International Journal of Organic Chemistry
Vol.3 No.2(2013), Article ID:33261,6 pages DOI:10.4236/ijoc.2013.32020
Enantioselective Aldol Reactions of Aliphatic Aldehydes with Singh’s Catalyst
Department of Chemistry, Aalto University, Espoo, Finland
Email: heli.kylmala@aalto.fi
Copyright © 2013 Heli Kylmälä, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 16, 2013; revised May 20, 2013; accepted May 28, 2013
Keywords: Aldol Reaction; Aldehydes; Enantioselectivity; Diastereoselectivity; Stereoselective Synthesis
ABSTRACT
Aldols from aliphatic aldehydes had been synthesized enantioselectively using Singh’s catalyst. Self and crossed aldol reactions with several linear aldehydes were performed.
1. Introduction
The aldol reaction is one of the most important carbon-carbon bond forming reactions [1]. There are only few studies where aldol reactions between two aliphatic aldehydes have been described [2-14]. In these reactions different amino acids [9], especially L-proline and its derivatives [15-18], diarylprolinols [11,14] and imidazolidinones [5] have been used as catalysts. Singh et al. designed an L-proline based chiral catalyst with a gemdiphenyl group at the β-carbon, which is essential for high enantioselectivities [17]. So far, this catalyst has been used in aldol reactions only between an aldehyde and a ketone [19-25]. To the best of our knowledge, this is the first time when enantioselective aldol reactions between two aliphatic aldehydes with Singh’s catalyst 1 are reported.
With Singh’s catalyst the stereochemistry of aldol products can be explained by their transition state (Figure 1), which is based on a model supported by DFT calculations [26]. Since aldehyde oxygen forms hydrogen bonds with the NH and OH groups of catalyst 1 the

Figure 1. Transition states with Singh’s catalyst.
new C-C bond is formed from its Re face [17]. The thermodynamically favorable E-enamine is mainly formed giving syn-aldol products [27].
2. Results and Discussion
Reaction conditions for self-aldol reactions (Table 1, Entries 1 - 8) were optimized with monoacetal protected glutaraldehyde [28] in a reaction that has been published earlier by us [29]. The aldol reactions presented in this article (Scheme 1) were reproducible and no water elimination was observed. The self-aldol reactions were done at 25˚C in DMSO for 20 - 21 h. Correspondingly, the conditions for cross-aldol reactions were 0˚C - 4˚C, DMF, 50 - 53 h.
Self-aldol reactions with linear aldehydes, i.e. butyraldehyde (Entry 1, Table 1), valeraldehyde (Entry 2, Table 1), hexanaldehyde (Entry 3, Table 1), heptanaldehyde (Entry 4, Table 1) and 3-phenylpropionaldehyde (Entry 5, Table 1) gave relatively good yields (45% - 71% yield, 2.9 - 12.5:1 dr, 80% - 89% ee). Since self-aldol reactions were successful, we also wanted to

Scheme 1. Aldol reaction of aliphatic aldehydes with Singh’s catalyst 1.
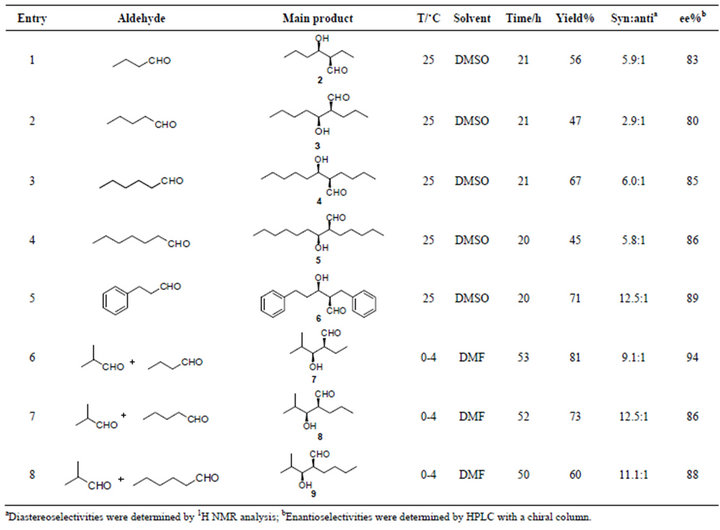
Table 1. Aldol reactions tested with Singh’s catalyst 1.
test some crossed aldol reactions. When isobutyraldehyde was treated with linear aldehydes (Entries 6 - 8, Table 1) the reactions gave as supposed even better yields and enantioselectivities (60% - 81% yield, 9.1:1 - 12.5:1dr, 86% - 94% ee).
