Journal of Electromagnetic Analysis and Applications
Vol. 2 No. 11 (2010) , Article ID: 3294 , 11 pages DOI:10.4236/jemaa.2010.211080
Inspections of Mobile Phone Microwaves Effects on Proteins Secondary Structure by Means of Fourier Transform Infrared Spectroscopy
![]()
1 Department of Physics, University of Messina, Messina, Italy.
Email: e.calabro@yahoo.com
Received September 10th, 2010; revised October15th, 2010; accepted October 18th, 2010.
Keywords: Mobile Phone, Microwaves, Protein, Infrared Spectrum, Secondary Structure, Amide I
ABSTRACT
In this study the effects of microwaves on the secondary structure of three typical proteins have been investigated. A set of samples of lysozyme, bovine serum albumin and myoglobin in D2O solutions were exposed for 8 hours to mobile phone microwaves at 900 MHz at a magnetic field intensity around 16 mA/m. The relative effects on the secondary structure of the proteins were studied by means of Fourier Transform Infrared Spectroscopy. An increase of the amide I band intensity in the secondary structure of the proteins was observed after the microwaves exposure. Furthermore, a weak shift of the amide I mode of bovine serum albumin and a heavier shift of the amide I of myoglobin occurred after the exposure. In addition, a clear increasing of the β-sheet components with respect to the α-helix content was observed in the spectra of bovine serum albumin and myoglobin after the exposure, suggesting the hypothesis of the formation of aggregates.
1. Introduction
Mobile phones and related telephony technologies transmit information that is encoded into electromagnetic waves in the microwave range around 900 MHz and 1800 MHz.
The Global System for Mobile Communications (GSM) radiation is characterized by a high frequency (HF) carrier wave periodically pulsed at low frequency (LF). Analogical telephones work with a frequency modulation system whereas digital telephones emit pulsed microwaves (MWs). Modulation refers to the patterns of change in the frequency or amplitude of the radiofrequency (RF) carrier wave.
The carrier wave has a specific impulse sequence. Every impulse sequence lasts 120 ms , it is made up by 26 impulses, and each one lasts 4.6 ms. The frequencies of these two periods are 8.3 Hz and 217 Hz. As cellular telephone technology has advanced, the modulation patterns have become increasingly complex using extremely low frequency (2 Hz-17.6 Hz).
The geographical area around a GSM base station for which it provides coverage is known as a cell that may be divided into sectors, in which case the base station transmits different frequencies into the different sectors.
The power level generated by a mobile phone to maintain a good connection depends on the distance from the base station. The power output is less when the base station is close to the phone user, so that base stations are being placed closer together to minimize the power output of a mobile phone. Hence, exposures at particular locations will be largely determined by the local power density, which can be measured.
People generally questions whether there are possible health consequences of this mushrooming mobile phone technology, particularly because the handset operates in close proximity to the human body.
Previous studies on the effects of mobile phone microwaves on human health have been carried up to now following the enormous increase of mobile telephony throughout the world.
Numerous observations indicate that in addition to heating, mobile phone microwave radiation might also induce non-thermal effects in narrow frequency windows that could not be explained by bulk heating.
Many scientific studies have investigated possible health effects of mobile phone radiations. An assessment published in 2007 by the European Commission Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR) regarding mobile phone radiation effects on human health concluded that despite no significant health effect has been demonstrated, more studies concerning health effects on children are needed [1].
As cellular telephone technology has advanced, the radiofrequency patterns used become more and more complex with lower frequencies, nearer to biological ones and so they may interfere with the sophisticated electromagnetic circuits of human body, for example in the brain. Most governments and cell-phone companies have claimed that the only possible biological effect of RF transmission is localized body heating.
RF-MW radiations produce a response in many types of neurones in the avian central nervous system [2], and can affect central cholinergic activity in the rat [3].
Mobile phone MWs have effects on blood-brain barrier permeability [4,5] and can produce oxidative damage in brains tissues [6].
In vitro tests demonstrated that RF-MWs can be carcinogens [7]. Transgenic mice most susceptible to cancer demonstrated a 2-times increase in tumor rate (B-cell lymphomas) after exposure to microwaves at a power density roughly equal to a cell-phone transmitting for two half-hour periods each day, compared to control mice unexposed to RF fields [8].
Further significant concern has been raised about the capacity of RF-MW radiations to cause DNA damage and chromosomes aberrations [9-13]. Hence, national radiation advisory authorities recommends measurements to minimize exposure to their citizens, but the exposure limits to electromagnetic fields recommended by the International Commission on Non-Ionizing Radiation Protection (I.C.N.I.R.P.) [14] can be exceeded during a call at a mobile phone as reported in [15].
Moreover, proteins are fundamentals in organic metabolism of livings. In the cells each protein must fold into the specific conformational state in a complex and highly crowded environment, and the folding process is aided by a range of auxiliary proteins [16,17].
Otherwise it was largely demonstrated that several type of environmental stress agents can alter the secondary structure of proteins.
Recently it was proved that also extremely low frequency electromagnetic fields (ELF-EMFs) can alter the secondary structure of proteins [18,19].
Resonance absorption by low-frequency vibrations was suggested by [20] as a molecular mechanism that would allow non-thermal coupling of microwave fields to protein dynamics.
Unfolding and refolding processes of β-lactoglobulin in urea solution measured using optical rotation dispersion has been observed to be accelerated by microwave irradiation at 2.45 GHz [21].
Small-angle X-ray scattering from solutions of bovine serum albumine was reported to show marked differences between samples that had been heated by microwaves at 2.45 GHz and those heated in a water bath to the same temperature [22], which was related to denaturation that could be obtained only by microwave heating.
X-ray diffraction measurements in tetragonal single crystals of hen egg-white lysozyme obtained under controlled continuous microwave irradiation at 8 GHz provided an increase of the average B factor (the mean-square displacements of individual atoms) with microwave power that can not be explained by heating [23].
Myoglobin solution at pH = 3 was exposed to MWs at 1.95 GHz and the heme site refolding was followed by measuring the molecular absorption, providing the result that the kinetics of exposed samples was slowered by MW [24].
The experimental data mentioned above led us to focus on the effects of mobile phone MWs on the secondary structure of three typical proteins: lysozyme, bovine serum albumin and myoglobin.
Lysozyme (LYS) is a residue protein found in secretions (e.g., saliva, sweat, and mucus) and more generally in leukocytes and kidneys.
Bovine serum albumin (BSA) is the most abundant of the proteins in blood plasma, which have the interesting properties in the binding a variety of hydrophobic ligands in the transport and deposition of a variety of endogenous and exogenous substances in blood.
Myoglobin (MB) is an iron-oxygen-binding protein found in the muscle tissue of vertebrates, related to hemoglobin particularly in the red blood cells, which is released in the blood stream following a muscle injury.
Fourier Transform Infrared (FTIR) Spectroscopy was used to investigate the structure of the proteins in solution, because it may be considered the most versatile spectroscopic technique for analyzing the secondary structure of a protein in diverse physiochemical environments.
2. Materials and Methods
2.1. Lysozyme, Bovine Serum Albumin and Myoglobin Sample
LYS, BSA and MB were purchased from Sigma Aldrich. Prior to infrared spectroscopy, the proteins were dissolved in D2O at 20°C at the concentration of 100 mg/ml.
H2O is the ideal solvent for biological sample, but the intense H2O bending mode at 1640 cm-1 coincides with the amide I region from 1610 to 1690 cm-1. To overcome H2O interference, the majority of FTIR investigations have focused on the amide I′ band in D2O solution. The deuterium-exchanged amide bands are referred to as amide I′, amide II′ etc., in the spectroscopy literature.
