Journal of Biomedical Science and Engineering
Vol.6 No.8A(2013), Article ID:35741,13 pages DOI:10.4236/jbise.2013.68A1001
Complement C3a signaling mediates production of angiogenic factors in mesenchymal stem cells

1Infectious and Inflammatory Disease Center, Sanford Burnham Institute, La Jolla, USA
2Torrey Pines Institute for Molecular Studies, San Diego, USA
Email: ischraufstatter@tpims.org
Copyright © 2013 Richard G. DiScipio et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 4 May 2013; revised 8 June 2013; accepted 22 June 2013
Keywords: MSC; C3a; C5a; Angiogenic Factor Production; Signaling Pathways
ABSTRACT
A major portion of the beneficial effect of mesenchymal stem cells (MSC) is due to the production of trophic and angiogenic factors by these cells, and one of the efforts to improve the therapeutic efficacy of these cells lies in enhancing this capacity. Since there is complement activation in all areas of tissue injury, and both C3a and C5a activate MSC, it was asked whether stimulation with C3a or C5a would upregulate the production of trophic factors by MSC. C3a caused significant up-regulation of various angiogenic factors, including VEGF, CXCL8/IL-8 and IL-6. In contrast there was no detectable production of the pro-inflammatory cytokines TNF-α and IL-1β in spite of nuclear translocation of NFκB. Although C5a also caused moderate up-regulation of angiogenic factors, the effect was borderline significant. Furthermore the production of angiogenic factors induced by C3a was of physiological relevance: Supernatants of MSCs cultured under serum-free conditions induced minimal tube formation of HUVECs as an in vitro measure of angiogenesis; tube formation was considerably enhanced, when supernatants from C3a-stimulated MSC were used, while C3a itself had no direct angiogenic effect on HUVECs. The signaling cascade responsible for the production of angiogenic factors by C3a or C5a could be defined as activation of the rho cascade which was necessary for nuclear translocation of NFκB p65 and of phospho-ERK1/2. Although rho was only transiently activated, inhibition of the rho or “downstream of it” of the NFκB pathway, prevented C3aand C5a-induced up-regulation of angiogenic factors.
1. INTRODUCTION
Mesenchymal stem cells (MSC) are rare cells found in all tissues which are able to differentiate into all types of connective tissue lineages including osteoblasts, adipocytes and chondrocytes. In addition, these cells produce a variety of trophic and angiogenic factors which contribute to tissue regeneration [1,2] and possess immune suppressive properties [3,4]. Because of these properties MSC are starting to find clinical application in a variety of diseases ranging from myocardial infarction [5] to graft versus host disease [6]. However, the full regenerative potential of these cells has not been realized due to poor tissue homing and limited cell survival following transplantation. Currently various means of improving MSC homing [7], growth and angiogenic factor production [8,9] and in vivo survival [10,11] are being pursued as ways to improve the therapeutic efficacy of MSC. Chemotactic factors for MSC include several growth factors (bFGF, PDGF, IGF-1) [12,13], some chemokines [14-16], and the anaphylatoxins C3a and C5a [17].
C3a and C5a are small (Mr 8700-11,000) polypeptides released from their precursor proteins, C3 and C5, respectively by C3/5 convertases during complement activation. Both C3a and C5a are well known as chemotactic, oxidant inducing and degranulating agents for myeloid cells [18-23], but the response of leukocytes to C3a is considerably weaker and more transient than that to C5a [20,21,24]. In particular C3a does not cause leukocyte accumulation in vivo [24] which contrasts with the strong inflammatory response to C5a [18,25]. However in MSC the response to C3a was at least as strong as that to C5a [17] and apart from serving as chemoattractants both C3a and C5a protected from oxidative injury in MSC [17].
The C3a receptor (C3aR) and the C5a receptor (C5aR/ CD88) are G-protein coupled receptors (GPCR) which usually couple to Gi. Activation of the C3aR tends to result in transient cellular responses in most cell types [1926]. However, there have been a few exceptions, where C3a elicited a prolonged and strong response including mast cells [17,27] and as already mentioned MSC [17]. The C3aR contains two interesting features. First, it has an unusual, very long second extracellular loop, of which sequences adjacent to the transmembrane domains are important for C3a binding [28]. Second, an apparent nuclear localization signal sequence is located near the Cterminus of the C3aR, FRKKAR starting at amino acid 442 [29], but a functional significance for this observation has not been discerned. It has been noted, however that the C3aR in MSC, but not in other cell types tested can be translocated to the nucleus following the addition of C3a [17]. While GPCR activation at the plasma membrane induces mostly short term signaling events, nuclear translocation results in long-term effects including prolonged nuclear ERK1/2 activation [30] leading to transcriptional activation [31] and cell proliferation [32].
In monocytic cells, C3a and C5a activate rho in a Gidependent fashion, which leads to activation of NFκB downstream [33,34]. This in turn is responsible for transcriptional activation leading to the production of various inflammatory cytokines including TNF-α, IL-1β, IL-6 and IL-8/CXCL8. It is shown here that this signaling cascade is the same in MSC, but with a different outcome, since these cells appear to have developed mechanisms that suppress expression and/or processing of TNF-α [35] and IL-1β [36], while at the same time supporting increased expression of several angiogenic factors including VEGF, IL-6 and CXCL-8, thus converting a normally inflammatory signaling pathway into one that may contribute to tissue healing.
2. METHODS
2.1. Materials and Cell Culture
Human bone marrow MSC were supplied by the Tulane Center for Gene Therapy and cultured up to passage 5 in alpha-MEM (Life Technologies, Carlsbad, CA) containing 16.5% FCS (Atlanta Biologicals, Lawrenceville, GE). HUVECs were purchased from Lonza (Allendale, NJ) and grown in complete EGM (Lonza).
Natural C3a and recombinant C5a were purified as described [17] and were used at concentrations that had been shown previously to induce a maximal response [17].
The inhibitors PD98059, U0126, LY294002 and BayII- 7082 were purchased from EMD Biosciences (Gibbstown, NJ), Y27632 was obtained from Biomol (Enzo Life Sciences International, Plymouth Meeting, PA), Cell Permeable C3 Transferase protein inhibitor (CT04-A, Rho inhibitor) was supplied by Cytoskeleton (Denver, CO), and pertussis toxin by List Biological Laboratories
(Campbell, CA). Inhibitor concentrations were as follows: 50 μM PD98059, 10 μM U0126, 10 μM LY294002, 10 μM BayII-7082, 10 μM Y27632, 1 μg/ml C3 Transferase inhibitor, and 100 ng/ml pertussis toxin. Inhibitor pre-incubation times were 30 min except for the C3 Transferase inhibitor and pertussis toxin, which were added 20 hrs before cell stimulation. ELISA kits were from Biolegend (CXCL-8, IL-6, TNF-α, and IL-1β, San Diego, CA) or Peprotech (VEGF, Rocky Hill, NJ). Antibodies against total ERK1, NFκB p65 and Lamin A were from Santa Cruz Biotechnology (Santa Cruz, CA), all other antibodies (phospho-ERK1/2, phospho-Elk and total Elk) were from Cell Signaling Technology (Danvers, MA). The antibody arrays (RayBio Human Angiogenesis Antibody Array 1) were obtained from RayBiotech (Norcross, GA).
