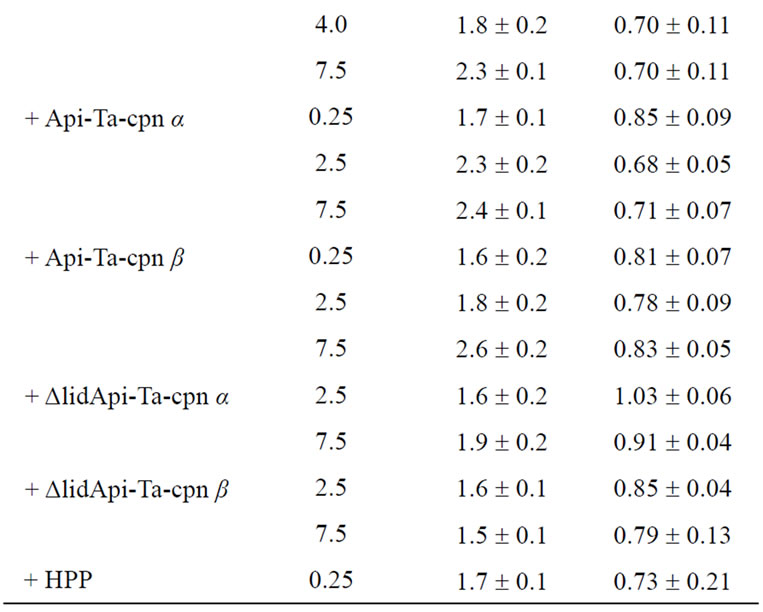
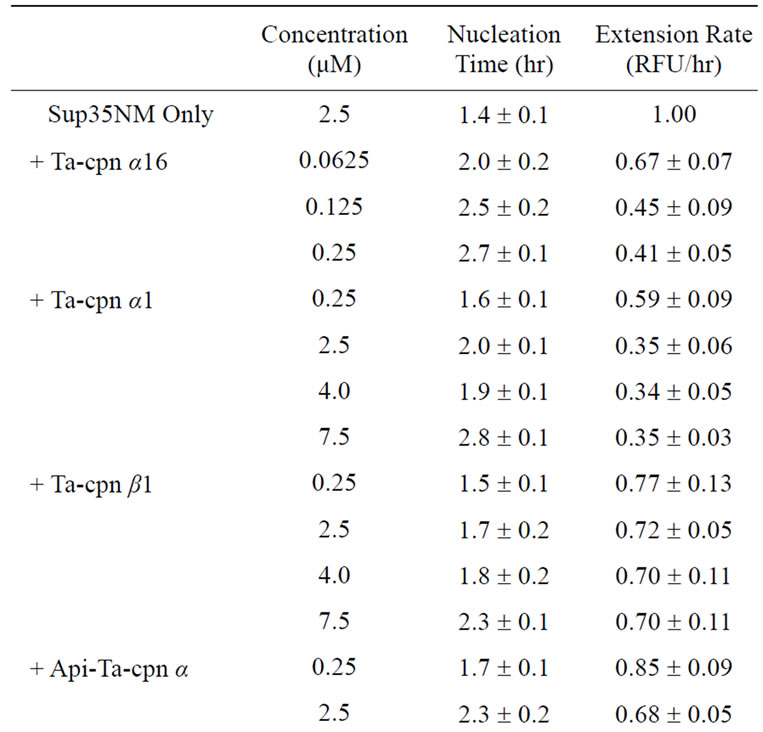
Table 1. Sup35NM fibrillation suppression parameters at various conditions.
sample was centrifuged and washed with phosphatebuffered saline and then applied to freshly cleaved mica, incubated for 30 min, washed with 150 μl of water, and dried [33] .
3. RESULTS
3.1. Suppression of Sup35NM Fibril Formation by Oligomeric and Monomeric Ta-Cpn
In previous studies, we found that recombinant homohexadecameric chaperonin (Ta-cpn α16) from T. acidophilum displays an ATPase activity and an ability to assist the refolding of proteins such as GFPuv and Thermus sp. MDH, although recombinant homo-hexadecameric chaperonin from β-subunit (Ta-cpn β16) could not be isolated [28]. Additionally, we reported that the monomeric forms of Ta-cpn α and β (Ta-cpn α1 and Ta-cpn β1) possessed a novel versatile nucleotide hydrolysis activity and an ability to protect proteins from thermal unfolding, reflecting an ability of these monomeric chaperonins to bind substrate proteins [29]. In the present study, we attempted to elucidate the ability of each of these chaperonin forms to interact with and suppress the fibrillation of yeast derived Sup35NM protein in vitro. Sup35NM forms fibrils rapidly, usually within several hours [34], and a real-time tracking of the reaction with ThioT fluorescence could be performed.