2.2. Experimental Design
The exposure system consisted of an operational mobile phone by which the microwaves were transmitted. The signal provider was Vodafone and the micro-cell was that relative to the Department of Physics of the University of Messina (Italy).
The exposure source was a mobile phone Nokia 1100, to which a sound was transmitted from a Nokia 1200. The relative sound spectrum analysis (at 1/3 octave band) of the energy average sound level Leq was acquired by a precision integrating sound level meter, the Larson Davis LxT. It was used to acquire the octave frequency analysis of the sound used to generate the mobile phone MWs during the entire exposure, and is represented in Figure 1.
Samples of 250 µL of the proteins LYS, BSA and MB were placed at three centimeters from the mobile phone, and exposed to the relative GSM 900 MHz microwaves.
The intensity of the magnetic component was in the range from 8 to 25 mA/m during the entire exposure, as can be observed in the Time Analysis represented in Figure 4.
Analogue unexposed samples at the same room temperature were used as the control.
2.3. Microwaves Measurements
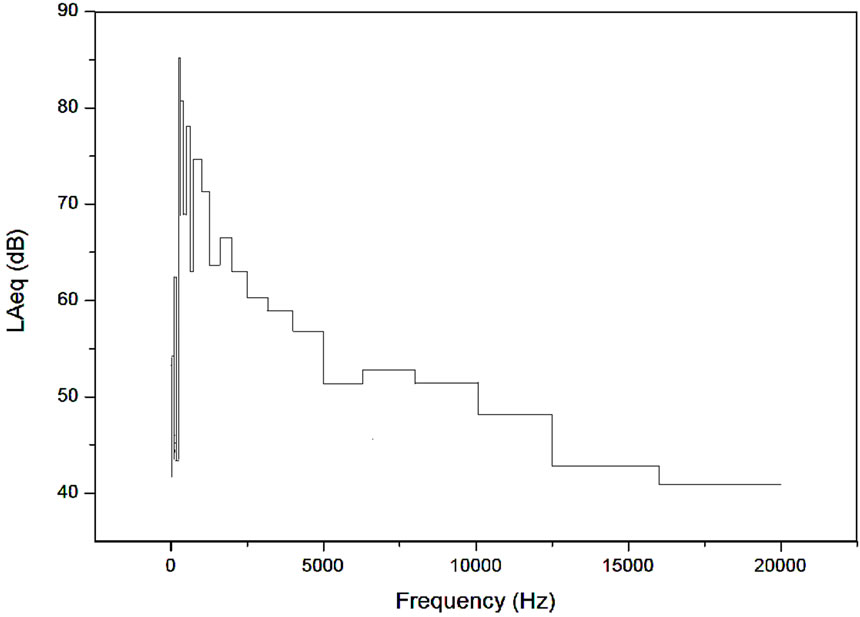
Figure 1. Spectrum analysis (1/3 octave band) of the energy average sound level Leq of a sound transmitted from a mobile phone Nokia 1200 to a Nokia 1100. The octave frequency spectrum analysis was acquired by a precision integrating sound level meter, the Larson Davis LxT, during the exposure.
A SRM-3000 instrument of Narda Safety Test Solutions was chosen to measure the electromagnetic field components emitted by the mobile phone. The device was linked through a cable to a Narda three axis antenna covering the frequency range from 75 MHz to 3 GHz. The Spectrum Analysis mode of the device was chosen as preliminary analysis to detect the MWs frequencies values impinging the three axis antenna, as accurately described in [15].
The different frequencies transmitted by the local base station corresponded to the values of 898.5, 899, 900.3, 900.8, 901.4 and 906.2 MHz, as represented in Figure 2.
Better results were obtained by a resolution bandwidth RBW = 6 MHz and the Average Mode of the measured values were taken over a number of 16 results.
The intensities of the power density and magnetic components of MWs were continuously monitored by Time Analysis mode, whose representative analyses are shown in Figure 3 and Figure 4, respectively. This operating mode allowed timer-controlled measurements at a defined frequency, monitoring the EMF level at the selected channel.
Spectrum and time analyses results were transferred to a PC where the electromagnetic components related to MWs emitted by the mobile phone were monitored during the entire exposure.
The mobile phones, Nokia 1200 and Nokia 1100, were used to generate the MW radiations to which the proteins were exposed.
2.4. Infrared Spectroscopy
FTIR spectra were recorded by a spectrometer, Vertex 80v, from Bruker Optics.
The attenuated total reflection (ATR) method was chosen for spectrum collection. In fact, FTIR tecnique requires the placement of proteins in solution between two calcium fluoride windows, with a potential for solvent interference and the ATR technique is the desired method to overcome solvent masking, since the penetration depth of infrared light is inherently limited to a fraction of the wavelength of about λ/10 [25] allowing secondary structure analyses on small volumes of protein in H2O or D2O solutions.
The proteins in D2O solution samples were placed between a pair of CaF2 windows separated with a 25 µm Teflon spacer.
For each spectrum, 64 interferograms were collected with a spectral resolution of 4 cm-1 in the range from 7500 to 350 cm-1.
IR spectra of the D2O solution were subtracted from the spectra of the protein at the corresponding temperature. Each measure was performed under vacuum to
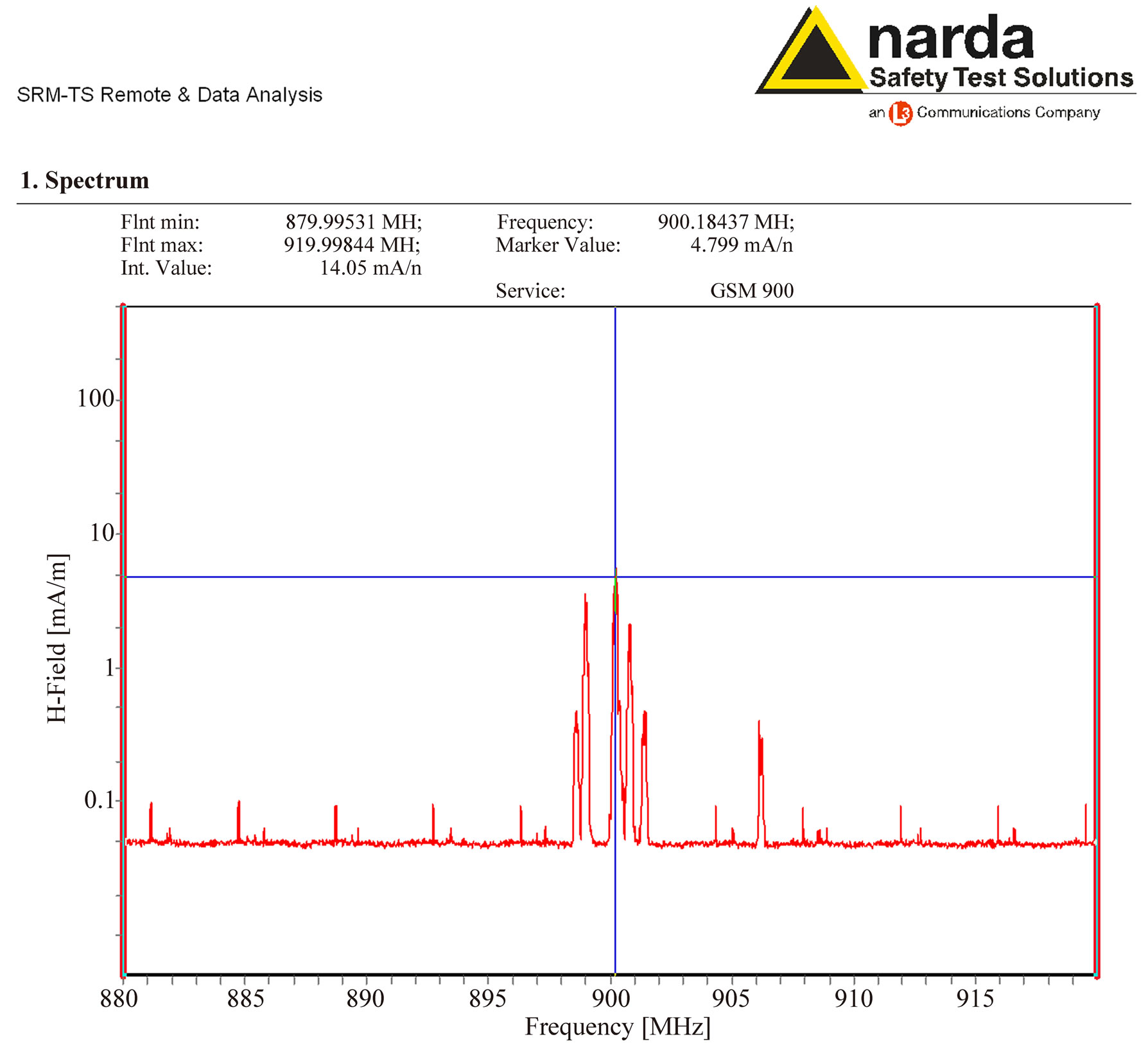
Figure 2. Spectrum analysis of the magnetic field component of the microwaves emitted from the mobile phone Nokia 1100 around 900 MHz, measured by a Narda SRM 3000 during the exposure. The frequency peaks at 898.5, 899, 900.3, 900.8, 901.4 and 906.2 MHz can be observed.