2.2. Subcellular Fractionation
To isolate nuclear fractions, cells were vortexed in hypotonic buffer (10 mM HEPES, pH 8.0, 10 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 1 mM DTT, 0.2 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin) containing 0.5% NP-40, then microfuged at highest speed for 1 min. Supernatants representing the cytoplasmic fraction were retained, the pellets were washed 2 × with the same buffer, then resuspended in buffer B (20 mM HEPES, pH 8.0, 250 mM NaCl, 2 mM EDTA, 2 mM EGTA, 2 mM Na3VO4, 1 mM DTT, 0.2 mM PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin), incubated on ice for 15 min followed by another 2 min centrifugation at 4˚C. The supernatants (nuclear fraction) were transferred to clean tubes containing 4 × gel sample buffer, boiled and separated on SDS-gels. The purity of the fractions was confirmed by Western blotting using lamin A as a nuclear marker and β-tubulin as a cytoplasmic marker.
2.3. Western Blots
The cellular fractions were resolved by SDS-PAGE, transferred to nitrocellulose membranes, blocked with 4% dry milk in TBS-Tween, and exposed to specific primary antibodies as described for each experiment. Antibody binding was detected using horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse secondary antibodies and enhanced chemi-luminescence (Super Signal West Dura, Thermo). Blots were re-probed with a second antibody, e.g. anti-ERK1 antibody to assure equal loading. ImageJ software was used to quantify results.
2.4. Rho Activation Assay
MSC at approximately 50% confluence were treated with 300 nM C3a or 50 nM C5a for the times indicated. Rho activity in total cell lysates was measured using a calorimetric ELISA-based assay (G-LISA, Cytoskeleton) according to the manufacturer’s instructions.
2.5. Fluorescence Microscopy
Cells were cultured on fibronectin-coated glass coverslips, stimulated for the indicated times with 300 nM C3a, 50 nM C5a or 50 ng/ml TNF-α, fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X100 in PBS. After blocking with 2% FCS for 30 min at room temperature, cells were incubated overnight with antiC3aR (BD Biosciences), anti-phospho-ERK1/2 antibodies (Cell Signaling) or anti-NFκB p65 (Santa Cruz Biotechnology) at 4˚C, followed by staining with Alexa Fluor 488 anti-rabbit IgG or Alexa Fluor 488 anti-mouse and Alexa Fluor 568 anti-rabbit IgG antibody (Life Technologies) in 2% FCS. Cells were then stained with DAPI (Sigma Chemical Co., St. Louis, MI) for 10 min and washed three times with PBS before mounting with AntiFade (Life Technologies). Images were taken on an Olympus FV1000 confocal microscope.
2.6. RT-PCR
MSC at about 60% confluence were cultured for 3 hrs in the presence or absence of 300 nM C3a or 50 nM C5a under serum-free conditions. RNA from MSCs was isolated with the RNeasy kit (Qiagen, Valencia, CA). Complementary DNA was synthesized from MSC RNA using Omniscript reverse transcriptase (Qiagen). The primers are listed under supplementary information. Amplification was performed for 35 cycles in a Perkin Elmer Cetus DNA thermal cycler.
2.7. ELISAs
MSC at about 60% confluence were cultured for 20 hrs in the presence or absence of inhibitors with or without 300 nM C3a or 50 nM C5a under serum-free conditions in α-MEM. Following the 20 hr incubation, cytokine concentrations were detected in the culture supernatants by commercial ELISA kits. Alternatively, the supernatants were used with an angiogenesis protein array (Ray Biotech, Norcross GA, angiogenesis array 1), which was developed according to the supplier’s manual.
2.8. In Vitro Angiogenesis Assay
HUVECs (passage 3-5) were used with the Millicell μ- Angiogenesis Activation Assay Kit (Millipore, Billerica, MA) following the kit’s instructions. Specifically, HUVECs were harvested with Accutase (Invitrogen) and resuspended in α-MEM containing 1% FCS and the same volume of test sample (serum-free α-MEM, serum-free α-MEM containing 300 nM C3a, serum-free α-MEM containing 50 nM C5a, serum-free α-MEM containing 100 ng/ml phorbol myristate acetate (PMA) as a positive control, the 20 hr supernatant of unstimulated MSC, the 20 hr supernatant of MSC stimulated with 300 nM C3a or the 20 hr supernatant of MSC stimulated with 50 nM C5a). The cells (10,000 cells/well) were layered over 10 μl of fibrin gel and incubated for 6 hr in a 37˚C tissue culture incubator. At this point the supernatant was carefully removed, the cells were overlaid with 10 ul of fibrin gel, the corresponding serum-free test sample was added again and the cells were incubated for another 24 hrs at 37˚C. Representative images were taken on a Nikon Eclipse TE200 microscope (Nikon Instruments, Melville, NY) with a Spot camera system (Diagnostics Instruments, Sterling Heights, MI).
2.9. Transwell Chemotaxis Assay
A single cell suspension of MSCs was loaded into the upper wells of 0.15% gelatin-coated Transwells (Costar, 8 μm pore-size, 2 × 104 cells/insert) and pre-incubated with inhibitors for 30 min prior to the addition of stimulus. The lower wells contained 0.2% BSA in α-MEM and either no stimulus or doses of C3a (100 nM ) or C5a (15 nM), which had been found previously to induce maximal chemotaxis [17]. The assembled wells were incubated for 8 hrs in a tissue culture incubator, cells in the upper compartment were carefully removed, the filters were stained with DAPI and the transmigrated cells were counted on a Nikon Eclipse TE200 inverted fluorescence microscope (Nikon Instruments, Melville, NY) with a Spot camera system (Diagnostics Instruments). Background chemotaxis of unstimulated cells was defined as 100%.