First, we examined whether oligomeric Ta-cpn α16 was able to interact with Sup35NM and suppress amyloid fibril formation. As shown in Figure 2(a), without chaperonin and under the conditions we used, Sup35NM (2.5 μM) formed fibrils after a 1.4 hr incubation, following a sigmoidal curve typical to the amyloid fibrillation mechanism; a nucleus formation step (detected as an initial lag phase that is sensitive to the addition of preformed fibril “seeds”) followed by a fibril extension step [34]. Specific values regarding fibril nucleus formation and extension that were determined from analysis of at least 3 independent experiments are summarized in Ta-
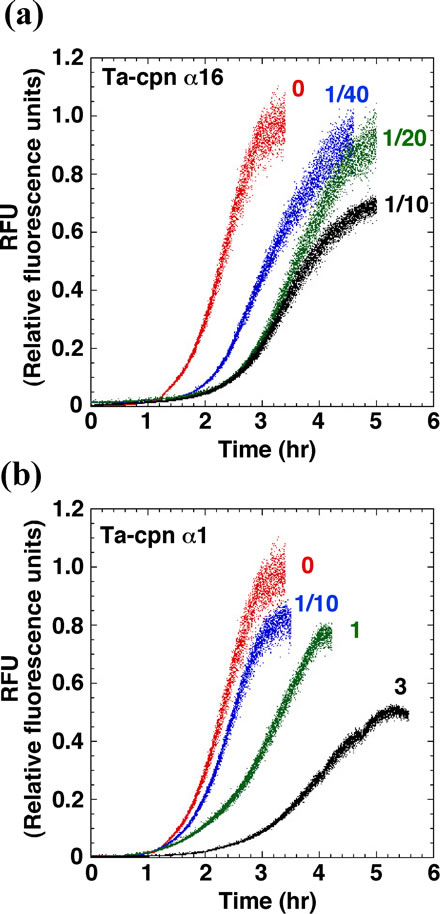
Figure 2. Suppression effects of Ta-cpn α16 and Ta-cpn α1 towards amyloid fibril formation of Sup35NM. (a) Effect of Ta-cpn α16. Fibril formation of 2.5 μM Sup35NM were followed by ThioT fluorescence in the absence (red) or presence of 62.5 nM (1/40 mol-eq) (blue), 125 nM (1/20 mol-eq) (green), and 250 nM (1/10 mol-eq) (black) Ta-cpn α16; (b) Effect of Ta-cpn α1. Fibril formation of 2.5 μM Sup35NM in the presence of 0 M (red), 250 nM (1/10 mol-eq) (blue), 2.5 μM (1 mol-eq) (green), and 7.5 μM (3 mol-eq) (black) Ta-cpn α1.
ble 1. Upon addition of a 1/40 (62.5 nM oligomer), 1/20 (0.125 μM), and 1/10 (0.25 μM) molar equivalent (mol-eq) of Ta-cpn α16 (as oligomer) relative to Sup35NM monomer, a concentration-dependent extension of the lag time and a corresponding decrease in fibril extension rate were observed. These effects were not observed when 1 mol-eq of bovine serum albumin was added (nucleation time = 1.4 hr, extension rate = 0.95 RFU/hr), suggesting that Ta-cpn molecule interacted specifically with Sup35- NM. Next, to probe which state of Sup35NM Ta-cpn α16 recognizes, we shifted the time at which Ta-cpn α16 was added to the Sup35NM fibril formation sample, i.e., the chaperonin was added at 0, 1.5, 2.2 or 2.8 hr after initiation of fibril formation. The fibril suppression effect was observed only when Ta-cpn α16 was added at 0 hr (data not shown). It should be noticed that Ta-cpn α16 was stable and active under all of the protein concentrations and incubation times we applied in the present experiments. This finding suggested that Ta-cpn α16 could interact with only soluble intermediate species of Sup35- NM at a very early stage.
Next, we repeated our experiments using monomeric Ta-cpn. As shown in Figure 2(b) and Table 1, an effect that was similar to Ta-cpn α16 was observed when Ta-cpn α1 was added, although the relative strength of this effect was diminished; data corresponding to the addition of 0.25 μM (1/10 mol-eq), 2.5 μM (1.0 mol-eq), and 7.5 μM (3.0 mol-eq) Ta-cpn α1 (as monomer) are presented in Figure 2(b). Since 4.0 μM monomeric Ta-cpn corresponds to 0.25 μM oligomeric Ta-cpn (Ta-cpn α16), we compared the concentration-specific effects on nucleation time and extension rate of Ta-cpn α16 and of Ta-cpn α1, as shown in Table 1; 2.7 hr and 0.41 RFU/hr for Ta-cpn α16, and 1.9 hr and 0.34 RFU/hr for Ta-cpn α1. The results in Table 1 demonstrate that Ta-cpn α16 was more effective in prolonging the fibril nucleation step, rather than in retarding the fibril extension rate. As also seen for Ta-cpn α16, the effect of Ta-cpn α1 in suppressing fibril formation was lost when addition of this chaperonin was delayed. Also, at the protein concentrations and incubation time we used, Ta-cpn α1 remained in a monomeric state, as confirmed by gel filtration experiment. When considered in a concentration-dependent context, our results showed that the ability of Ta-cpn to suppress the formation of Sup35NM fibrils was strengthened by the formation of the chaperonin oligomeric state. This is reasonable when we consider that a more effective concentration of binding sites may be formed in the chaperonin oligomer. Almost similar results were obtained for Ta-cpn β1 in retarding the formation of Sup35- NM fibril nuclei. However, the effect of this subunit in slowing the fibril extension rate was less than that of Ta-cpn α1 (Table 1).