eliminate minor spectral contributions due to residual water vapor.
IR spectra were baseline-corrected and area-normalized for exposed proteins solutions and control samples. In particular, vector normalization was used, calculating the average value of the spectrum and subtracting from the spectrum decreasing the mid-spectrum. The sum of the squares of all values was calculated, and the spectrum was divided by the square root of this sum. The automatic baseline scattering correction function was used to subtract baselines from spectra, which allows getting spectra with band edges of up to the theoretical baseline.
In addition, interactive baseline rubber band correction was used. This method also uses a rubber band that is stretched from one spectrum end to the other, and the band is pressed onto the spectrum from the bottom up with varying intensity. The resulting spectrum will be the original spectrum minus the baseline points manually set and a subsequent concave rubber-band correction. We used the default value of n = 64 baseline points and 80 iterations.
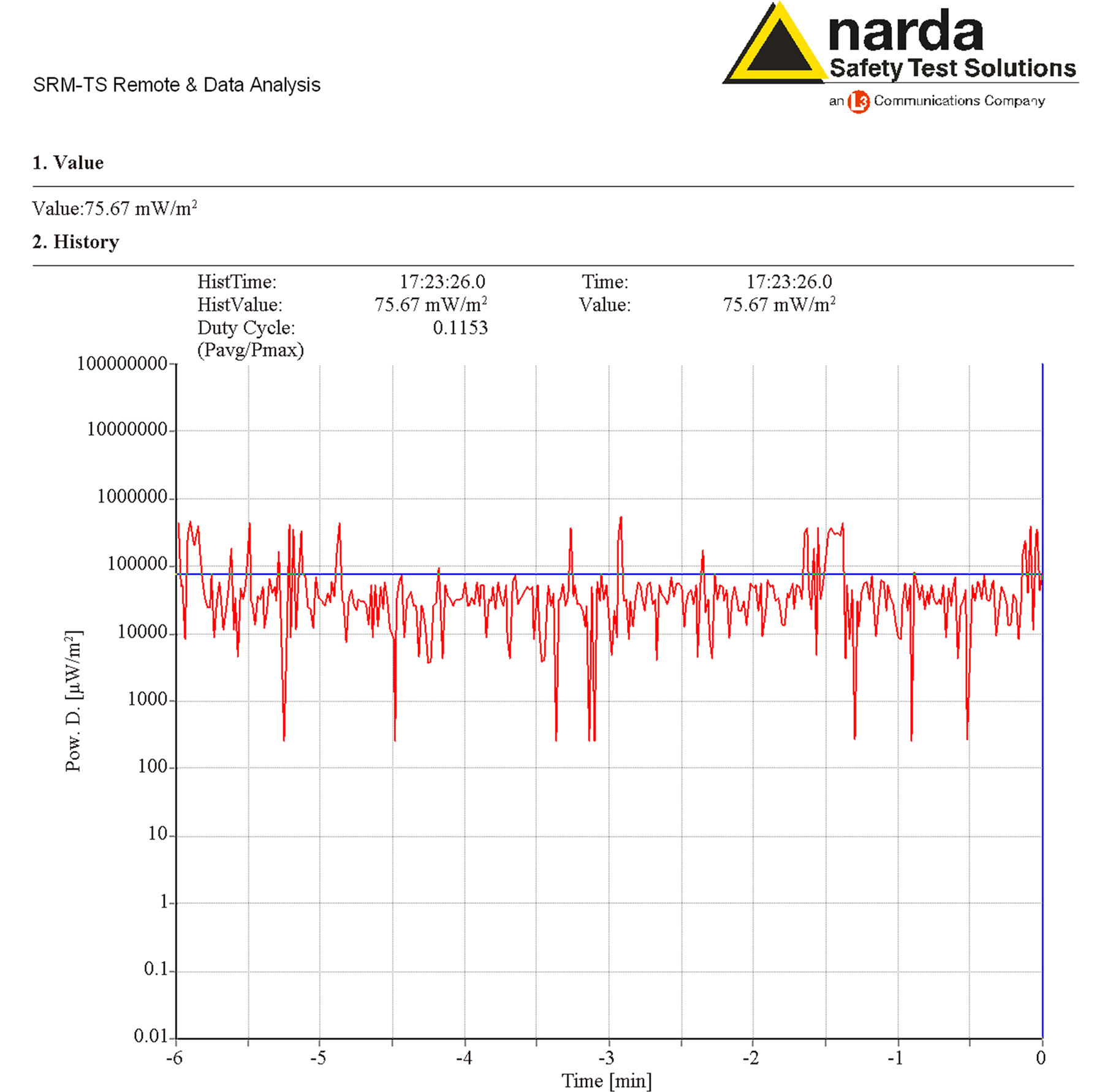
Figure 3. Time analysis of the power density measured by a Narda SRM 3000 during the exposure, whose average intensity value was 90 mW/m2.
To enhance the fine spectral structure, the second-derivative analysis of infrared spectra was performed. In the second-derivative spectrum, the intrinsic shape of an infrared absorbance is approximated by a Lorentzian function, and the peak frequency is practically identical to the original peak frequency, but the half-bandwidth is reduced [26].
The height of a second-derivative peak is proportional to the original peak height with an opposite sign, and the half-bandwidth of the second-derivative peak is proportional to the original half-bandwidth [27].
Either exposed or control samples were located in the same room at a temperature of 20°C.