3. Conclusions
In conclusion, enantioselective aldol reactions of aliphatic aldehydes have been obtained in good enantiomeric excess (80% - 94%).
4. Experimental Section
4.1. General
All solvents and reagents were used as obtained from supplier. Analytical TLC was performed using Merck siliga gel F254 (230 - 400 mesh) plates and analyzed by heating upon staining with KMnO4 solution. For silica gel chromatography, the flash chromatography technique was used, with Merck silica gel 60 (230 - 400 mesh) and p.a. grade solvents were used. 1H (399.98 MHz) and 13C NMR (100.59 MHz) spectra were recorded on a Bruker Avance 400 spectrometer in CDCl3. The chemical shifts are reported in ppm relative to TMS (δ 0.00) for 1H NMR and in ppm relative to CDCl3 (δ 77.0) for 13C NMR. IR spectra were recorded on a Perkin-Elmer Spectrum One FTIR spectrometer. High resolution mass spectrometric data were obtained using MicroMass LCT Premier Spectrometer. The enantiomeric excess (ee) values of the products were determined by HPLC analysis.
4.2. General Procedures for Aldol Preparation
In self-aldol reactions (Entries 1 - 5, Table 1) aldehyde (2 mmol) was dissolved in DMSO (2 mL). H2O (0.04 mL), catalyst 1 (0.15 mmol) and AcOH (0.15 mmol) were added. The mixture was stirred at room temperature for 20 - 21 h. The reaction mixture was quenched with saturated aqueous NH4Cl solution. The layers were separated and the aqueous layer was extracted with Et2O. Combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Crude products were purified with flash chromatography (20% Et2O/hexane).
In crossed aldol reactions (Entries 6 - 8, Table 1) isobutyraldehyde (6 mmol) was dissolved in DMF (2 mL). H2O (0.04 mL), catalyst 1 (0.15 mmol) and AcOH (0.15 mmol) were added. The linear aldehyde (1 mmol) in DMF (5 mL) was added during 48 h at 0˚C - 4˚C and the mixture was stirred further 2 - 5 h. The reaction mixture was quenched with saturated aqueous NH4Cl solution. The layers were separated and the aqueous layer was extracted with Et2O. Combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure. Crude products were purified with flash chromatography (20% Et2O/hexane).
4.2.1. 2-Ethyl-3-hydroxyhexanal (2)
Colorless oil. Yield: 56%. IR: 3425, 2875, 2731, 1719 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.74 (1 H, d, J = 3.0 Hz, CHO), 3.90 - 3.86 (1 H, m, CHOH), 2.52 (1 H, brs, OH), 2.29 - 2.23 (1 H, m, CHCHO), 1.77 (1 H, dddd, J = 7.5, 7.5, 7.5, 14.0 Hz, CH3CH2CHCHO), 1.66 (1 H, dddd, J = 7.5, 7.5, 7.5, 14.0 Hz, CH3CH2CHCHO), 1.58 - 1.45 (4 H, m, CH3CH2CH2- CHOH), 0.95 (3 H, t, J = 7.5 Hz, CH3), 0.94 (3 H, t, J = 7.5 Hz, CH3); 13C NMR (100 MHz, CDCl3) δ: 206.0, 70.9, 58.7, 37.1, 19.3, 18.6, 13.8, 11.4; HRMS m/z [M + Na]+ calcd for C8H16O2: 167.1048, found: 167.1046. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.79 (1 H, d, J = 3.5 Hz, CHO), 8.03 - 8.00 (2 H, m, Ph), 7.60 - 7.55 (1 H, m, Ph), 7.47 - 7.42 (2 H, m, Ph), 5.26 (1 H, dt J = 4.5, 8.5 Hz, CHOAr), 2.54 - 2.48 (1 H, m, CHCHO), 1.85 - 1.75 (1 H, m, CH2CHCHO), 1.74 - 1.52 (3 H, m, CH2CHCHO, CH2CHOAr), 1.47 - 1.35 (2 H, m, CH3CH2CH2CHOAr), 0.97 (3 H, t, J = 7.5, CH3), 0.94 (3 H, t, J = 7.5, CH3); 13C NMR (CDCl3): δ 203.0, 166.0, 133.2, 129.7, 128.9, 128.5, 73.4, 57.1, 34.6, 19.2, 18.7, 13.8, 11.7] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 6.2 min, minor isomer: tr = 6.7 min.