3.2. Importance of Helical Protrusion Segment in the Apical Domain of Ta-Cpn for Suppression of Sup35NM Fibril Formation
As shown in Figure 2(b), Ta-cpn monomers were capable of altering both the fibril nucleation and fibril extension steps of Sup35NM fibril formation. As refolding protein intermediates are trapped by the apical domains of group II oligomeric chaperonin [32], it seemed rational to expect that the apical domain of Ta-cpn monomer was closely involved in the interactions with Sup35- NM intermediates. In the group I chaperonin GroEL, it was reported that the isolated apical domain was able to bind substrate intermediates and facilitate the refolding [35] . In order to confirm that the apical domain interacts with Sup35NM and suppresses the fibril formation, we prepared isolated apical domain proteins of Ta-cpn α1 and β1 (hereafter called Api-Ta-cpn α and Api-Ta-cpn β, respectively) and performed experiments using these proteins. CD spectra of purified Api-Ta-cpn α and other derivative protein (ΔlidApi-Ta-cpn α, see below) are shown in Figure 3(a), demonstrating that the proteins assumed a folded structure, although CD intensities were small compared to that of Ta-cpn α1 [29].
As expected, as shown in Figure 3(b) and Table 1, Sup35NM fibril formation could be suppressed by the addition of Api-Ta-cpn α; nucleation times were prolonged in experiments where 0.25 μM (1/10 mol-eq), 2.5 μM (1.0 mol-eq), and 7.5 μM (3.0 mol-eq) Api-Ta-cpn α was added, and the extent of these effects was roughly similar to that of Ta-cpn α1. In contrast, the effect of Api-Ta-cpn α on the fibril extension rate was small compared to that of Ta-cpn α1 (Figures 2(a), 3(b) and Table 1). A roughly similar suppression effect was observed for Api-Ta-cpn β, although individual values differed from those of Api-Ta-cpn α (Table 1). Thus, although the ability to slow fibril extension was relatively inferior to Ta-cpn α1 or Ta-cpn β1, isolated apical domain proteins of Ta-cpn α and β subunits of were found to interact with Sup35NM intermediates and suppress the fibril formation.
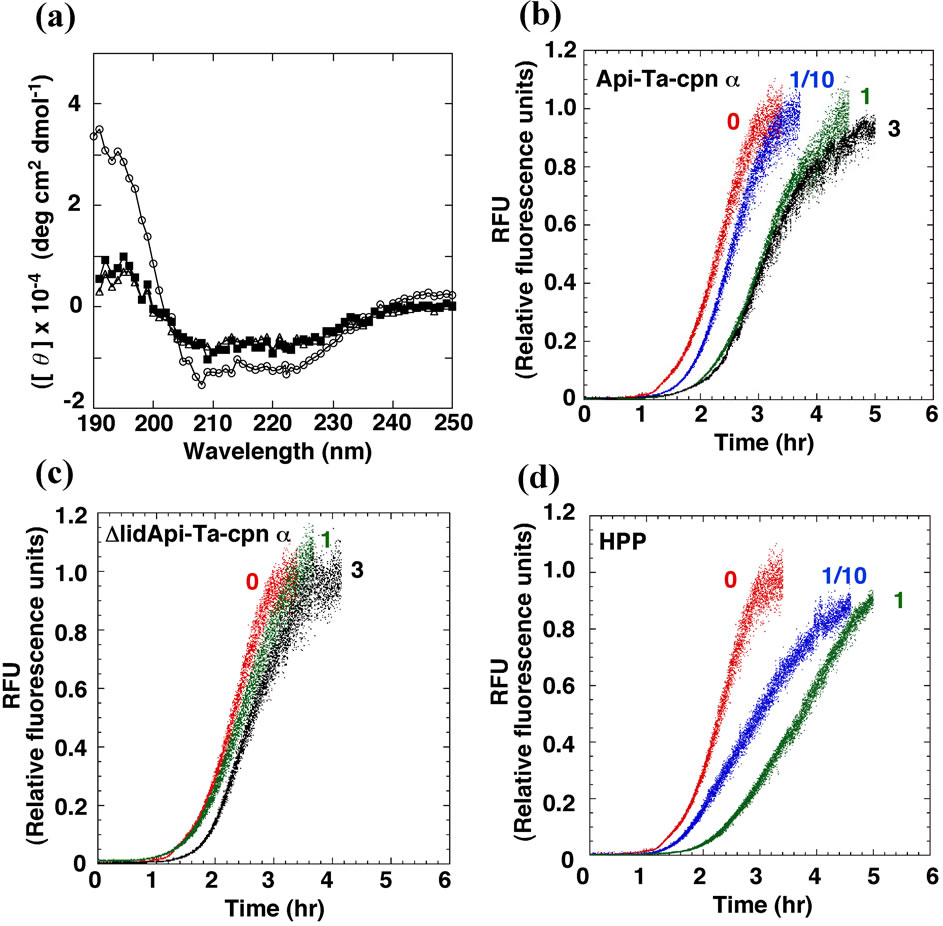
Figure 3. CD spectra and suppression effects of Api-Ta-cpn α, ΔlidApi-Ta-cpn α, and HPP towards fibril formation of Sup35NM. (a) CD spectra of Ta-cpn α1 (open circles), Api-Ta-cpn α (open triangles), and ΔlidApi-Ta-cpn α (closed squares) were measured; (b) Effect of Api-Ta-cpn α. Fibril formations of 2.5 μM Sup35NM were followed by ThioT fluorescence in the presence of 0 M (red), 250 nM (1/10 mol-eq) (blue), 2.5 μM (1 mol-eq) (green), and 7.5 μM (3 mol-eq) (black) Api-Ta-cpn α; (c) Effect of ΔlidApi-Ta-cpn α. Fibril formations of 2.5 μM Sup35NM in the presence of 0 M (red), 2.5 μM (1 mol-eq) (green), and 7.5 μM (3 mol-eq) (black) ΔlidApi-Ta-cpn α; (d) Effect of HPP. Fibril formations of 2.5 μM Sup35NM in the absence of 0 M (red), 250 nM (1/10 mol-eq) (blue), and 2.5 μM (1 mol-eq) (green) HPP.