3. Results and Discussion
A typical house consists of a matrix of wires throughout the walls and ceilings. Moreover home devices or appliances are particularly MW sources, above all mobile phones
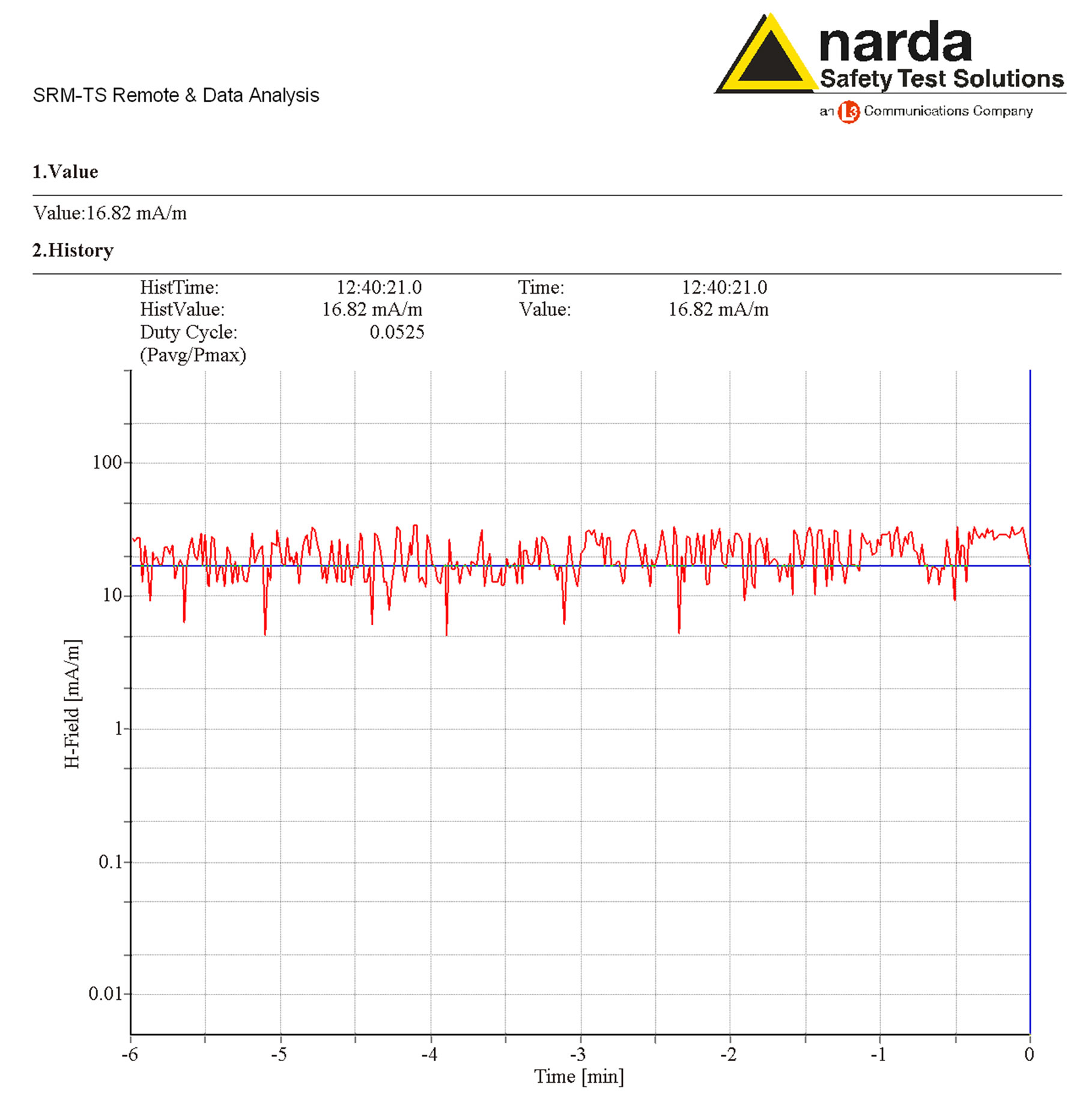
Figure 4. Time analysis of the magnetic field component measured by a Narda SRM 3000 during the exposure. The magnetic field intensity varied in the range from 8 to 25 mA/m during the exposure and the average value was 16 mA/m.
and transmission masts, Wi-Fi systems and cordless phones. When energy is absorbed by an organic system, it is converted in heat causing an increase in dipolar molecules activity and so in tissues temperature, altering the biological function.
However non-thermal effects seem to appear even if RF/MW intensity is not enough to cause detectable thermal effects.
The Specific Absorption Rate (SAR) is a parameter of potential biological significance which is the energy absorbed by the irradiated biological system (W/kg).
SAR and energy distribution into an organisms depend on many aspects including organs and tissues composition, such as the water content, the body structure and the distance from the EMF source. Therefore the distribution of the absorbed energy into the organism is not uniform.
Nevertheless, every mobile telephony company provides a representative value of SAR relative to each mobile phone model.
However, the SAR to which the proteins were exposed (0.85 W/Kg) was lesser than the limits of 1.6 W/kg and 2 W/kg recommended in the USA and Europe, respectively.
The structural changes of the vibration bands of LYS, BSA and MB induced by the exposure to mobile phone microwaves were studied by FTIR spectroscopy.
Measurements were conducted after exposures of 8 h.
Less reliable results would be provided after longer time of exposure because the relatively low surface tension of the investigated system, the hydration value of the investigated water solutions [28-29].
Time analysis of output power density showed that the exposure levels were well below the limit of 4.5 W/m2 (f / 200) recommended by the I.C.N.I.R.P. [14], as represented in Figure 3.
The magnetic flux density to which the proteins were exposed varied in the range from 8 to 25 mA/m during the exposure, as can be observed in the representative time analysis in Figure 4. This value was much lesser than the reference level of 111 mA/m derived from the expression H < 0.0037 f ½ for public exposure, reported on Table 1 of the Guidelines of [14].
FTIR-ATR spectra were collected for several exposed and not exposed samples of LYS, BSA and MB, as described in the preceding section.
The spectra of the proteins exhibited an intense amide I band centered around 1650 cm-1, corresponding mainly to an α-helix structure content due to C = O stretching vibration and a N―H bending mode, a low intensity amide II around 1545 cm-1, coupling of the N―H bending and C―N stretching modes, whereas the amide A and B regions, centered at 3293 and 3080 cm-1 respectively, that are due mostly to the peptide linkage N―H stretching mode, appeared as weak bands.
The first spectra of LYS, BSA and MB acquired after 4 h of exposure, showed an initial increase of their Amide I band intensity.