3. RESULTS
3.1. Production of Angiogenic Factors by MSC Stimulated with C3a or C5a
Since the production of trophic and angiogenic factors is a major mechanism by which MSC support tissue repair, it was determined whether stimulation with C3a or C5a would up-regulate production of such factors by MSC. Semi-quantitative RT-PCR indicated that stimulation with C3a induced up-regulation of expression of genes encoding several angiogenic and growth factors including VEGF, IL-8/CXCL8, bFGF and TGF-β1 (Figure 1A). Up-regulation of several angiogenic and growth factors could further be verified on the protein expression level: This was first performed semi-quantitatively using a protein array that detects 20 angiogenic factors. C3a caused consistently increased expression of angiogenin, bFGF, gro-α/CXCL1, IL-6, CXCL8, MCP-1/CCL2, PDGF-BB, and VEGF (Figure 1B). Minor up-regulation was also seen for ENA78/CXCL5, leptin, the active form of TGF- β1, and VEGF-D (Figure 1B). No consistent effect was
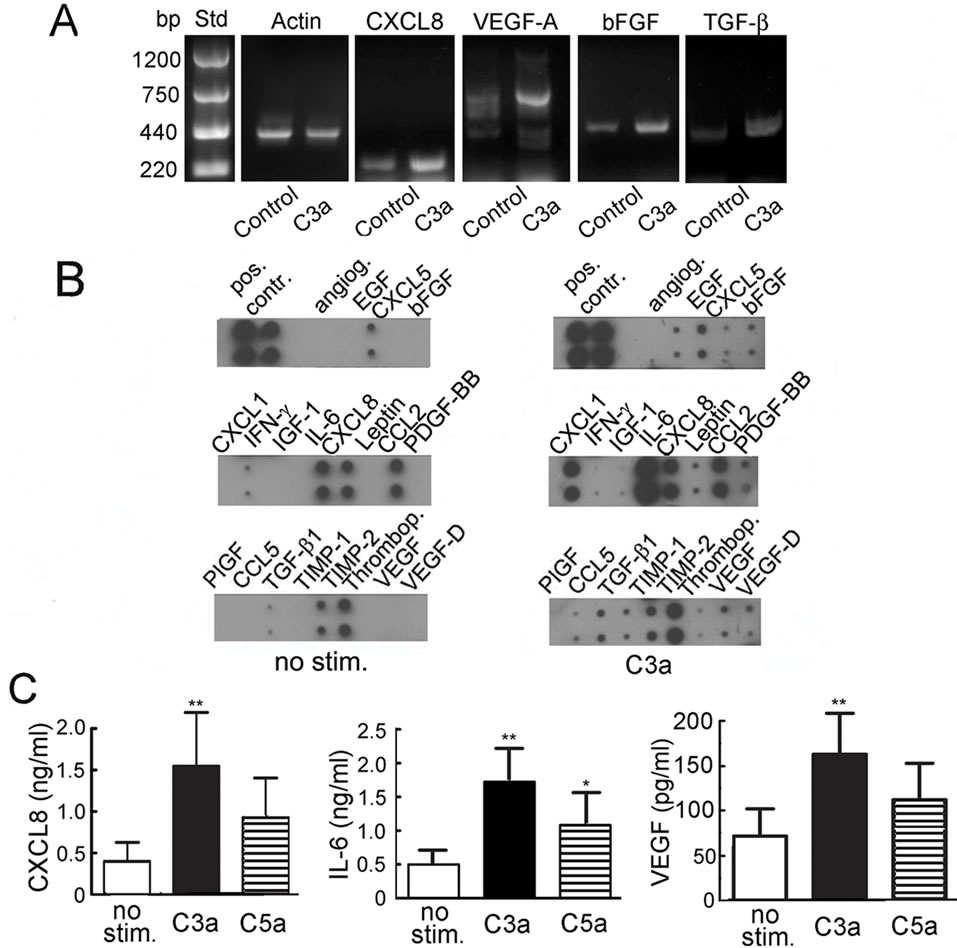
Figure 1. Production of angiogenic and growth factors by MSC stimulated with 300 nM C3a or 50 nM C5a. A: RT-PCR showing semi-quantitative detection of CXCL-8, VEGF-A, bFGF, TGF-β and β-actin in unstimulated or MSC stimulated with C3a for 3 hrs. One experiment representative of 3; B: Protein expression of angiogenic factors in 20 hr supernatants of MSC stimulated with or without C3a as detected by antibody blot. One experiment representative of 4 is shown. Angiogenin (angiog.), CXCL1, IL-6, CXCL8, MCP-1 and VEGF were consistently up-regulated in the presence of C3a, but IFNγ, IGF-1 and thrombopoietin (thrombop.) were not. Only very small amounts of bFGF and the active form of TGF-β1 could be detected, but they were slightly increased following C3a stimulation; C: Quantitation of CXCL-8, IL-6 and VEGF by ELISA. Mean and S.D., n = 4 - 8 (*p ≤ 0.05, **p ≤ 0.01).
seen for IFN-γ, IGF-I, PIGF, RANTES/CCL5, TIMP1 and TIMP2.
These semi-quantitative results were quantified by ELISA for CXCL8, IL-6 and VEGF as shown in Figure 1C. While C5a, which was used in parallel, also caused up-regulation of CXCL8, IL-6 and VEGF (Figure 1C), the increase was only statistically significant for IL-6.
Interestingly, this increase in angiogenic factor production was not accompanied by a concomitant increase in secreted TNF-α or IL-1β under any of the conditions used. TNF-α or IL-1β concentrations in MSC supernatants never reached the detection limit of the assay (5 pg/ml for TNF-α and 2 pg/ml for IL-1β).
3.1.1. Effect of C3aand C5a-Conditioned MSC Media on in Vitro Angiogenesis
In order to show the physiological relevance of the increased production of angiogenic factors by MSC stimulated with C3a or C5a, the same MSC supernatants were used in an in vitro angiogenesis assay, in which tube formation by HUVECs was evaluated. In the presence of α-MEM media, which was not conditioned by culture with MSC, the endothelial cells appeared as individual, separated cells (Figure 2A). The addition of PMA, which was used as a positive control, induced tube formation as expected (Figure 2B). Conditioned media derived from unstimulated MSC induced moderate alignment of cells into clusters (Figure 2C). Supernatants from MSC stimulated with C3a showed more pronounced capillary tube formation (Figure 2D) that was at least as prominent as seen with PMA (Figure 2B). The effect of the C3a-conditioned supernatant was not due to any direct effect of C3a on HUVECs, as C3a added to unconditioned α- MEM media showed no such effect (Figure 2E). The supernatant of C5a-stimulated MSC also caused moderate tube formation (Figure 2F). Again, direct addition of C5a had no such effect (results not shown).
3.1.2. Effect of Kinase Pathway Inhibitors on Production of Angiogenic Factors by MSC
In order to determine which signaling pathways were involved in C3aand C5a-mediated up-regulation of angiogenic factors, various pharmacological inhibitors which are known to block signaling pathways of C3a or C5a in other cell types were added prior to the addition of C3a or C5a to produce MSC conditioned media. Interestingly, a rho kinase inhibitor (Y27632) completely inhibited upregulation of VEGF and CXCL8 in MSC stimulated with either C3a (Figures 3A and B) or C5a (Supplementary
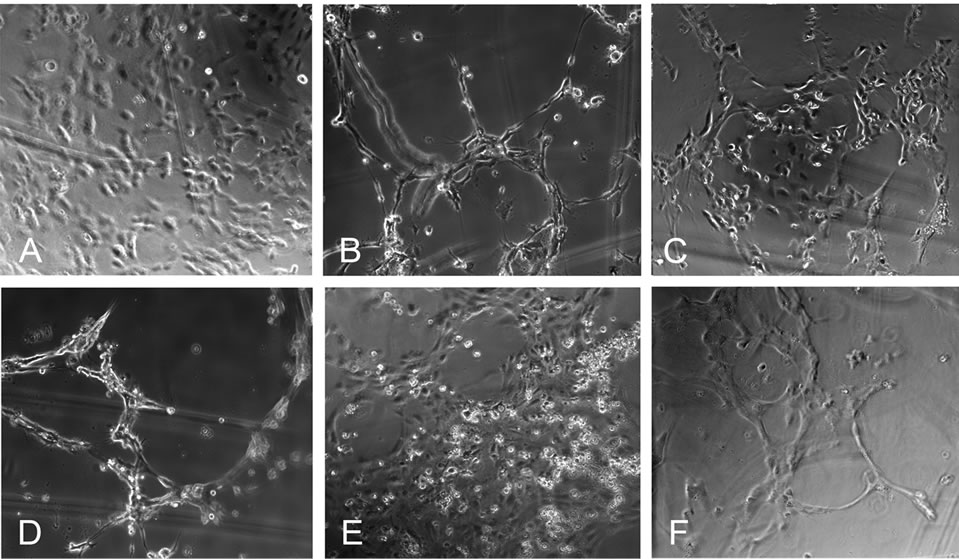
Figure 2. Conditioned media from C3a-stimulated MSC is angiogenic. Tube formation by HUVECs on a fibrin gel was used to detect whether the angiogenic factors produced by C3astimulated MSCs were present in sufficient concentration to promote angiogenic tube formation. A: There is no alignment of the endothelial cells in the negative control cultured with unconditioned α-MEM media; B: In the presence of PMA (positive control) tube formation is observed; C: MSC-conditioned media caused slight alignment of HUVECs, indicating weak angiogenic stimulation; D: Conditioned media from C3astimulated MSC showed clear capillary tube formation. This was not mediated by a direct effect of C3a on endothelial cells, as C3a directly added to unconditioned α-MEM media did not show this effect E; F: Conditioned media from C5a-stimulated MSC less pronounced tube formation. One experiment representative of 3 is shown.