Next, in order to further define the specific portion of the apical domain that was especially important for interaction with Sup35NM, we attempted to focus on the helical protrusion segment [36,37] . We first prepared helical protrusion deleted mutants of Api-Ta-cpn α and Api-Ta-cpn β (hereafter called ΔlidApi-Ta-cpn α and ΔlidApi-Ta-cpn β, respectively). No drastic secondary structural changes were caused by the deletion of helical protrusion, as confirmed by CD measurements of ΔlidApiTa-cpn α and ΔlidApi-Ta-cpn β. Interestingly, as shown in Figure 3(c) and Table 1, both ΔlidApi-Ta-cpn α and ΔlidApi-Ta-cpn β failed to show any significant suppression effects towards Sup35NM fibril formation even at a 7.5 μM (3.0 mol-eq) concentration. This result suggested strongly that the helical protrusion segment is directly involved in suppressing Sup35NM fibril formation. Furthermore, we performed the same suppression experiments using a helical protrusion deleted variant of the initial Ta-cpn α subunit. After expression in E. coli cells, oligomeric (ΔlidTa-cpn α16) forms were purified, and we determined that ΔlidTa-cpn α16 had lost its ability to suppress nucleus formation and retard fibril extension of Sup35NM. These results demonstrated that the helical protrusion segment of the apical domain is the site that is used in suppressing the fibrillation of Sup35NM.
Finally, we performed experiments using a synthesized helical protrusion peptide (HPP) of Ta-cpn α subunit. Very interestingly, as shown in Figure 3(d) and Table 1, addition of 0.25 μM (1/10 mol-eq) HPP decreased the fibril extension rate to about 70% of the original values and adding 2.5 μM (1.0 mol-eq) HPP resulted in a extension in the time required for nucleus formation (from 1.4 hr to 2.3 hr) and a decrease in the fibril extension rate to 60% of the original values. This result indicated that helical protrusion segment itself could suppress Sup35NM fibril formation quite effectively.
3.3. Amyloid Fibril Observation by AFM
As shown above, although Ta-cpn proteins were capable of suppressing amyloid fibril formation of Sup35NM to a certain extent, but not inhibit it completely; a prolonged lag-time and/or a decreased fibril extension rate were observed, but fibrils were eventually formed. This suggests that either Ta-cpn interacted only tentatively or weakly with Sup35NM protein during the fibril formation, or, that strong binding of Ta-cpn to Sup35NM protein was insufficient to stop fibril formation completely. In order to arrive at a mechanism that explains this chaperonin-derived suppression effect, it would be very important to clarify this point.
We therefore obtained AFM images of amyloid fibrils that had been formed in the absence and presence of Ta-cpn proteins and compared these images to observe differences in fibril morphology. However, we could not find any significant differences in morphology. Typical images are shown in Figures 4(a) and (b). AFM images of Sup35NM fibrils formed without chaperonin (Figure 4(a)) were visually very similar to fibrils formed after reaction with Ta-cpn α16 (Figure 4(b)) (fibrils were ~30 nm in width and ~10 nm in height in both samples). However, in images taken of fibrils formed in the presence of chaperonin, some small aggregated particles were also observed alongside the fibrils (Figure 4(b)). The presence of these aggregates may be relevant to the suppression effect of Ta-cpn α16.
Next, in order to investigate whether Ta-cpn molecules were still bound to Sup35NM fibrils after the conclusion of the fibril extension experiment, we performed immuno-AFM measurements using gold-conjugated antibody. If fibrils formed in the presence of Ta-cpn protein still retained chaperonin molecules attached to the fibril structure, gold-conjugated antibodies that recognize ApiTa-cpn α protein would cluster in the vicinity of the fibrils. Figure 4(c) shows an AFM image of Sup35NM fibril formed in the presence of Api-Ta-cpn α. As seen in

Figure 4. AFM images of Sup35NM amyloid fibrils formed in the absence and presence of Ta-cpn and derivatives. Sup35NM amyloid fibrils formed (a) in the absence of chaperonin, and (b) in the presence of 250 nM Ta-cpn α16. The scale bars in (a) and (b) represent 1 μm; (c) AFM measurements of immuno-goldtreated Sup35NM amyloid fibrils formed in the presence of 7.5 μM Api-Ta-cpn α; (d) Pre-formed Sup35NM amyloid fibrils were mixed with 7.5 μM Api-Ta-cpn α, and AFM images were measured. Api-Ta-cpn α was labeled with 40-nm gold particle attached to secondary antibody. The arrows indicate positions of attached gold particles. The scale bars in (c) and (d) represent 200 nm.
the figure, the dimension of the fibrils seen when antibody was added was increased apparently (to ~80 nm in width) compared to the image of Figure 4(b), due to binding of immuno-gold (40 nm) particles, indicating that Api-Ta-cpn α remained bound to Sup35NM amyloid fibrils. It was difficult to observe any differences in the height of fibrils, probably due to the AFM measurements in air atmosphere.