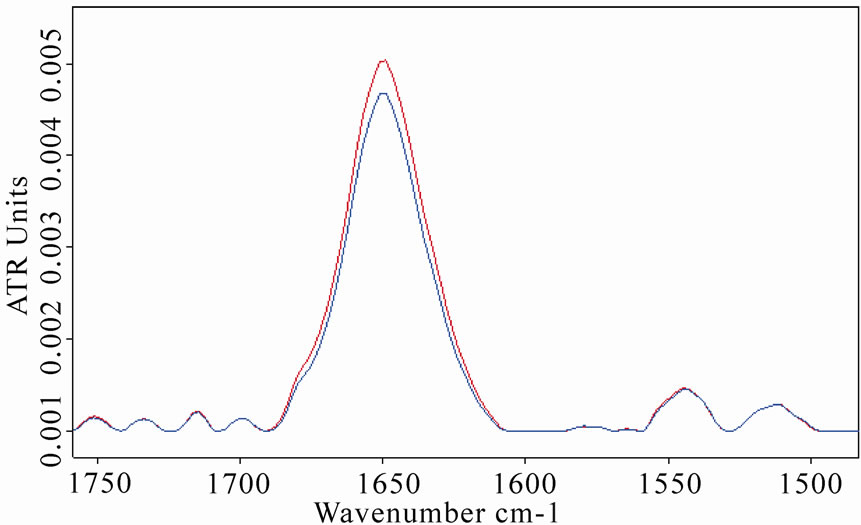
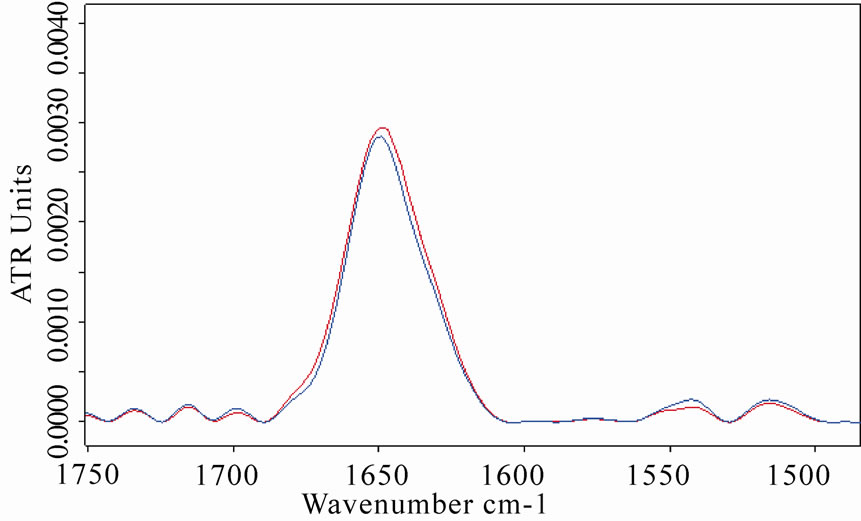
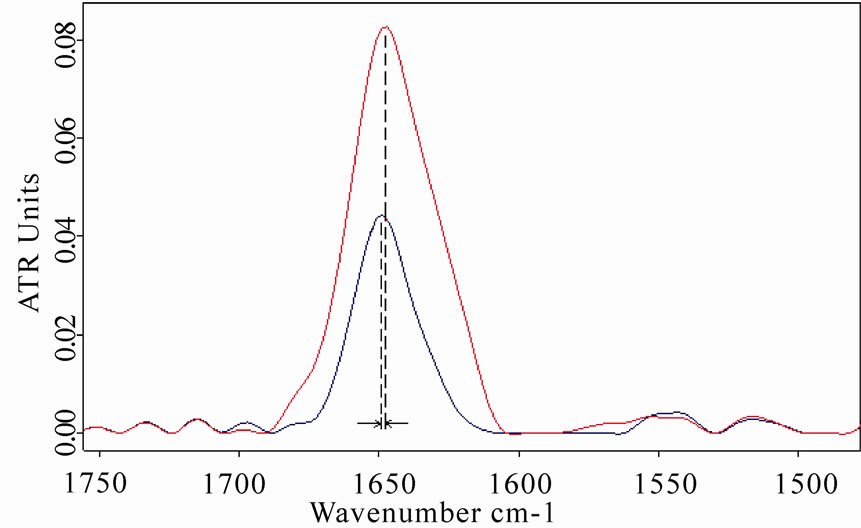
Amide I and amide II spectral regions of LYS, BSA and MB after 8 h of exposure are represented in Figures 5(a), 6(a), 7(a), respectively (red lines refer to exposed samples). It appears that a clear increase in amide I mode occurred after the exposure.
In particular, a weak shift of the amide I band (1 cm-1) was observed in BSA spectra after the exposure, whereas a heavier shift (2.5 cm-1) of the amide I in MB spectra was detected, giving a clear proof of a structural change of the protein after the exposure.
An increase of amide A vibration band was also observed in LYS, BSA and MB spectra after the exposure, as represented in Figures 5(b), 6(b), 7(b), respectively. This result is in agreement with the explanation of the observed amide A band as a combination of amide I and amide II that interacts with the strong NH fundamental through a cubic anharmonic potential, acquiring enough intensity to be observable through a Fermi resonance and resulting in two bands (amide A and amide B) whose separation is proportional to the interaction strength [30-31].
Furthermore, manipulations of BSA and MB spectra were performed by a spectrum deconvolution.
The concept of Fourier self deconvolution is based on the assumption, which a spectrum of single narrow bands is broadened in the liquid or solid state and can not be distinguished in the amide envelope. A curve fitting procedure can be applied to estimate quantitatively the area
 (a)
(a)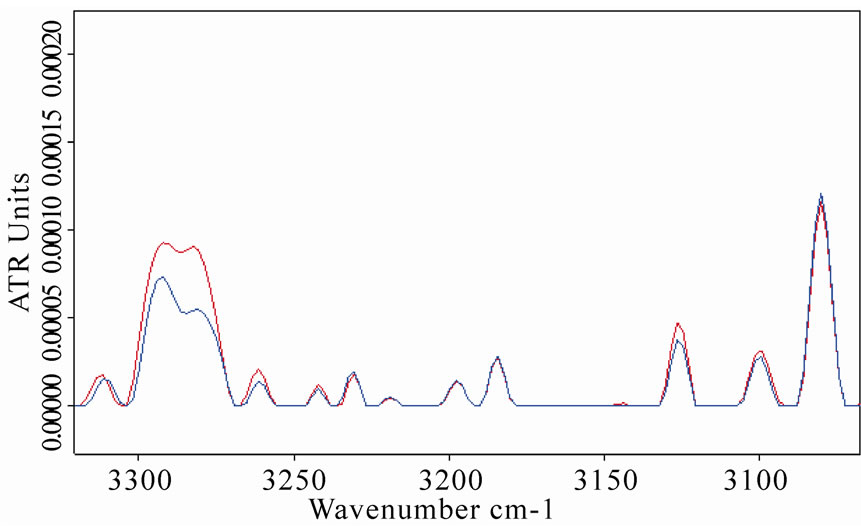 (b)
(b)
Figure 5. (a) Infrared spectrum from 1700 to 1500 cm-1 of amide I and amide II regions of lysozyme in D2O solution acquired after 8 h of exposure (red line refers to exposed sample); (b) The same spectra zoomed in from 3400 to 3050 cm-1, in which the amide A and amide B bands are shown. The increases in amide I and amide A mode are clearly evidenced after the exposure.
 (a)
(a)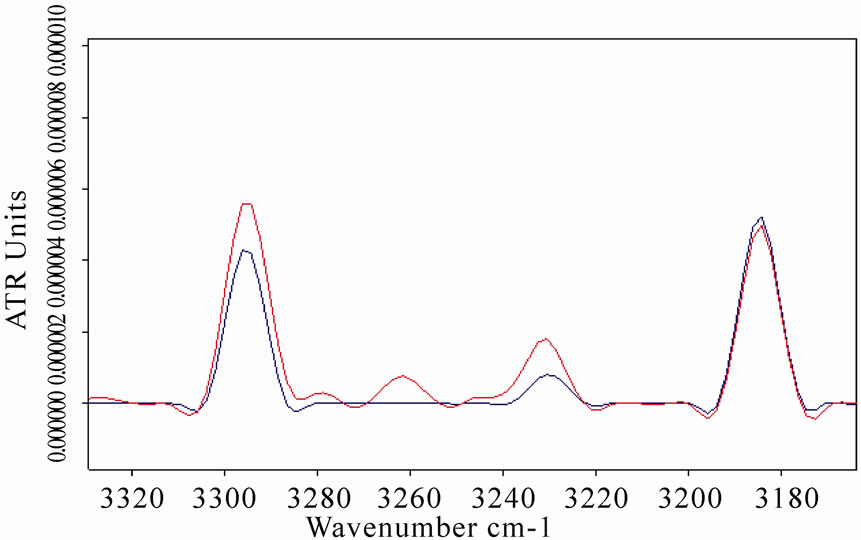 (b)
(b)
Figure 6. (a) Infrared spectrum from 1700 to 1500 cm-1 of amide I and amide II regions of bovine serum albumin in D2O solution acquired after 8 h of exposure (red line refers to exposed sample); (b) The same spectra zoomed in from 3400 to 3100 cm-1, in which the amide A band is represented. The ATR-FTIR analysis allowed evidence of the increases in amide I and amide A intensities and the shift of amide I band after the exposure.
of each component representing a type of secondary structure.