4.2.2. 3-Hydroxy-2-propylheptanal (3)
Colorless oil. Yield: 47%. IR: 3425, 2873, 2730, 1721 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.75 (1 H, d, J = 3.0 Hz, CHO), 3.87 - 3.81 (1 H, m, CHOH), 2.35 (1 H, dddd, J = 3.0, 5.5, 5.5, 8.0 Hz, CHCHO), 2.10 (1 H, brs, OH), 1.77 - 1.67 (1 H, m, CH3- CH2CH2CHCHO), 1.61 - 1.42 (3 H, m, CH3CH2CH2- CHCHO, CH3CH2CH2CH2CHOH), 1.40 - 1.29 (6 H, m, CH3CH2CH2CHCHO, CH3CH2CH2CH2CHOH), 0.94 (3 H, t, J = 7.0, CH3), 0.92 (3 H, t, J = 7.0, CH3); 13C NMR (100 MHz, CDCl3) δ: 205.8, 71.6, 57.0, 34.8, 28.5, 27.7, 22.6, 20.4, 14.1, 14.0; HRMS m/z [M + Na]+ calcd for C10H20O2: 195.1361, found: 195.1356. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.78 (1 H, d, J = 3.5 Hz, CHO), 8.03 - 8.00 (2 H, m, Ph), 7.60 - 7.55 (1 H, m, Ph), 7.47 - 7.43 (2 H, m, Ph), 5.44 (1 H, dt J = 5.0, 8.0 Hz, CHOAr), 2.66 - 2.57 (1 H, m, CHCHO), 1.81 - 1.60 (4 H, m, CH3CH2CH2CHCHO, CH3CH2CH2- CH2CHOH), 1.56 - 1.26 (6 H, m, CH3CH2CH2CHCHO, CH3CH2CH2CH2CHOH), 0.97 (3 H, t, J = 7.0, CH3), 0.91 (3 H, t, J = 7.0, CH3); 13C NMR (CDCl3): δ 203.0, 169.5, 133.2, 129.9, 129.7, 128.5, 73.9, 55.3, 35.0, 28.1, 27.5, 22.5, 20.4, 13.9, 13.7] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 6.3 min, minor isomer: tr = 6.8 min.
4.2.3. 2-Butyl-3-hydroxyoctanal (4)
Colorless oil. Yield: 67%. IR: 3426, 2860, 2728, 1720 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.74 (1 H, d, J = 3.0 Hz, CHO), 3.86 - 3.82 (1 H, m, CHOH), 2.33 (1 H, dddd, J = 3.0, 5.5, 5.5, 8.5 Hz, CHCHO), 2.06 (1 H, brs, OH), 1.78 - 1.68 (1 H, m, CH2CHCHO), 1.63 - 1.45 (3 H, m, CH2CHCHO, CH2CHOH), 1.38 - 1.26 (10 H, m, CH2), 0.91 (3 H, t, J = 7.0, CH3), 0.90 (3 H, t, J = 7.0, CH3); 13C NMR (100 MHz, CDCl3) δ: 205.8, 71.6, 57.2, 35.1, 31.7, 29.3, 26.1, 25.2, 22.8, 22.6, 14.0, 13.8; HRMS m/z [M + Na]+ calcd for C12H24O2: 223.1674, found: 223.1664. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.77 (1 H, d, J = 3.5 Hz, CHO), 8.04 - 7.99 (2 H, m, Ph), 7.61 - 7.55 (1 H, m, Ph), 7.48 - 7.43 (2 H, m, Ph), 5.44 (1 H, ddd, J = 5.0, 5.0, 8.0 Hz, CHOAr), 2.58 (1 H, dddd, J = 3.5, 5.0, 5.0, 9.0 Hz, CHCHO), 1.84 - 1.64 (3 H, m, CH2CHCHO, CH2CHOAr), 1.63 - 1.46 (1 H, m, CH2CHCHO), 1.41 - 1.21 (10 H, m, CH2), 0.93 - 0.82 (6 H, m, CH3); 13C NMR (CDCl3): δ 203.0, 165.9, 133.2, 129.9, 129.7, 128.5, 73.9, 55.4, 32.4, 31.5, 29.3, 25.7, 25.0, 22.6, 22.4, 13.9, 13.8] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 5.7 min, minor isomer: tr = 6.0 min.