In order to see if Api-Ta-cpn α was able to interact with preformed Sup35NM fibrils, we observed AFM images of samples where Api-Ta-cpn α was added after completion of Sup35NM fibrillation and then stained by immuno-gold particles. As shown by arrows in Figure 4(d), a significant number of immuno-gold particles were observed along the fibril structure, which indicated that Api-Ta-cpn α was able to bind to the surface of preformed Sup35NM fibrils. However, the number of colloidal gold particles (i.e., Api-Ta-cpn α molecules) was much less than the number of observed attached to fibrils formed in the presence of Api-Ta-cpn α (Figure 4(c)). From this it seems that Ta-cpn was able to interact and bind to many sites of Sup35NM molecule prior to fibril formation, that are not accessible in the final fibril form. This difference in interaction may offer a possible explanation for the ability of Ta-cpn to suppress the fibril formation of Sup35NM.
4. DISCUSSION
Although chaperonins are strongly involved in keeping cells healthy by maintaining the integrity of cellular proteins in the face of stress, a clear and broad role for this group of molecular chaperones in protecting cells from various long-term amyloidogenic disorders is not obvious. In order to determine if such a role exists for chaperonins found in various species, we have probed the effects of the group II chaperonin from T. acidophilum on the in vitro fibrillation reaction of Sup35NM. Sup35 is a yeast prion protein and forms a typical amyloid fibril in vitro [34]. Interactions between Sup35NM amyloid fibrils with various molecular chaperones have been reported in many studies involving Hsp104, Hsp70, and Hsp40 [1,38.39] , but reports involving chaperonins have been scarce to date. Related studies that have been reported include effects of the eukaryotic group II chaperonin, TRiC/CCT, on the aggregation of huntingtin (Htt) protein by binding to a short Htt sequence element located in the N-terminal of poly-Q tracts [5] and the productive assembly was promoted by cooperation with the Hsp70 system [6]. Also, it has been reported that TRiC/ CCT inhibited the aggregation of VHL tumor suppressor [32] and Gβ WD40 proteins [40] .
Our experiments showed that the chaperonin from T. acidophilum displayed a suppressing effect toward Sup- 35NM amyloid fibril formation. Not only oligomeric Ta-cpn (Ta-cpn α16) but also monomeric Ta-cpn (Ta-cpn α1 and Ta-cpn β1) displayed these suppression effects. The effectiveness of monomeric chaperonins was low compared to the Ta-cpn oligomer (Figure 2 and Table 1). An apical domain fragment of Ta-cpn (Api-Ta-cpn) as well as a synthetic peptide corresponding to the helical protrusion region also showed similar inhibitory effects. Contrariwise, Ta-cpn subunits which lacked the helical protrusion (ΔlidApi-Ta-cpn) were ineffective in suppressing fibril formation (Figure 3 and Table 1). These results were explained straightforwardly, i.e., all chaperonin proteins and derivatives that contained the helical protrusion segment were effective in suppressing Sup35NM fibril formation, although effectiveness differed among the variants. Our finding demonstrates that group II chaperonin, both oligomeric and monomeric forms, may be actively involved in suppressing amyloid fibril formation, in a manner similar to other molecular chaperones such as Hsp70, Hsp40, and Hsp104 [1,38,39,41] .
One thing to be noted was that the Ta-cpn could not inhibit the Sup35NM fibril formation completely. Only transient suppression effects were observed, and the effects were seen only when chaperonins and derivatives were added at the very beginning of the amyloid fibril formation reaction. In addition, after retarding the formation of fibrils, Api-Ta-cpn remained attached to the surface of the fibrils that had been eventually formed, as demonstrated by the localization of gold-labeled antibodies that recognized chaperonin protein (Figure 4(c)). Taken together with the fact that Ta-cpn also has the ability to bind to surface of preformed fibrils of Sup35 (Figure 4(d)), the suppression effects are likely due to dynamic binding to an early intermediate of Sup35NM during fibril formation. The dynamic interactions between Ta-cpn and Sup35NM protein, which is mainly mediated by the helical protrusion segment of the apical domain, could conceivably lead to a longer fibril nucleus formation time and a decrease in the fibril extension rate. Accordingly, a schematic model for the suppression mechanism of Sup35NM fibril formation by Ta-cpn is shown in Figure 5. As shown in Figure 5, group II chaperonin (Ta-cpn α16, Ta-cpn monomer, Api-Ta-cpn, and HPP) binds to the early soluble intermediates of Sup35NM protein dynamically. The binding affinity depends on the specific form of chaperonin protein used in the assays but the helical protrusion segment of group II chaperonin is the main interface involved in binding. The dynamic binding suppresses fibril formation but eventually the reaction proceeds to form mature fibrils. After fibril formation, some chaperonin interacts remain bound to the surface of the fibrils.