In [27] the amide I was deconvoluted with a Lorentzian line shape function and the deconvoluted spectrum was fitted with Gaussian band shapes by an iterative curve fitting procedure. The results are in good agreement with the information obtained from X-ray crystallographic structures of the proteins under study.
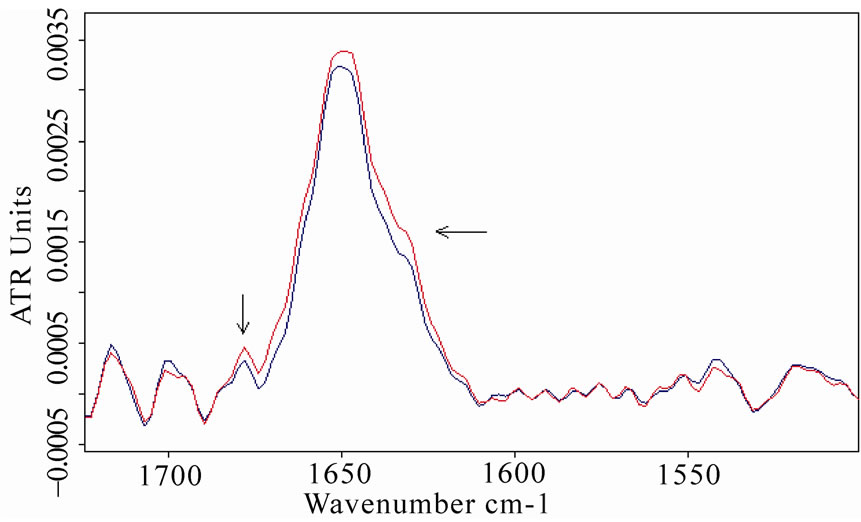
This analysis was applied to the acquired spectra, revealing the presence of a band close to 1650 cm-1 that is due to α-helix structures, and around 1635 and 1685 cm-1, that can be associated with β-sheet structures [32-33]. A clear increase in β-sheet bands appeared in BSA and MB spectra, comparing exposed and unexposed spectra, as shown in Figure 8. These features can be attributed to an unfolding process of the protein and the formation of
 (a)
(a)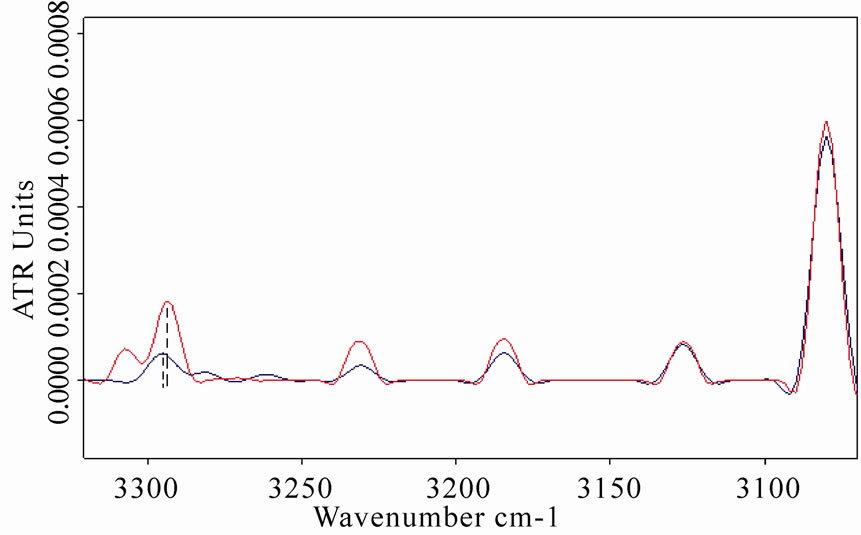 (b)
(b)
Figure 7. (a) Infrared spectrum from 1700 to 1500 cm-1 of amide I and amide II regions of myoglobin in D2O solution acquired after 8 h of exposure. Red line refers to the exposed sample spectrum, which represents as the amide I mode intensity increased and shifted after the exposure; (b) The same spectra zoomed in from 3400 to 3050 cm-1, in which the amide A and amide B bands are evidenced. The concave rubberband correction allowed a clear evidence of the double peaks of the amide A band close to 3293 and 3307 cm-1 in the exposed sample spectrum, whereas their intensities appear weakly in the control.
aggregates [34].
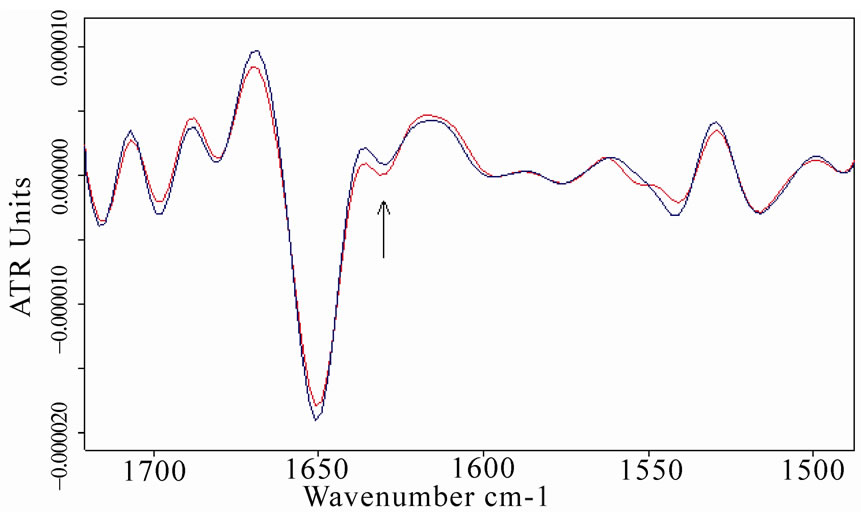
The second-derivative spectra of BSA and MB confirmed this analysis, as represented in Figure 9.
Such result is in agreement with spectroscopic measurements in the visible region of MB [24], where the increasing absorption of the Soret band indicating the refolding process of the reconstitution of heme pocket of MB from acidic conditions, was decreased by the exposure to MWs in comparison to the control.
Also previous measurements of light scattering from BSA solutions exposed to microwaves at 1.0 GHz showed enhanced aggregation for microwave-exposed samples [35].