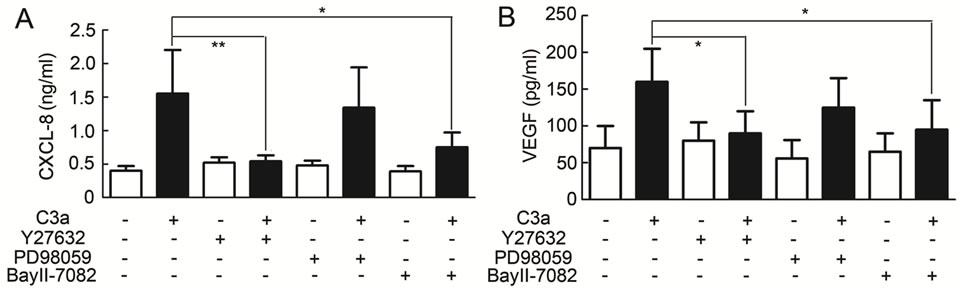
Figure 3. Effect of inhibitors on angiogenic factor production by MSC stimulated with 300 nM C3a. MSC supernatants were produced as described for Fig. 1 in the presence or absence of various signaling pathway inhibitors and CXCL-8 (A) and VEGF (B) were detected by ELISA. The Rho kinase inhibitor Y27632 and the NFκB inhibitor BayII-7082 both induced statistically significant inhibition of C3a-dependent CXCL-8 and VEGF production, while inhibition of the ERK1/2 MAP kinase pathway with PD98059 only slightly reduced the production of IL-8 and VEGF. Mean and S.D., n = 5 - 6.
Table 1). The NFκB inhibitor BayII-7982 also largely inhibited increased production of VEGF and CXCL-8 (Figures 3A and B, and Supplementary Table 1), while inhibition of the ERK pathway with PD98059 (Figures 3A and B, and Supplementary Table 1) showed only a minor inhibitory effect.
3.2. C3a and C5a Activate Rho in MSC
Since Y27632 blocked C3aand C5a-mediated production of angiogenic factors in MSC, and since rho kinase is downstream of rho activation, rho activity was determined in MSC stimulated with C3a or C5a. Rho was consistently activated following addition of C3a (Figure 4A) or C5a (Figure 4B). The activation was instantaneous (Figure 4) and preceded any detectable ERK1/2 phosphorylation [17] suggesting that activation of the Rho cascade may be upstream of ERK1/2 phosphorylation.
We previously reported that activation of the C3aR and C5aR in MSC is mediated by Gi, as pertussis toxin blocked both anaphylatoxin-dependent chemotaxis and ERK1/2 phosphorylation [17]. However, rho pathway activation is usually seen downstream of GPCRs that couple to Gα12, Gα13 or Gq [37]. To determine whether rho activation was due to coupling of the C3aR or C5aR to Gi or to one of the other G-proteins, rho activation assays were repeated following Gi inhibition by pertussis toxin. Pertussis toxin completely inhibited C3aand C5amediated rho activation in MSC (Figure 4), indicating that rho activation in these cells is Gi-dependent rather than downstream of Gα12, Gα13 or Gq.
Nuclear Translocation of Phospho-ERK1/2 and of NFkBp65 Is Rho Pathway Dependent
Since rho activation preceded ERK1/2 phosphorylation, the effect of inhibition of the rho pathway on ERK1/2 phosphorylation was determined using Y27632 to inhibit rho kinase and Cell Permeable C3 Transferase protein
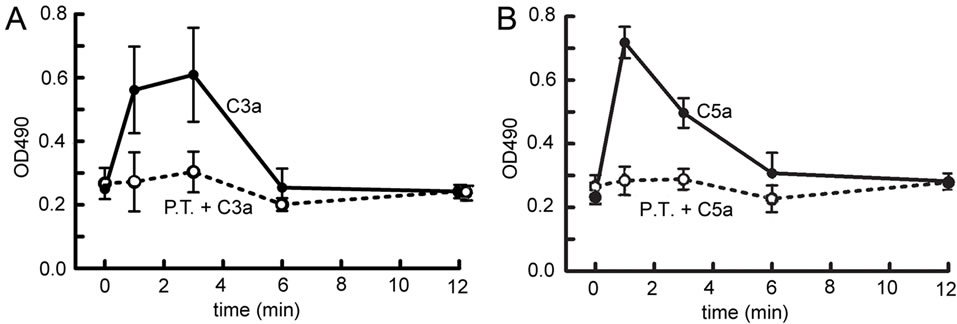
Figure 4. Time-course of rho activity detected in MSC stimulated with C3a or C5a. Rho activity was determined in MSC lysates following stimulation with 300 nM C3a (A) or 50 nM C5a (B) as described in the method section (filled circles). To determine whether rho activation occurred downstream of coupling of the receptor to Gi, the experiment was also performed in the presence of pertussis toxin (P.T.) (open circles). Mean and S.D. of 3 experiments in duplicate. Both C3a and C5a induced transient, but significant rho activation, which was Gidependent.
inhibitor (CT04-A) to block rho itself. Inactivation of rho kinase showed no effect on ERK1/2 phosphorylation in whole cell lysates (results not shown), although the rho kinase inhibitor tended to slightly increase baseline ERK1/ 2 phosphorylation in the absence of cell stimulation. However, when the C3a-stimulated cells were fractionated into nuclear and cytoplasmic fractions, it became evident that inhibition of the rho pathway prevented the translocation of phospho-ERK1/2 to the nucleus: Y27632 as well as the rho inhibitor prevented the accumulation of phosphorylated ERK1/2 in the nucleus (Figures 5A and C), which was associated with increased amounts of cytoplasmic phospho-ERK1/2 in the presence of the rho/ rho kinase pathway inhibitor (Figures 5B and D). A second rho kinase inhibitor with a different chemical structure (Rho kinase inhibitor IV, Calbiochem) had the same effect (results not shown). The results for C3a-mediated ERK1/2 phosphorylation of nuclear and cytoplasmic fraction in the presence and absence of Y27632 are quantified in Figures 5E and F. Consistent with the inhibitory effect of Y27632 on nuclear translocation of phosphoERK1/2, Elk, a downstream nuclear target of phosphoERK1/2, which is phosphorylated following C3a stimulation, failed to be phosphorylated in the presence of Y27632 (Figure 5G), indicating that activation of the rho kinase pathway is important for transcriptional activation by C3a.
Stimulation of the C5aR in MSC causes robust ERK1/ 2 phosphorylation, which remains however largely cytoplasmic as previously described by us [17], but the very limited nuclear translocation of phospho-ERK1/2 induced by C5a was similarly blocked by Y27632 (results not shown).