The mechanism by which group II chaperonins recognize and bind to unfolded protein molecules is a recent
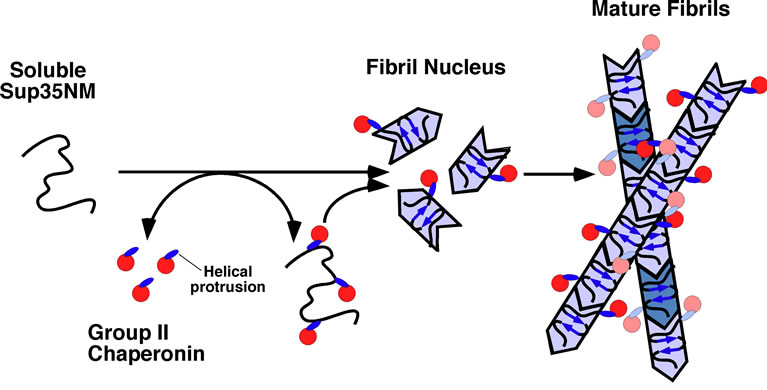
Figure 5. A schematic model for the mechanism of Sup35NM fibril suppression by Ta-cpn. The helical protrusion segment of Ta-cpn mainly binds to monomeric or soluble oligomeric forms of Sup35NM protein dynamically. Such dynamic interactions contribute to suppression of Sup35NM amyloid fibril formation. After completion of fibrillation, Ta-cpn is still able to bind to the surface of Sup35NM fibrils. See text for details.
topic of study. In the group I chaperonin GroEL, it is generally accepted that two α-helices located in the apical domain, helix H and helix I, recognize and bind various refolding intermediates of substrate proteins, mainly through hydrophobic interaction [35,42-44] . In group II chaperonins, an analogous pair of α-helices, helix 10 and helix 11, exists, similar to group I chaperonins. However, a prominent difference in the group II chaperonins is that helix 10 is greatly extended and together with a long loop region, this helix forms the helical protrusion, which acts as a lid of central cavity [21]. The role of the helical protrusion motif in protein molecule recognition and binding is presently being disputed. In one study, it was argued that helices 10 and 11 of TRiC/CCT recognized non-native conformations of substrate proteins through hydrophobic interaction, in a manner similar to that of GroEL [32,45-47] . In other studies, it has been reported that the helical protrusion motif recognizes a quasi-native conformation of β-tubulin, mainly by electrostatic interactions [37]. In the present study, the helical protrusion motif was found to be indispensable in suppressing Sup35NM fibril formation. Since a large number of charged amino acid residues reside in the helical protrusion of Ta-cpn α (249IKKTEIEAKVQISDPSKIQDFLNQETN275), these charges may contribute to binding affinity for Sup35NM protein, of which M-domain region is highly charged [41] . Our results are therefore in support of a binding mechanism where the helical protrusion motif actively assists molecular recognition and binding of denatured proteins.
Finally, we indulge in some speculation regarding our experimental results and the curious finding that a significant fraction of group II chaperonin subunits (~50%) may be found in monomeric form in cells of T. acidophilum [29]. As our results have shown, the ability of Ta-cpn to suppress the fibril formation of Sup35NM is not restricted to the oligomeric form, but is observed for the monomer as well. This was due to the fact that the interactions responsible for fibril suppression were highly localized in the helical protrusion motif of the apical domain, as experiments using the synthetic peptide have shown (Figure 3(d)). It is reasonable therefore to consider that monomeric chaperonin is able to assert a similar effect on proteins in T. acidophilum cells.
In this context, we note with interest that the ability of Ta-cpn to assist protein folding and prevent aggregation differs, depending on its quaternary structure; oligomeric Ta-cpn displays both refolding activity [28] and the ability to suppress fibril formation, whereas the monomeric form is only capable of fibril suppression. The fact that the helical protrusion motif, a region with a high concentration of electrostatic charges, is integral to fibril suppression indicates that the latter ability depends on localized, electrostatic interactions with the target protein, rather than a relatively delocalized, hydrophobic interaction such as that probably used by the group I and group II chaperonins in the orthodox mechanism of folding assistance. Hydrophobic interactions would be strengthened by consolidating binding sites for putative targets, for example by associating multiple subunits into a cohesive oligomer rather than a stand-alone monomeric state. Perhaps in T. acidophilum cells, monomeric variants of Ta-cpn assert an aggregation suppression effect based on electrostatic interactions, and a more elaborate chaperoning effect as an oligomer. This “double duty” role of chaperonins based upon different molecular interactions may act as an additional versatile mechanism that is more capable of maintaining the structural integrity of many important proteins in the archeal cell.
5. ACKNOWLEDGEMENTS
This work was partly supported by grants-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and a grant from the Venture Business Laboratory of Tottori University.
REFERENCES
- Glover, J.R. and Lindquist, S. (1998) Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell, 94, 73-82. doi:10.1016/S0092-8674(00)81223-4
- Chai, Y., Koppenhafer, S.L., Bonini, N.M. and Paulson, H.L. (1999) Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. Journal of Neuroscience, 19, 10338-10347.
- Muchowski, P.J., Schaffar, G., Sittler, A., Wanker, E.E., Hayer-Hartl, M.K. and Hartl, F.U. (2000) Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proceedings of the National Academy of Sciences USA, 97, 7841-7846. doi:10.1073/pnas.140202897
- Stockel, J. and Hartl, F.U. (2001) Chaperonin-mediated de novo generation of prion protein aggregates. Journal of Molecular Biology, 313, 861-872. doi:10.1006/jmbi.2001.5085
- Tam, S., Spiess, C., Auyeung, W., Joachimiak, L., Chen, B., Poirier, M.A. and Frydman, J. (2009) The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nature Structural and Molecular Biology, 16, 1279-1285. doi:10.1038/nsmb.1700
- Behrends, C., Langer, C.A., Boteva, R., Bottcher, U.M., Stemp, M.J., Schaffar, G., Rao, B.V., Giese, A., Kretzschmar, H., Siegers, K. and Hartl, F.U. (2006) Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Molecular Cell, 23, 887-897. doi:10.1016/j.molcel.2006.08.017
- Frydman, J., Nimmesgern, E., Erdjument-Bromage, H., Wall, J.S., Tempst, P. and Hartl, F.U. (1992) Function in protein folding of TRiC, a cytosolic ring complex containing TCP-1 and structurally related subunits. EMBO Journal, 11, 4767-4778.