 (a)
(a)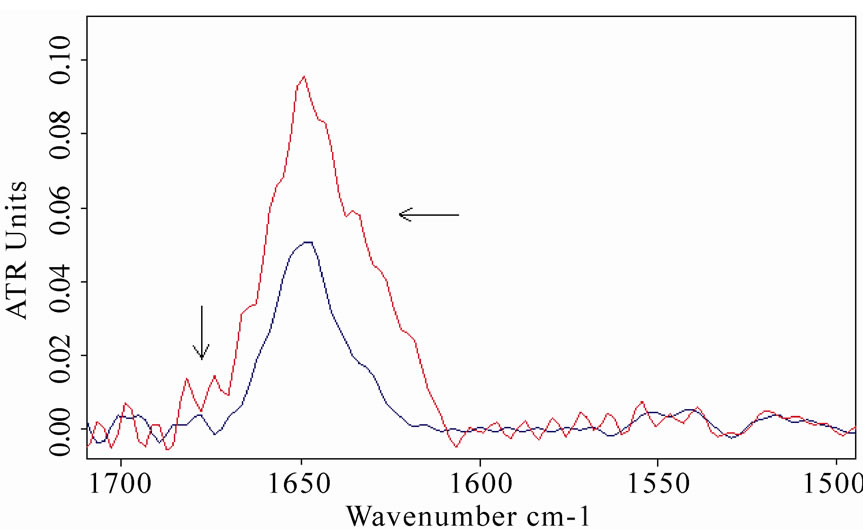 (b)
(b)
Figure 8. Fourier self deconvolution analysis applied to the spectra of bovine serum albumin (a) and myoglobin; (b) The increasing of β-sheet structures around 1635 and 1685 cm-1 (both localized by arrows) after the exposure is evidenced (the red lines refer to exposed sample spectra).
Computations of the integrated area of amide I and amide A bands and of β-sheet contents were conducted for a relevant number of exposed and not exposed samples spectra, whose averaged values are reported on Table 1, providing that the values of exposed samples were significantly different in comparison to the controls (p < 0.05 for LYS and BSA spectra, and p < 0.001 for MB spectra).
4. Conclusions
The exposure of samples of lysozyme, bovine serum albumin and myoglobin in D2O solutions to mobile phone microwaves at 900 MHz at magnetic field intensity around 16 mA/m, provided significant alterations of their secondary structure evidenced by FTIR techniques.
The intensity of the amide I mode of all the proteins increased after 8 h of exposure.
In particular, a shift of the amide I of bovine serum albumin and myoglobin was observed after the exposure,
 (a)
(a)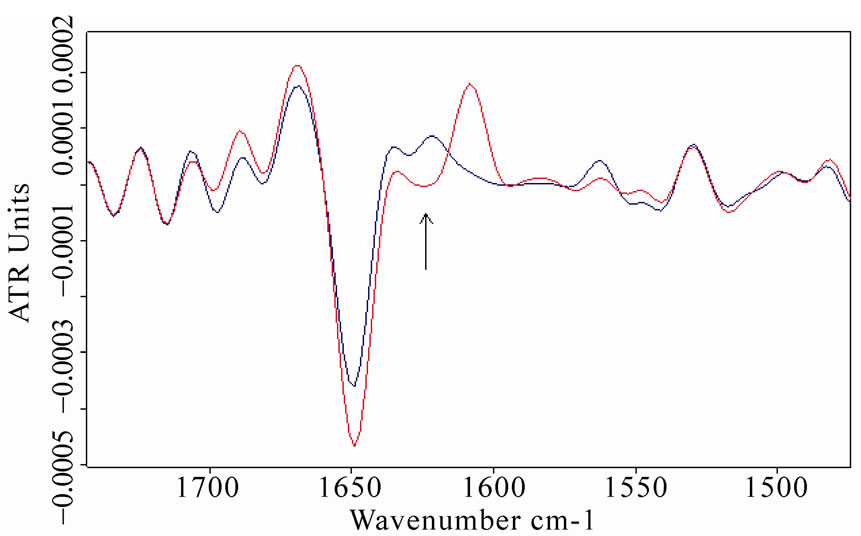 (b)
(b)
Figure 9. Second-derivative spectra of amide I and amide II regions of bovine serum albumin (a) and myoglobin (b) After the exposure (red lines represent the spectra of exposed samples). The relative increase in β-sheet content close to 1635 cm-1 (localized by an arrow) as to the α-helix component is visible for both proteins.
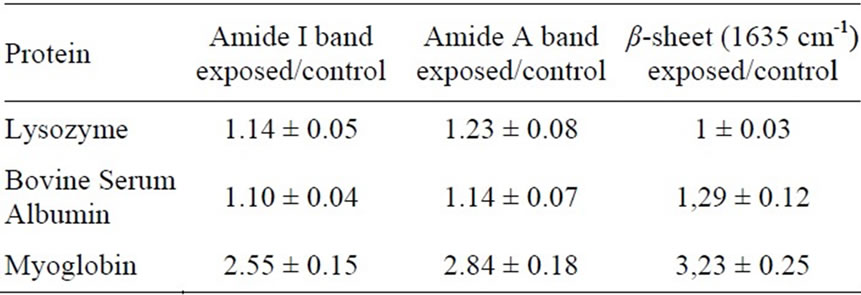
Table 1. Integrated area ratio exposed / control of characteristic vibration bands of proteins secondary structure after the exposure
which confirmed a clear change in the proteins conformation.
In addition, an increase of the β-sheet components with respect to the α-helix content was also observed in the amide I region of bovine serum albumin and myoglobin, leading to the possibility that the microwaves exposure can produce an unfolding process of the protein and the formation of aggregates.
These results led us to conclude that mobile phone microwaves can affect the secondary structure of lysozyme, bovine serum albumin and myoglobin.
REFERENCES
- European Commission, “Possible Effects of Electromagnetic Fields (EMF) on Human Health,” Scientific Committee on Emerging and Newly Identified Health Risks on Human Health, 19 July 2006, pp. 1-58.
- R. C. Beasond and P. Semm, “Responses of Neurons to an Amplitude Modulated Microwave Stimulus,” Neuroscience Letters, Vol. 333, No. 3, 2002, pp.175-178.
- H. Lai, A. Horita, C. K. Chou and A. W. Guy, “Low-Level Microwave Irradiations Affect Central Cholinergic Activity in the Rat,” Journal of Neurochemistry, Vol. 48, No. 1, 1987, pp. 40-45.
- K. Fritze, C. Sommer, B. Schmitz, G. Mies, K. Hossman, M. Kiessling et al., “Effect of Global System for Mobile Communication (GSM) Microwave Exposure On Blood-Brain Barrier Permeability in Rat,” Acta Neuropathol, Vol. 94, No. 5, 1997, pp. 465-470.
- F. Tore, P. E. Dulou, E. Haro, B. Veyret and P. Aubineau,“Two-Hour Exposure to 2 W/kg, 900 MHz GSM Microwaves Induces Plasma Protein Extravasation in Rat Brain,” Proceedings from the 5th International Congress of the European Bioelectromagnetics Association, Helsinki, 6 September 2001, pp. 43-45.
- L. G. Salford, A. E. Brun, J. L. Eberhardt, L. Malmgren and B. R. R. Persson, “Nerve Cell Damage in Mammalian Brain after Exposure to Microwaves from GSM Mobile Phones,” Environmental Health Perspectives, Vol. 111, No. 7, 2003, pp. 881-883.
- E. K. Balcer-Kubiczek and G. H. Harrison, “Neoplastic Transformation of C3H/10T1/2 Cells Following Exposure to 120 Hz Modulated 2.45 GHz Microwaves and Phorbol Ester Tumour Promoter,” Radiation Research, Vol. 126, No. 1, 1991, pp.65-72.