Since NFκB inhibition also blocked the production of angiogenic factors by MSC, and since NFκB has to be translocated to the nucleus in order to acquire functionality, it was further determined, whether Y27632 would inhibit nuclear translocation of NFκBp65. Although nu-
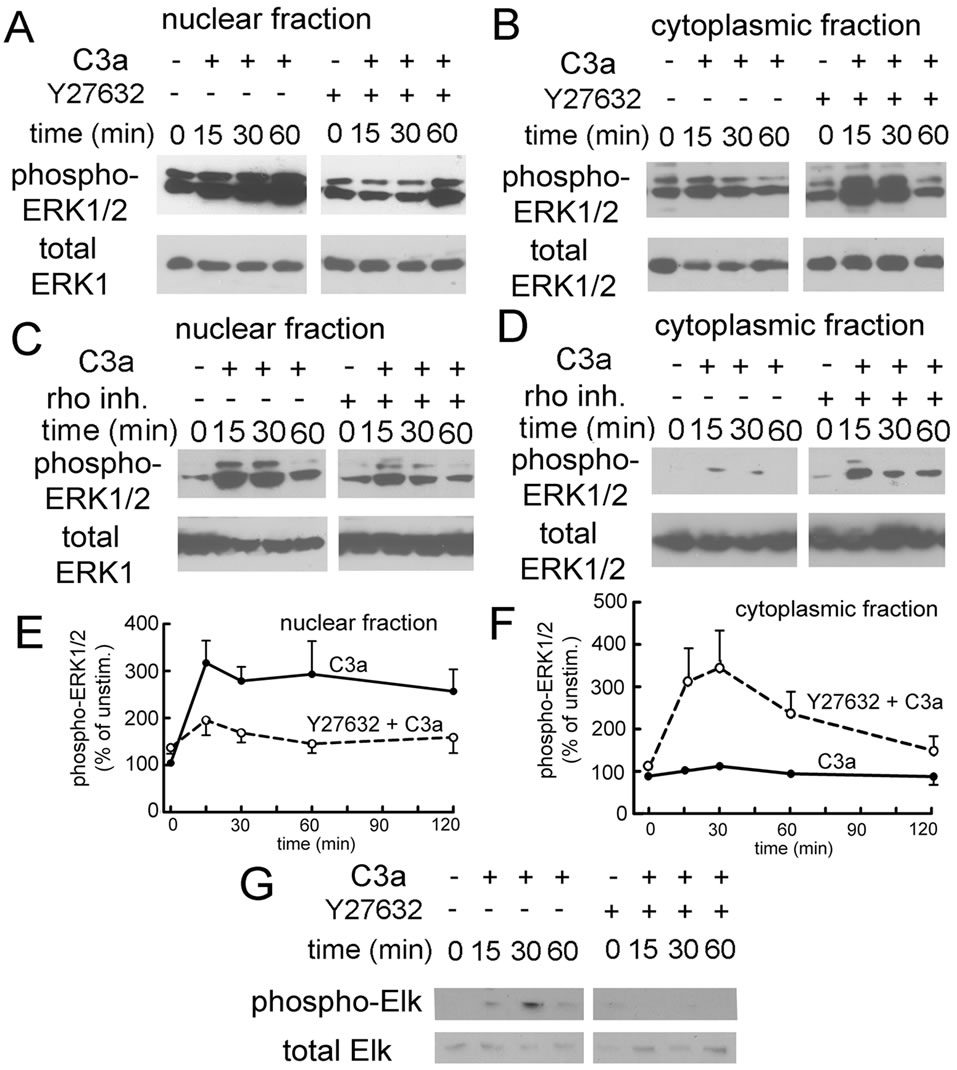
Figure 5. Effect of rho pathway inhibition on nuclear and cytoplasmic ERK1/2 phosphorylation as determined by Western blotting. A: Effect of Y27632 on ERK1/2 phosphorylation in nuclear fractions of MSC stimulated with 300 nM C3a. Nuclear ERK1/2 phosphorylation was largely attenuated in the presence of Y27632; B: Effect of Y27632 on ERK1/2 phosphorylation in cytoplasmic fractions of the same cells. Following C3a stimulation, cytoplasmic ERK1/2 phosphorylation was enhanced in cells exposed to Y27632. Inhibition of rho kinase prevented translocation of phospho-ERK1/2 to the nucleus, but not ERK1/2 phosphorylation per se; C: Effect of the rho inhibitor CT04-A on ERK1/2 phosphorylation in nuclear fractions of MSC stimulated with 300 nM C3a; D: Effect of the rho inhibitor CT04-A in cytoplasmic fractions of the same cells. Results seen in the presence of the rho inhibitor were the same as seen with rho kinase inhibition supporting the previous conclusion independently; E: Quantitation of the results seen in nuclear fractions of cells treated with Y27632 (●: no inhibitor, DMSO only; ○: 10 μM Y27632, mean and S.D. of 5 experiments) F: Quantitation of the results seen in cytoplasmic fractions of cells treated with Y27632 (●: no inhibitor, DMSO only; ○: 10 μM Y27632, mean and S.D. of 4 experiments). Error bars are only shown if they were outside the limit of the symbol; G: Effect of Rho kinase inhibition on C3a-dependent Elk phosphorylation. Y27632 blocked Elk phosphorylation as predicted. One experiment representative of 3.
clear translocation of NFκB was not as prominent following stimulation with C3a or C5a as it was with TNF- α used as a positive control, it was seen reproducibly with both anaphylatoxins (Figures 6A and B), but the response to C3a was again more pronounced. Similarly, inhibition of rho kinase inhibited nuclear translocation of NFκB in response to C3a and C5a (Figure 6C).
The results obtained by cell fractionation and Western blotting could furthermore be verified by immune-fluo-
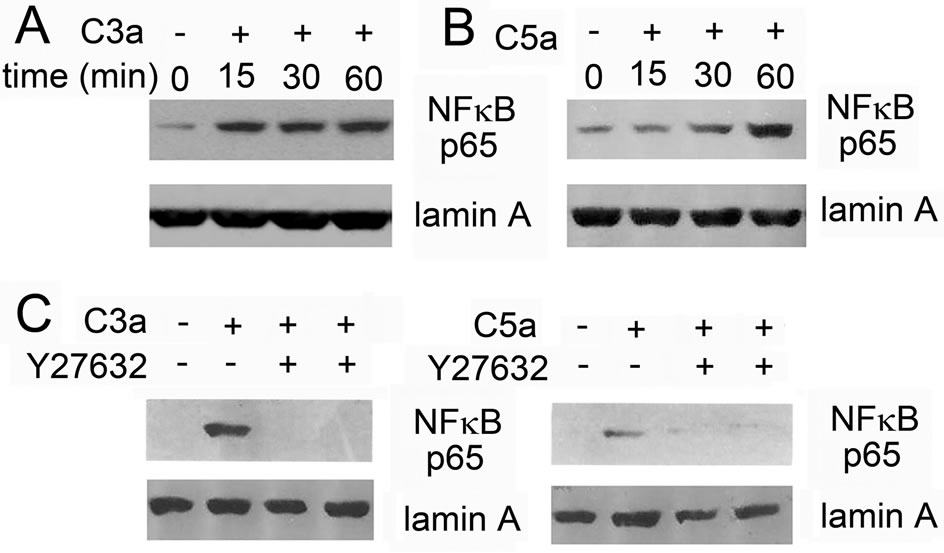
Figure 6. Effect of rho kinase inhibition on nuclear translocation of NFκBp65 as determined by Western blotting: MSC were stimulated with 300 nM C3a or 50 nM C5a for the indicated times and nuclear extracts were prepared and used in Western blots: A: Time-course of nuclear translocation of NFκBp65 in MSC stimulated with C3a; B: Time-course of nuclear translocation of NFκB in MSC stimulated with C5a, but with 4 times longer exposure time than used for A; C: Effect of the rho kinase inhibitor Y27632 on nuclear translocation of NFκBp65 in MSC stimulated with C3a or C5a for 1 hour, equal exposure time for C3a and C5a. The nuclear translocation of NFκBp65 observed following anaphylatoxin stimulation was blocked in the presence of the rho kinase inhibitor. One experiment representative of 3.
rescent staining and confocal microscopy. While NFκBp65 was excluded from the nucleus in unstimulated cells (Figure 7A), TNF-α induced complete nuclear translocation of NFκB (Figure 7B), which was almost as strong in cells stimulated with C3a (Figure 7C), while the effect of C5a was more moderate (Figure 7E). Again, this translocation was blocked by inhibition of rho kinase as shown for C3a in Figure 7D.