4.2.4. 3-Hydroxy- 2-pentylnonanal (5)
Colorless oil. Yield: 45%. IR: 3426, 2858, 2728, 1720 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.74 (1 H, d, J = 3.0 Hz, CHO), 3.86 - 3.82 (1 H, m, CHOH), 2.33 (1 H, dddd, J = 3.0, 5.5, 5.5, 8.0 Hz, CHCHO), 2.04 (1 H, brs, OH), 1.77 - 1.67 (1 H, m, CH2CHCHO), 1.63 - 1.45 (3 H, m, CH2CHCHO, CH2CHOH), 1.37 - 1.25 (14 H, m, CH2), 0.89 (6 H, t, J = 7.0, CH3) ); 13C NMR (100 MHz, CDCl3) δ: 205.9, 71.6, 57.3, 35.1, 31.9, 31.8, 29.2, 26.8, 26.3, 25.5, 22.6, 22.4, 14.0, 13.9; HRMS m/z [M + Na]+ calcd for C14H28O2: 251.1987, found: 251.1988. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.77 (1 H, d, J = 3.5 Hz, CHO), 8.03 - 7.99 (2 H, m, Ph), 7.60 - 7.55 (1 H, m, Ph), 7.49 - 7.43 (2 H, m, Ph), 5.44 (1 H, ddd, J = 5.0, 5.0, 8.0 Hz, CHOAr), 2.58 (1 H, dddd, J = 3.5, 5.0, 5.0, 8.5 Hz, CHCHO), 1.82 - 1.65 (3 H, m, CH2CHCHO, CH2CHOAr), 1.64 - 1.48 (1 H, m, CH2CHCHO), 1.41 - 1.16 (12 H, m, CH2), 0.88 (3 H, t, J = 7.0 Hz, CH3), 0.86 (3 H, t, J = 7.0 Hz, CH3); 13C NMR (CDCl3): δ 203.0, 166.0, 133.1, 130.0, 129.7, 128.5, 73.9, 55.5, 32.5, 31.7, 31.6, 29.0, 26.8, 26.0, 25.3, 22.5, 22.4, 14.2, 14.0] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 5.5 min, minor isomer: tr = 5.7 min.
4.2.5. 2-Benzyl-3-hydroxy-5-phenylpentanal (6)
Colorless oil. Yield: 71%. IR: 3441, 2861, 2733, 1720 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.80 (1 H, d, J = 2.0 Hz, CHO), 7.31 - 7.26 (4 H, m, Ph), 7.25 - 7.14 (6 H, m, Ph), 3.81 (1 H, ddd, J = 4.0, 6.0, 8.0 Hz, CHOH), 3.04 (1 H, dd, J = 8.0, 14.0 Hz, CH2- CHCHO), 2.95 (1 H, dd, J = 7.0, 14.0 Hz, CH2CHCHO), 2.81 (1 H, ddd, J = 6.0, 9.0, 13.5 Hz, CH2CH2CHOH), 2.77 - 2.71 (1 H, m, CHCHO), 2.66 (1 H, ddd, J = 7.0, 9.0, 14.0 Hz, CH2CH2CHOH), 2.17 (1 H, d, J = 6.0 Hz, OH), 1.94 - 1.87 (2 H, m, CH2CH2CHOH); 13C NMR (100 MHz, CDCl3) δ: 205.1, 141.4, 138.2, 129.0, 128.7, 128.5, 128.4, 126.6, 126.0, 70.7, 58.1, 37.0, 32.6, 32.1; HRMS m/z [M + Na]+ calcd for C18H20O2: 291.1361, found: 291.1368. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.87 (1 H, d, J = 2.5 Hz, CHO), 8.02 - 7.99 (2 H, m, Ph), 7.63 - 7.57 (1 H, m, Ph), 7.49 - 7.44 (2 H, m, Ph), 7.32 - 7.08 (10 H, m, Ph), 5.50 (1 H, ddd, J = 4.0, 4.5, 8.5 Hz, CHOAr), 3.12 (1 H, dd, J = 8.0, 14.0 Hz, CHOCHCH2), 3.05 - 2.99 (1 H, m, CHOCHCH2), 2.86 (1 H, dd, J = 6.0, 14.0 Hz, CHOCHCH2), 2.79 - 2.66 (2 H, m, CH2CH2CHOAr), 2.27 - 2.16 (1 H, m, CH2CH2CHOAr), 2.10 - 2.00 (1 H, m, CH2CH2CHOAr); 13C NMR (CDCl3): δ 201.7, 165.9, 142.8, 138.1, 133.3, 130.6, 129.7, 129.0, 128.9, 128.7, 128.5, 128.3, 126.6, 126.2, 73.1, 56.8, 34.2, 31.9, 29.6] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 9.4 min, minor isomer: tr = 11.0 min.