- Gao, Y., Thomas, J.O., Chow, R.L., Lee, G.H. and Cowan, N.J. (1992) A cytoplasmic chaperonin that catalyzes beta-actin folding. Cell, 69, 1043-1050. doi:10.1016/0092-8674(92)90622-J
- Yaffe, M.B., Farr, G.W., Miklos, D., Horwich, A.L., Sternlicht, M.L. and Sternlicht, H. (1992) TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature, 358, 245-248. doi:10.1038/358245a0
- Marco, S., Carrascosa, J.L. and Valpuesta, J.M. (1994) Reversible interaction of beta-actin along the channel of the TCP-1 cytoplasmic chaperonin. Biophysical Journal, 67, 364-368. doi:10.1016/S0006-3495(94)80489-8
- Farr, G.W., Scharl, E.C., Schumacher, R.J., Sondek, S. and Horwich, A.L. (1997) Chaperonin-mediated folding in the eukaryotic cytosol proceeds through rounds of release of native and nonnative forms. Cell, 89, 927-937. doi:10.1016/S0092-8674(00)80278-0
- Ditzel, L., Lowe, J., Stock, D., Stetter, K.O., Huber, H., Huber, R. and Steinbacher, S. (1998) Crystal structure of the thermosome, the archaeal chaperonin and homolog of CCT. Cell, 93, 125-138. doi:10.1016/S0092-8674(00)81152-6
- Bukau, B. and Horwich, A.L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell, 92, 351-366. doi:10.1016/S0092-8674(00)80928-9
- Gething, M.J. and Sambrook, J. (1992) Protein folding in the cell. Nature, 355, 33-45. doi:10.1038/355033a0
- Yifrach, O. and Horovitz, A. (2000) Coupling between protein folding and allostery in the GroE chaperonin system. Proceedings of the National Academy of Sciences USA, 97, 1521-1524. doi:10.1073/pnas.040449997
- Ranson, N.A., Farr, G.W., Roseman, A.M., Gowen, B., Fenton, W.A., Horwich, A.L. and Saibil, H.R. (2001) ATP-bound states of GroEL captured by cryo-electron microscopy. Cell, 107, 869-879. doi:10.1016/S0092-8674(01)00617-1
- Hartl, F.U. and Hayer-Hartl, M. (2002) Molecular chaperones in the cytosol: from nascent chain to folded protein. Science, 295, 1852-1858. doi:10.1126/science.1068408
- Taniguchi, M., Yoshimi, T., Hongo, K., Mizobata, T. and Kawata, Y. (2004) Stopped-flow fluorescence analysis of the conformational changes in the GroEL apical domain: relationships between movements in the apical domain and the quaternary structure of GroEL. Journal of Biological Chemistry, 279, 16368-16376. doi:10.1074/jbc.M311806200
- Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.C., Joachimiak, A., Horwich, A.L. and Sigler, P.B. (1994) The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature, 371, 578-586. doi:10.1038/371578a0
- Hunt, J.F., Weaver, A.J., Landry, S.J., Gierasch, L. and Deisenhofer, J. (1996) The crystal structure of the GroES co-chaperonin at 2.8 Å resolution. Nature, 379, 37-45. doi:10.1038/379037a0
- Meyer, A.S., Gillespie, J.R., Walther, D., Millet, I.S., Doniach, S. and Frydman, J. (2003) Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell, 113, 369-381. doi:10.1016/S0092-8674(03)00307-6
- Nitsch, M., Klumpp, M., Lupas, A. and Baumeister, W. (1997) The thermosome: alternating alpha and beta-subunits within the chaperonin of the archaeon Thermoplasma acidophilum. Journal of Molecular Biology, 267, 142-149. doi:10.1006/jmbi.1996.0849
- Ruepp, A., Graml, W., Santos-Martinez, M.L., Koretke, K.K., Volker, C., Mewes, H.W., Frishman, D., Stocker, S., Lupas, A.N. and Baumeister, W. (2000) The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature, 407, 508-513. doi:10.1038/35035069
- Bosch, G., Baumeister, W. and Essen, L.O. (2000) Crystal structure of the beta-apical domain of the thermosome reveals structural plasticity in the protrusion region. Journal of Molecular Biology, 301, 19-25. doi:10.1006/jmbi.2000.3955
- Heller, M., John, M., Coles, M., Bosch, G., Baumeister, W. and Kessler, H. (2004) NMR studies on the substratebinding domains of the thermosome: structural plasticity in the protrusion region. Journal of Molecular Biology, 336, 717-729. doi:10.1016/j.jmb.2003.12.035
- Koradi, R., Billeter, M. and Wuthrich, K. (1996) MOLMOL: a program for display and analysis of macromolecular structures. Journal of Molecular Graphics, 14, 51-55.