Similarly, nuclear translocation of phospho-ERK1/2 could be visualized following stimulation with C3a (Figure 8A, bottom panel), which was prevented in the presence of Y27632 (Figure 8B), where phospho-ERK1/2 remained in the cytoplasm. We previously reported that the C3aR in MSC is translocated to the nucleus following stimulation with C3a, especially, if these cells are grown at low cell density [17]. However, if the Rho kinase pathway or the ERK1/2 cascade were inhibited by Y27632 or PD98059 respectively, no such nuclear translocation of the receptor could be observed (Figures 8B and C). Under these conditions the C3aR in C3a-stimulated MSC appeared localized in endosomal vesicles, the most common route of receptor uptake for activated GPCRs.
3.3. Signaling Pathways Involved in C3aand C5a-Mediated Chemotaxis of MSC
To determine whether the same signaling pathways were important for MSC migration in the presence of C3a or C5a, the same inhibitors were used in chemotaxis assays. C3a-mediated chemotaxis of MSCs was fully blocked in
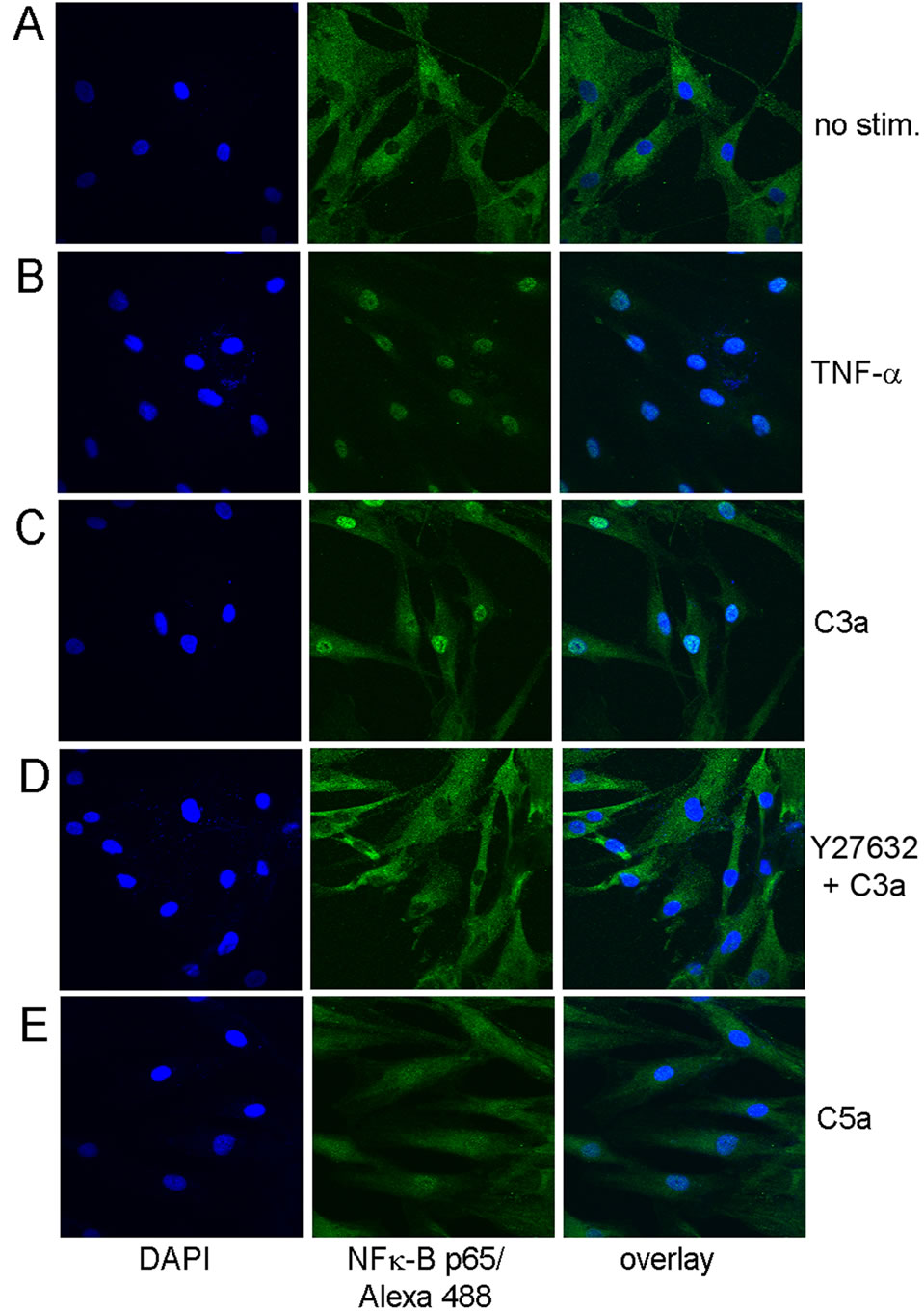
Figure 7. Immune-fluorescent detection of NFκBp65 in MSC stimulated with C3a or C5a for 1 hr. A: Cytoplasmic distribution of NFκBp65 in unstimulated MSC; B: Nuclear distribution of NFκBp65 in MSC stimulated with 50 ng/ml TNF-α used as a positive control; C: Largely nuclear distribution of NFκBp65 in MSC stimulated with 300 nM C3a; D: Inhibition of nuclear translocation of NFκBp65 in MSC stimulated with 300 nM C3a in the presence of Y27632; E: Partial nuclear distribution of NFκBp65 in MSC stimulated with 50 nM C5a. One experiment representative of 3 similar experiments.