4.2.6. 2-Ethyl-3-hydroxy-4-methylpentanal (7)
Colorless oil. Yield: 81%. IR: 3449, 2876, 2724, 1719 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.77 (1 H, d, J = 3.0 Hz, CHO), 3.59 (1 H, t, J = 6.0 Hz, CHOH), 2.40 (1 H, dddd, J = 3.0, 5.5, 6.0, 8.5 Hz, CHCHO), 1.87 - 1.79 (1 H, m, (CH3)2CH), 1.78 - 1.59 (2 H, m, CH2CHCHO), 0.98 (3 H, d, J = 6.5 Hz, (CH3)2CH), 0.95 (3 H, t, J = 6.5 Hz, CH3CH2CHCHO), 0.94 (3 H, d, J = 6.5 Hz, (CH3)2CH); 13C NMR (100 MHz, CDCl3) δ: 206.1, 76.1, 56.0, 30.9, 19.7, 19.6, 16.7, 11.5; HRMS m/z [M + Na]+ calcd for C8H16O2: 167.1048, found: 167.1052. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.77 (1 H, d, J = 4.0 Hz, CHO), 8.07 - 8.01 (2 H, m, Ph), 7.65 - 7.59 (1 H, m, Ph), 7.50 - 7.43 (2 H, m, Ph), 5.28 (1 H, dd, J = 5.0, 7.0 Hz, CHOAr), 2.60 - 2.54 (1 H, m, CHCHO), 2.10 (1 H, oct, J = 7.0 Hz, (CH3)2CH), 1.81 - 1.71 (1 H, m, CHOCHCH2), 1.65 - 1.55 (1 H, m, CHOCHCH2), 1.02 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.99 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.96 (3 H, t, J = 7.5 Hz, CH3CH2); 13C NMR (CDCl3): δ 203.2, 162.3, 133.2, 129.8, 129.7, 128.5, 78.1, 55.4, 30.6, 19.8, 19.2, 17.9, 11.6] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 6.0 min, minor isomer: tr = 5.5 min.
4.2.7. 3-Hydroxy-4-methyl-2-propylpentanal (8)
Colorless oil. Yield: 73%. IR: 3452, 2874, 2729, 1717 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.76 (1 H, d, J = 3.0 Hz, CHO), 3.56 (1 H, q, J = 6.0 Hz, CHOH), 2.48 (1 H, dddd, J = 3.0, 5.5, 6.0, 8.5 Hz, CHCHO), 1.99 (1 H, d, J = 6.0 Hz, OH), 1.88 - 1.78 (1 H, m, (CH3)2CH), 1.78 - 1.66 (1 H, m, CH3CH2CH2), 1.62 - 1.49 (1 H, m, CH3CH2CH2), 1.35 (2 H, sext, J = 7.5 Hz, CH3CH2), 0.97 (3 H, d, J = 6.0 Hz, (CH3)2CH), 0.95 (3H, d, J = 6.0 Hz, (CH3)2CH), 0.94 (3 H, t, J = 7.5 Hz, CH3CH2); 13C NMR (100 MHz, CDCl3) δ: 205.9, 76.5, 54.4, 31.0, 28.8, 20.3, 19.6, 16.7, 14.1; HRMS m/z [M+Na]+ calcd for C9H18O2: 181.1204, found: 181.1202. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.76 (1 H, d, J = 4.0 Hz, CHO), 8.05 - 8.01 (2 H, m, Ph), 7.60 - 7.55 (1 H, m, Ph), 7.49 - 7.43 (2 H, m, Ph), 5.26 (1 H, dd, J = 5.0, 7.0 Hz, CHOAr), 2.70 - 2.61 (1 H, m, CHCHO), 2.10 (1 H, oct, J = 7.0 Hz, (CH3)2CH), 1.77 - 1.66 (1 H, m, CHOCHCH2), 1.55 - 1.45 (1 H, m, CHOCHCH2), 1.41 - 1.30 (2 H, m, CH3CH2CH2), 1.02 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.98 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.91 (3 H, t, J = 7.0 Hz, CH3CH2CH2); 13C NMR (CDCl3): δ 203.3, 166.1, 133.2, 129.8, 129.7, 128.5, 78.3, 53.6, 30.6, 28.6, 20.3, 19.2, 17.9, 13.9] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 6.0 min, minor isomer: tr = 5.4 min.