- Gutsche, I., Mihalache, O., Hegerl, R., Typke, D. and Baumeister, W. (2000) ATPase cycle controls the conformation of an archaeal chaperonin as visualized by cryo-electron microscopy. FEBS Letters, 477, 278-282. doi:10.1016/S0014-5793(00)01811-1
- Hirai, H., Noi, K., Hongo, K., Mizobata, T. and Kawata, Y. (2008) Functional characterization of the recombinant group II chaperonin alpha from Thermoplasma acidophilum. Journal of Biochemistry, 143, 505-515. doi:10.1093/jb/mvm241
- Noi, K., Hirai, H., Hongo, K., Mizobata, T. and Kawata, Y. (2009) A potentially versatile nucleotide hydrolysis activity of group II chaperonin monomers from Thermoplasma acidophilum. Biochemistry, 48, 9405-9415. doi:10.1021/bi900959c
- Cavicchioli, R., Pilak, O., Harrop, S.J., Siddiqui, K.S., Chong, K., De Francisci, D., Burg, D., Williams, T.J. and Curmi, P.M.G. (2011) Chaperonins from an Antarctic archaeon are predominantly monomeric: crystal structure of an open state monomer. Environmental Microbiology, 13, 2232-2249. doi:10.1111/j.1462-2920.2011.02477.x
- Stansfield, I., Jones, K.M., Kushnirov, V.V., Dagkesamanskaya, A.R., Poznyakovski, A.I., Paushkin, S.V., Nierras, C.R., Cox, B.S., Ter-Avanesyan, M.D. and Tuite, M.F. (1995) The products of the SUP45 (eRF1) and SUP35 genes interact to mediate translation termination in Saccharomyces cerevisiae. EMBO Journal, 14, 4365- 4373.
- Spiess, C., Miller, E.J., McClellan, A.J. and Frydman, J. (2006) Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Molecular Cell, 24, 25-37. doi:10.1016/j.molcel.2006.09.003
- Yagi, H., Kusaka, E., Hongo, K., Mizobata, T. and Kawata, Y. (2005) Amyloid fibril formation of alpha-synuclein is accelerated by preformed amyloid seeds of other proteins: implications for the mechanism of transmissible conformational diseases. Journal of Biological Chemistry, 280, 38609-38616. doi:10.1074/jbc.M508623200
- Glover, J.R., Kowal, A.S., Schirmer, E.C., Patino, M.M., Liu, J.J. and Lindquist, S. (1997) Self-seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion-like factor of S. cerevisiae. Cell, 89, 811- 819. doi:10.1016/S0092-8674(00)80264-0
- Zahn, R., Buckle, A.M., Perrett, S., Johnson, C.M., Corrales, F.J., Golbik, R. and Fersht, A.R. (1996) Chaperone activity and structure of monomeric polypeptide binding domains of GroEL. Proceedings of the National Academy of Sciences USA, 93, 15024-15029. doi:10.1073/pnas.93.26.15024
- Klumpp, M., Baumeister, W. and Essen, L.O. (1997) Structure of the substrate binding domain of the thermosome, an archaeal group II chaperonin. Cell, 91, 263-270. doi:10.1016/S0092-8674(00)80408-0
- Jayasinghe, M., Tewmey, C. and Stan, G. (2010) Versatile substrate protein recognition mechanism of the eukaryotic chaperonin CCT. Proteins, 78, 1254-1265. doi:10.1002/prot.22644
- Tipton, K.A., Verges, K.J. and Weissman, J.S. (2008) In vivo monitoring of the prion replication cycle reveals a critical role for Sis1 in delivering substrates to Hsp104. Molecular Cell, 32, 584-591. doi:10.1016/j.molcel.2008.11.003
- Shorter, J. and Lindquist, S. (2008) Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO Journal, 27, 2712-2724. doi:10.1038/emboj.2008.194
- Kubota, S., Kubota, H. and Nagata, K. (2006) Cytosolic chaperonin protects folding intermediates of Gbeta from aggregation by recognizing hydrophobic beta-strands. Proceedings of the National Academy of Sciences USA, 103, 8360-8365. doi:10.1073/pnas.0600195103
- Krishnan, R. and Lindquist, S.L. (2005) Structural insights into a yeast prion illuminate nucleation and strain diversity. Nature, 435, 765-772. doi:10.1038/nature03679
- Fenton, W.A., Kashi, Y., Furtak, K. and Horwich, A.L. (1994) Residues in chaperonin GroEL required for polypeptide binding and release. Nature, 371, 614-619. doi:10.1038/371614a0
- Xu, Z., Horwich, A.L. and Sigler, P.B. (1997) The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature, 388, 741-750. doi:10.1038/41944
- Tanaka, N. and Fersht, A.R. (1999) Identification of substrate binding site of GroEL minichaperone in solution. Journal of Molecular Biology, 292, 173-180. doi:10.1006/jmbi.1999.3041
- Llorca, O., McCormack, E.A., Hynes, G., Grantham, J., Cordell, J., Carrascosa, J.L., Willison, K.R., Fernandez, J.J. and Valpuesta, J.M. (1999) Eukaryotic type II chaperonin CCT interacts with actin through specific subunits. Nature, 402, 693-696. doi:10.1038/45294
- Dekker, C., Stirling, P.C., McCormack, E.A., Filmore, H., Paul, A., Brost, R.L., Costanzo, M., Boone, C., Leroux, M.R. and Willison, K.R. (2008) The interaction network of the chaperonin CCT. EMBO Journal, 27, 1827-1839. doi:10.1038/emboj.2008.108
- Yam, A.Y., Xia, Y., Lin, H.T., Burlingame, A., Gerstein, M. and Frydman, J. (2008) Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nature Structural and Molecular Biology, 15, 1255-1262. doi:10.1038/nsmb.1515
NOTES
*Corresponding author.