- H. Repacholi, A. Basten, V. Gebski, D. Noonan, J. Finnie and A. W. Harris, “Lymphomas in E mu-Piml Transgenic Mice Exposed to Pulsed 900 MHz Electromagnetic Fields,” Radiation Research, Vol. 147, No. 5, 1997, pp. 631-640.
- A. Maes, M. Collier, U. Van Gorp et al., “Cytogenetic Effects of 935.2 MHz (GSM) Microwaves Alone and in Combination with Mitomycin C,” Mutation Research, Vol. 393, 1997, No. 1-2, pp. 151-156.
- V. Garaj-Vrhovac, D. Horvat and Z. Koren, “The Effect of Microwave Radiation on the Cell Genome,” Mutation Research, Vol. 243, No. 2, 1990, pp. 87-93.
- I. Y. Belyaev, C. B. Koch, O. Terenius, K. RoxstromLindquist, L. O. Malmgren, W. H. Sommer, L. G. Salford and B. R. Persson, “Exposure of Rat Brain to 915 MHz GSM Microwaves Induces Changes in Gene Expression but not Double Stranded DNA Breaks or Effects on Chromatin Conformation,” Bioelectromagnetics, Vol. 27, No. 4, May 2006, pp. 295-306.
- E. Diem, C. Schwarz, F. Adlkofer, O. Jahn and H. Rudiger, “Non-thermal DNA Breakage by Mobile-Phone Radiation (1800MHz) in Human Fibroblasts and in Transformed GFSH-R17 Rat Granulosa Cells in Vitro,” Mutation Research, Vol. 583, No. 2, 6 June 2005, pp. 178-183.
- R. R. Tice, G. G. Hook, M. Donner, D. I. McRee and A. W. Guy, “Genotoxicity of Radiofrequency Signals. I. Investigation of DNA Damage and Micronuclei Induction in Cultured Human Blood Cells,” Bioelectromagnetics, Vol. 23, No.2, 2002, pp.113-126.
- International Commission on Non-Ionizing Radiation Protection, “Guidelines for Limiting Exposure to Time -Varying Electric, Magnetic, and Electromagnetic Fields (up to 300 GHz),” Health Physics, Vol. 74, No. 7, April 1998, pp. 494-522.
- E. Calabrò and S. Magazù, “Monitoring Electromagnetic Field Emitted by High Frequencies Home Utilities,” Journal of Electromagnetic Analysis & Applications, Vol. 2, No. 9, 2010, pp. 571-579.
- R. J. Ellis and F. U. Hartl, “Principles of Protein Folding in the Cellular Environment,” Current Opinion in Structural Biology, Vol. 9, No. 1, February 1999, 102-110.
- M. J. Gething and J. Sambrook, “Protein Folding in the Cell,” Nature, Vol. 355, No. 6355, 1992, PP. 33-45.
- S. Magazù, E. Calabrò and S. Campo, “FTIR Spectroscopy Studies on the Bioprotective Effectiveness of Trehalose on Human Hemoglobin Aqueous Solutions under 50 Hz Electromagnetic Field Exposure,” The Journal of Physical Chemistry B, Vol.114, No. 37, 2010, pp. 12144-12149.
- S. Magazù and E. Calabrò, “Studying the Electromagnetic-Induced Changes of the Secondary Structure of Bovine Serum Albumin and the Bioprotective Effectiveness of Trehalose by FTIR Spectroscopy,” submitted, 2010.
- H. Fröhlich, “Long-Range Coherence and Energy Storage in Biological Systems,” International Journal of Quantum Chemistry, Vol. 2, No. 5, September 1968, pp. 641-649.
- H. Bohr and J. Bohr, “Microwave-Enhanced Folding and Denaturation of Globular Proteins,” Physical Review E, Vol. 61, No. 4, 2000, pp. 4310-4314.
- I. V. Lerina, V. A. Kovalenko, A. M. Belous, Y. D. Rozanova, B. N. Leonov and T. F. Morozova, “Structural Changes in Serum Albumin Induced by Microwave Heating,” Biophysics, Vol. 42, No. 5, 1997, pp. 1043-1047.
- R. Weissenborn, K. Diederichs, W. Welte, G. Maret and T. Gisler, “Non-Thermal Microwave Effects on Protein Dynamics? An X-Ray Diffraction Study on Tetragonal Lysozyme Crystals,” Biological Crystallography (Acta Crystallographica Section D), Vol. 61, 2005, pp.163-172.
- F. Mancinelli, M. Caraglia, A. Abbruzzese, G. d’Ambrosio, R. Massa and E. Bismuto, “Non-Thermal Effects of Electromagnetic Fields at Mobile Phone Frequency on the Refolding of an Intracellular Protein: Myoglobin,” Journal of Cellular Biochemistry, Vol. 93, No. 1, 1 September 2004, pp. 188-196.
- B. M. Smith and S. Franzen, “Single-Pass Attenuated Total Reflection Fourier Transform Infrared Spectroscopy for the Analysis of Proteins in H2O Solution,” Analytical Chemistry, Vol. 74, No. 16, 2002, pp. 4076-4080.
- W. K. Surewicz and H. H. Mantsch, “New Insight into Protein Secondary Structure from Resolution-Enhanced Infrared Spectra,” Biochim et Biophys Acta, Vol. 952, No. 2, 1988, pp. 115-130.
- H. Susi and D. M. Byler, “Resolution-Enhanced Fourier Transform Infrared Spectroscopy of Enzymes,” Methods Enzymol., Vol. 130, 1986, pp. 290-311.
- M. P. Jannelli, S. Magazù, G. Maisano, D. Majolino and P. Migliardo, “Hydration Phenomena and Cooperative Diffusion in Polymer Water Solutions,” Physica Scripta, Vol. 50, 1994, pp. 215-217.
- S. Magazù, “NMR, Static and Dynamic Light and Neutron Scattering Investigations on Polymeric Aqueous Solutions,” Journal of Molecular Structure, Vol. 523, No. 1-3, 2000, pp. 47-59.
- T. Miyazawa, “The Characteristic Band of Secondary Amides at 3100 cm-1,” Journal of Molecular Spectroscopy, Vol. 4, No. 1-6, 1960, pp. 168-172.
- S. Krimm and A. M. Dwivedi, “Vibrational Analysis of Peptides, Polypeptides and Proteins. XII. Fermi Resonance Analysis of the Unperturbed ND Stretching Fundamental in Polypeptides,” Journal of Raman Spectroscopy, Vol. 12, No. 2, 1982, pp.133-137.
- A. A. Ismail, H. H. Mantch and P. T. T. Wong, “Aggregation of Chymotrypsinogen: Portrait by Infrared Spectroscopy,” Biochim et Biophys Acta, Vol. 1121, No. 1-2, 1992, pp. 183-188.
- T. Lefevre and M. Subirade, “Molecular Differences in the Formation and Structure of Fine-Stranded and Particulate β-Lactglobulin Gels,” Biopolymers, Vol. 54, 2000, pp. 578-586.
- R. Bauer, R. Carrotta, C. Rischel and L. Ogendal, “Characterization and Isolation of Intermediates in β-Lactoglobulin Heat Aggregation at High pH,” Biophysical Journal, Vol. 79, No. 2, 2000, pp. 1030-1038.
- D. I. de Pomerai, B. Smith, A. Dawe, K. North, T. Smith, D. B. Archer, I. R. Duce, D. Jones and P.M. Candido, “Microwave Radiation can Alter Protein Conformation without Bulk Heating,” FEBS Letters, Vol. 543, No. 1-3, 2003, pp. 93-97.