4.2.8. 2-(1-Hydroxy-2-methylpropyl)hexanal (9)
Colorless oil. Yield: 60%. IR: 3453, 2873, 2729, 1720 cm−1; for main diastereomer: 1H NMR (400 MHz, CDCl3) δ: 9.75 (1 H, d, J = 3.0 Hz, CHO), 3.57 (1 H, t, J = 5.5 Hz, CHOH), 2.46 (1 H, dddd, J = 3.0, 5.5, 5.5, 8.5 Hz, CHCHO), 1.88 - 1.78 (1 H, m, (CH3)2CH), 1.77 - 1.67 (1 H, m, CH2CHCHO), 1.63 - 1.54 (1 H, m, CH2CHCHO), 1.38 - 1.25 (4 H, m, CH3CH2CH2), 0.97 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.94 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.91 (3 H, t, J = 7.0 Hz, CH2CH3); 13C NMR (100 MHz, CDCl3) δ: 206.0, 76.5, 54.6, 31.0, 29.1, 26.4, 22.8, 19.6, 16.7, 13.8; HRMS m/z [M + Na]+ calcd for C10H20O2: 195.1361, found: 195.1361. Data of 9 in accordance with literature values [2a]. The enantiomeric purity was determined by HPLC analysis of the corresponding benzoate ester [1H NMR (CDCl3): δ 9.76 (1 H, d, J = 4.0 Hz, CHO), 8.04 - 8.01 (2 H, m, Ph), 7.60 - 7.55 (1 H, m, Ph), 7.48 - 7.43 (2 H, m, Ph), 5.27 (1 H, dd, J = 5.0, 7.0 Hz, CHOAr), 2.68 - 2.61 (1 H, m, CHCHO), 2.10 (1 H, oct, J = 7.0 Hz, (CH3)2CH), 1.78 - 1.64 (1 H, m, CHOCHCH2), 1.58 - 1.48 (1 H, m, CHOCHCH2), 1.38 - 1.22 (4 H, m, CH3CH2CH2), 1.02 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.98 (3 H, d, J = 7.0 Hz, (CH3)2CH), 0.92 - 0.86 (3 H, m, CH3CH2CH2); 13C NMR (CDCl3): δ 203.3, 161.4, 133.2, 129.8, 129.7, 128.5, 78.3, 53.8, 30.6, 29.2, 22.6, 22.2, 19.2, 17.8, 13.9] using Daicel Chiralpak IA column (25 cm) along with precolumn (5 cm). Eluent: 0.5% ethanol in hexane; Flow rate: 1.0 mL/min; λ = 220 nm; Major isomer: tr = 5.8 min, minor isomer: tr = 5.4 min.