the presence of the MAPK pathway inhibitor PD98059 (Figure 9A), which also led to partial inhibition of C5adependent chemotaxis (Figure 9B). A similar reduction of C3aor C5a-mediated cell migration was seen in the presence of the rho kinase inhibitor Y27632 (Figures 9A and B). LY294002, an inhibitor of PI-3K, which frequently blocks chemotaxis, showed no effect on C3amediated chemotaxis (Figure 9A), but caused partial inhibition of cell migration induced by C5a (Figure 9B), consistent with what is known for leukocyte chemotaxis [38]. Concomitant inhibition of both the ERK1/2 and PI- 3K pathways showed an additive effect resulting in complete inhibition of C5a-mediated chemotaxis (Figure 9B). Since NFκB activation is not a pathway associated with cell migration, no effect was expected, when NFκB was inhibited, which was indeed the case (results not shown). Thus signaling pathways for chemotaxis and angiogenic
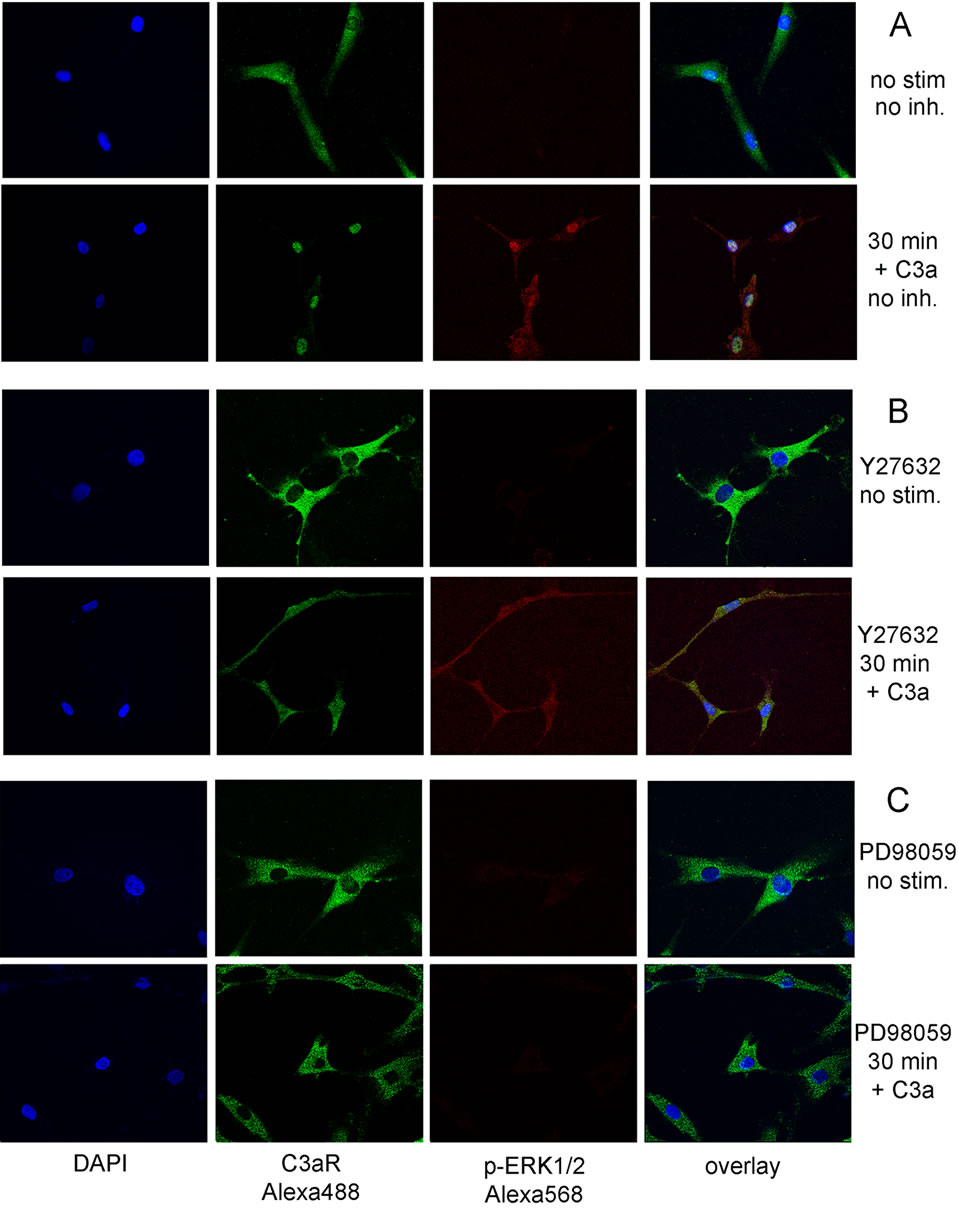
Figure 8. Immune-fluorescent detection of phospho-ERK1/2 (red) and of the C3aR (green) in MSC stimulated with 300 nM C3a for 30 min. In the absence of an inhibitor both phosphoERK1/2 and the C3aR are translocated to the nucleus following stimulation with C3a (A). In the presence of the rho kinase inhibitor Y27632, ERK1/2 was still phosphorylated, but was not translocated to the nucleus (B). Similarly the C3aR was not translocated to the nucleus, in the presence of Y27632 or PD989059 but accumulated in endosomal vesicles (B and C).
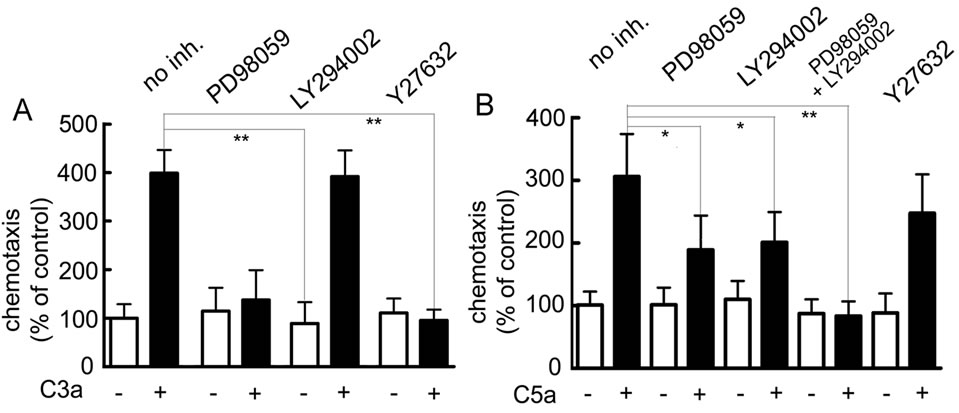
Figure 9. Effect of inhibitors on cell migration of MSC stimulated with the anaphylatoxins C3a or C5a. MSC were preincubated with inhibitors for 30 min in Transwell inserts prior to the addition of 100 nM C3a or 15 nM C5a to the bottom wells. A: Effect of inhibitors on chemotaxis induced by C3a: Inhibition of either the ERK1/2 cascade (PD98059) or the rho cascade (Y27632) blocked C3a-mediated cell migration, while inhibition of PI-3K (LY294002) was ineffective. Mean and S.D. of 3 experiments in triplicate; B: Effect of inhibitors on chemotaxis induced by C5a: Inhibition of the ERK1/2 cascade or of PI-3K partially blocked cell migration, while inhibition of both these pathways abolished C5a-mediated cell migration. Rho kinase inhibition showed only a minor effect. Mean and S.D. of 3 experiments in triplicate.
factor production are partially shared and partially different.
4. DISCUSSION
These results indicate that activation of the anaphylatoxin receptors on MSC can increase the production of angiogenic factors by these cells. This is achieved by using the same signaling pathways that are used, when monocytic cells are activated by C3a or C5a, which is primarily activation of NFκB downstream of rho activation [34], but in contrast to the response in monocytes/ macrophages, this does not lead to the concomitant release of the inflammatory cytokines TNF-α and IL-1β, thus converting a normally inflammatory pathway into one that supports regenerative processes.
Several pathways can lead to increased production of CXCL-8, IL-6 and VEGF; these include NFκB [39], AP-1 [40] and HIF1α [41] activation/up-regulation. Inhibitor studies indicated that NFκB activation was the major pathway, but a small contribution of other pathways cannot be excluded, e.g. nuclear phospho-ERK1/2 can activate AP1 and up-regulate its expression, resulting in increased CXCL-8 and VEGF expression [42,43].