REFERENCES
- T. D. Machajewski and C.-H. Wong, “The Catalytic Asymmetric Aldol Reaction,” Angewandte Chemie International Edition, Vol. 39, No. 8, 2000, pp. 1352-1375. doi:10.1002/(SICI)1521-3773(20000417)39:8<1352::AID-ANIE1352>3.0.CO;2-J
- A. B. Northrup and D. W. C. MacMillan, “The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes,” Journal of the American Chemical Society, Vol. 124, No. 24, 2002, pp. 6798-6799. doi:10.1021/ja0262378
- P. Pihko and A. Erkkilä, “Enantioselective Synthesis of Prelactone B Using a Proline-Catalyzed Crossed-Aldol Reaction,” Tetrahedron Letters, Vol. 44, No. 41, 2003, pp. 7607-7609. doi:10.1016/j.tetlet.2003.08.060
- C. Pidathala, L. Hoang, N. Vignola and B. List, “Direct Catalytic Asymmetric Enolexo Aldolizations,” Angewandte Chemie International Edition, Vol. 42. No. 24, 2003, pp. 2785-2788. doi:10.1002/anie.200351266
- I. K. Mangion, A. B. Northrup and D. W. C. MacMillan, “The Importance of Iminium Geometry Control in Enamine Catalysis: Identification of a New Catalyst Architecture for Aldehyde-Aldehyde Couplings,” Angewandte Chemie International Edition, Vol. 43, No. 48, 2004, pp. 6722-6724. doi:10.1002/anie.200461851
- R. I. Storer and D. W. C. MacMillan, “Enantioselective Organocatalytic Aldehyde-Aldehyde Cross-Aldol Couplings. The Broad Utility of α-Thioacetal Aldehydes,” Tetrahedron, Vol. 60, No. 35, 2004, pp. 7705-7714. doi:10.1016/j.tet.2004.04.089
- A. B. Northrup, I. K. Mangion, F. Hettche and D. W. C. MacMillan, “Enantioselective Organocatalytic Direct Aldol Reactions of α-Oxyaldehydes: Step One in a TwoStep Synthesis of Carbohydrates,” Angewandte Chemie International Edition, Vol. 43, No. 16, 2004, pp. 2152- 2154. doi:10.1002/anie.200453716
- A. Córdova, “Direct Catalytic Asymmetric Cross-Aldol Reactions in Ionic Liquid Media,” Tetrahedron Letters, Vol. 45, No. 20, 2004, pp. 3949-3952. doi:10.1016/j.tetlet.2004.03.080
- S. Aratake, T. Itoh, T. Okano, T. Usui, M. Shoji and Y. Hayashi, “Small Organic Molecule in Enantioselective, Direct Aldol Reaction “in Water”,” Chemical Communications, No. 24, 2007, pp. 2524-2526.
- Y. Hayashi, S. Samanta, T. Itoh and H. Ishikawa, “Asymmetric, Catalytic, and Direct Self-Aldol Reaction of Acetaldehyde Catalyzed by Diarylprolinol,” Organic Letters, Vol. 10, No. 24, 2008, pp. 5581-5583. doi:10.1021/ol802438u
- Y. Chi, S. T. Scroggins, E. Boz and J. M. J. Fréchet, “Control of Aldol Reaction Pathways of Enolizable Aldehydes in an Aqueous Environment with a Hyperbranched Polymeric Catalyst,” Journal of the American Chemical Society, Vol. 130, No. 51, 2008, pp. 17287- 17289. doi:10.1021/ja806584q
- M. Markert, U. Scheffler and R. Mahrwald, “Asymmetric Histidine-Catalyzed Cross-Aldol Reactions of Enolizable Aldehydes: Access to Defined Configured Quaternary Stereogenic Centers,” Journal of the American Chemical Society, Vol. 131, No. 46, 2009, pp. 16642-16643. doi:10.1021/ja907054y
- Y. Hayashi, Y. Yasui, T. Kawamura, M. Kojima and H. Ishikawa, “One-Pot Synthesis of Chiral α-Substituted β,γ-Epoxy Aldehyde Derivatives through an Asymmetric Aldol Reaction of Chloroacetaldehyde,” Angewandte Chemie International Edition, Vol. 50, No. 12, 2011, pp. 2804-2807. doi:10.1002/anie.201005577
- T. Kano, H. Sugimoto and K. Maruoka, “Efficient Organocatalytic Cross-Aldol Reaction between Aliphatic Aldehydes through Their Functional Differentiation,” Journal of the American Chemical Society, Vol. 133, No. 45, 2011, pp. 18130-18133. doi:10.1021/ja208873k
- M. S. Sarkar, Y. Taira, A. Nakano, K. Takashaki, J. Ishihara and S. Hatakeyama, “Organocatalytic Asymmetric Synthesis of Quinine and Quindine,” Tetrahedron Letters, Vol. 52, No. 8, 2011, pp. 923-927. doi:10.1016/j.tetlet.2010.12.066
- C.-S. Da, L.-P. Che, Q.-P. Guo, F.-C. Wu, X. Ma and Y.-N. Jia, “2,4-Dinitrophenol as an Effective Cocatalyst: Greatly Improving the Activities and Enantioselectivities of Primary Amine Organocatalysts for Asymmetric Aldol Reactions,” Journal of Organic Chemistry, Vol. 74, No. 6, 2009, pp. 2541-2546. doi:10.1021/jo802758b
- M. Raj, Vishnumaya, S. K. Ginotra and V. K. Singh, “Highly Enantioselective Direct Aldol Reaction Catalyzed by Organic Molecules,” Organic Letters, Vol. 8, No. 18, 2006, pp. 4097-4099. doi:10.1021/ol0616081
- For a review: S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, “Asymmetric Enamine Catalysis,” Chemical Reviews, Vol. 107, No. 12, 2007, pp. 5471-5569. doi:10.1021/cr0684016
- V. Maya, M. Raj and V. K. Singh, “Highly Enantioselective Organocatalytic Direct Aldol Reaction in an Aqueous Medium,” Organic Letters, Vol. 9, No. 13, 2007, pp. 2593-2595. doi:10.1021/ol071013l
- L. Song, X. Chen, S. Zhang, H. Zhang, P. Li, W. Duan and W. Wang, “An Organocatalytic Approach to the Construction of Chiral Oxazolidinone Rings and Application in the Synthesis of Antibiotic Linezolid and Its Analogues,” Organic Letters, Vol. 10, No. 23, 2008, pp. 5489-5492. doi:10.1021/ol802333n
- M. Raj, V. Maya and V. K. Singh, “Highly Efficient Small Organic Molecules for Enantioselective Direct Aldol Reaction in Organic and Aqueous Media,” Journal of Organic Chemistry, Vol. 74, No. 11, 2009, pp. 4289-4297. doi:10.1021/jo900548f
- M. Raj and V. K. Singh, “Organocatalytic Reactions in Water,” Chemical Communications, No. 44, 2009, pp. 6687-6703.
- K. Baer, M. Kraußer, E. Burda, W. Hummel, A. Berkessel and H. Gröger, “Sequential and Modular Synthesis of Chiral 1,3-Diols with Two Stereogenic Centers: Access to All Four Stereoisomers by Combination of Organoand Biocatalysis,” Angewandte Chemie International Edition, Vol. 48, No. 49, 2009, pp. 9355-9358. doi:10.1002/anie.200900582
- H. Zhang, S. Zhang, L. Liu, G. Luo, W. Duan and W. Wang, “Synthesis of Chiral 3-Substituted Phthalides by a Sequential Organocatalytic Enantioselective Aldol-Lactonization Reaction. Three-Step Synthesis of (S)-(–)-3-Butylpthalide,” Journal of Organic Chemistry, Vol. 75, No. 2, 2010, pp. 368-374. doi:10.1021/jo902118x
- G. Rulli, N. Duangdee, K. Baer, W. Hummel, A. Berkessel and H. Gröger, “Direction of Kinetically versus Thermodynamically Controlled Organocatalysis and Its Application in Chemoenzymatic Synthesis,” Angewandte Chemie International Edition, Vol. 50, No. 34, 2011, pp. 7944-7947. doi:10.1002/anie.201008042
- Z. Tang, F. Jiang, L.-T. Yu, X. Cui, L.-Z. Gong, A.-Q. Mi, Y.-Z. Jiang and Y.-D. Wu, “Novel Small Organic Molecules for a Highly Enantioselective Direct Aldol Reaction,” Journal of the American Chemical Society, Vol. 125, No. 18, 2003, pp. 5262-5263. doi:10.1021/ja034528q
- S. Sulzer-Mossé and A. Alexakis, “Chiral Amines as Organocatalysts for Asymmetric Conjugate Addition to Nitroolefins and Vinyl Sulfones via Enamine Activation,” Chemical Communications, No. 30, 2007, pp. 3123-3135.
- H. Flink, T. Putkonen and R. Jokela, “Microwave-Assisted Selective Protection of Glutaraldehyde and Its Symmetrical Derivatives as Monoacetals and Thioacetals,” Tetrahedron, Vol. 66, No. 4, 2010, pp. 887-890. doi:10.1016/j.tet.2009.11.107
- H. Flink and R. Jokela, “Microwave-Assisted Total Synthesis of Tangutorine,” Tetrahedron, Vol. 68, No. 20, 2012, pp. 3811-3814. doi:10.1016/j.tet.2012.03.068