Inhibition of rho kinase had no effect on overall ERK1/ 2 phosphorylation, but prevented translocation of phosphoERK1/2 to the nucleus (Figures 5 and 8). Thus activation of the rho kinase pathway provided the means to regulate the spatial distribution of phospho-ERK1/2, and such signaling compartmentalization is a major mechanism by which non-specific ERK1/2 phosphorylation induced by different stimuli and in various cellular contexts can achieve specific outcomes [37]. A similar role for rho kinase in nuclear translocation of phospho-ERK1/ 2 has been reported previously [44]. The exact mechanism involved in these instances is still not understood, but since the nuclear translocation of phospho-ERK1/2, NFκB and of the C3aR was prevented, when the rho cascade was inhibited, it seems to be a general inhibitory effect on nuclear import perhaps related to cytoskeletal function. Rho kinase activation is known to result in increased actin polymerization/stress fiber formation [45] and consecutive activation of the transcription factor serum response factor (SRF) [46]. MSC appear to have a high baseline level of rhoA [47] and polymerized F-actin, as even prolonged serum starvation fails to block stress fiber formation in these cells [17]. Since unpolymerized/ G-actin inhibits SRF, an inhibition, which is relieved when G-actin levels are depleted due to actin polymerization, any additional rho-dependent F-actin formation, e.g. as seen with C3a or C5a stimulation is likely to tilt the equilibrium towards activation of SRF. This is indeed the case, as C3a induced rho-kinase dependent phosphorylation of Elk (Figure 5H), which is part of the SRF complex [48].
It is less obvious, how nuclear translocation of phospho-ERK1/2 would translate into an effect on chemotaxis, but there is precedence for this observation [44,49, 50]. This effect of inhibition of the rho pathway on cell migration would appear not to be a direct effect of rho/ rho kinase activation on cell motility because of its short duration (5 min).
It has been shown that constitutively active rhoA or rho kinase supports osteogenesis in MSC, while dominant negative rhoA will result in adipogenic differentiation [51]. C3a or C5a in the absence of osteogenic cell culture medium does not cause any osteogenic differentiation of MSC (results not shown) perhaps due to the short duration of the rho cascade signal, but it remains to be seen, whether they have more subtle effects on MSC differentiation.
It has been reported previously that activation of the ERK1/2 cascade and the rho pathway are important for recruitment of MSC by sphingosine-1-phosphate (S1P) [52], and our results indicate that this is similarly true for C3a (Figure 9). However, it would be expected that the direction of migration in vivo would differ for the two stimuli: S1P reaches micromolar concentrations in plasma, but its concentration is 1000-fold lower in interstitial fluid [53] causing a chemotactic gradient by which MSC may be recruited to the bloodstream. In contrast C3a is inactivated by serum carboxypeptidase N [54], which will limit the action of C3a to the interstitial space, and it is therefore expected that C3a will play a role in the local recruitment of MSC to a site of tissue injury, where complement is activated without entering the vasculature. In addition interstitial C3a may recruit MSCs that have lodged on the lumen side of the microvasculature due to other stimuli.
Rho activation is generally associated with activation of GPCRs coupled to Gα12 or Gα13, which directly interact with the Rho guanine nucleotide exchange factor p115RhoGEF [55]. However, C3aor C5a-mediated Rho activation in MSC was completely Gi-dependent (Figure 4) indicating that Gi-coupling should not be a priori excluded, when rho activation is observed.
Complement activation leading to the production of the anaphylatoxins C3a and C5a occurs, wherever there is tissue injury. It is accepted that much of the therapeutic effect of MSC is due to the production of trophic factors by these cells rather than to tissue integration [56]. Our in vitro angiogenesis results showing endothelial cell tube formation in conditioned media from C3a-stimulated MSCs (Figure 2) suggest that C3a-activated MSC may constitute a naturally occurring repair mechanism used under pathophysiological conditions to improve tissue repair through the production of angiogenic factors in the interstitial space, and the perivascular location of MSC in vivo [57] situates them ideally to fulfill this function. In contrast conditioned media from C5a-stimulated showed less consistent tube formation (Figure 2F) in agreement with the less pronounced production of angiogenic factors induced by C5a (Figure 1C).
Others have shown that conditioned medium from MSC contains VEGF, IL-6 and CCL2, and that this medium can support angiogenesis [58,59]. We have confirmed that MSC produce and secrete angiogenic factors, and we have extended these observations to show that complement activation peptides C3a and C5a can augment the production of these factors.
There is no evidence that C3a or C5a induce in vitro angiogenesis directly, although endothelial cells express C3aRs and C5aRs [26]. However, C5a is a chemotactic factor for endothelial cells [26], and C3a can induce stress fiber formation in these cells through a rho-dependent pathway [26]. Furthermore, it has been described that both anaphylatoxins can induce chemokines such as CXCL8, an angiogenic factor, in cultured endothelial cells [60]. Therefore the anaphylatoxins may influence angiogenesis by activation of MSC as well as by additional subtle effects on endothelial cells.
Although the C3aR contains a possible nuclear import sequence, stimulation of this receptor in other cell types results in the usual receptor internalization to the endosomal compartment [17]. It is not clear what determines translocation of the C3aR to the nucleus in MSCs, but these cells appear to have a particularly pronounced nuclear uptake machinery [61,62], which has been implied to play a role in the control of self-renewal and differentiation. Embryonic stem cells undergo active remodeling of nuclear transport infrastructure to support nuclear translocation of cardiac transcription factors and engagement of cardiac differentiation [63], and it stands to be seen whether a similar situation exists in MSC.
In summary, C3a and C5a induce transient rho pathway activation in MSC, which precedes nuclear translocation of phospho-ERK1/2 and NFκB leading to the production of a variety of angiogenic factors by these cells, which may contribute to the regenerative function of these cells.
These results coupled with published findings about the effect of C3a on stem cells (17, 67) suggest a new realization as to the dominant biological role of this anaphylatoxin: Historically C3a and C5a have been discussed together as proinflammatory mediators, and whereas this is certainly correct for C5a, studies dealing with stem cells suggest that the true biological role of C3a may not be that of an inflammatory mediator but rather a local wound healing factor. The effect of C3a to potentiate bone marrow retention of hematopoietic stem cells [64], its neuroprotective effect on neural progenitors [62], the ability of C3a to chemo-attract MSC [17] and the findings presented here all point to a reparative function for this activation peptide derived from complement C3.
5. ACKNOWLEDGEMENTS
This work was funded by grants R21 HL094878, DOD/BCRP BC097127 and CBCRP 161B0086 to I.U.S., grants R43AI082759 and R21NS062428 to SKK, and grant 4061 from the National Multiple Sclerosis Foundation to RGD. The hMSC employed in this work were provided by the Tulane Center for Regenerative Medicine, now the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from NCRR of the NIH, Grant # P40RR017447. 
REFERENCES
- Caplan, A.I. and Dennis, J.E. (2006) Mesenchymal stem cells as trophic mediators. Journal of Cellular Biochemistry, 98, 1076-1084. doi:10.1002/jcb.20886
- Phinney, D.G. and Prockop, D.J. (2007) Concise review: Mesenchymal stem/multi-potent stromal cells (MSCs): The ctate of transdifferentiation and modes of tissue repair— Current views. Stem Cells, 25, 2896-2902. doi:10.1634/stemcells.2007-0637
- Aggarwal, S. and Pittenger, M.F. (2005) Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood, 105, 1815-1822. doi:10.1182/blood-2004-04-1559
- Maggini, J., Mirkin, G., Bognanni, I., Holmberg, J., Piazzon, I.M., Nepomnaschy, I., et al. (2010) Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile, PLoS ONE, 5, e9252. doi:10.1371/journal.pone.0009252
- Hare J.M., Traverse J.H., Henry T.D., Dib N., Strumpf R.K., Schulman S.P., et al. (2009) A randomized, doubleblind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. Journal of the American College for Cardiology, 54, 2277-2286. doi:10.1016/j.jacc.2009.06.055
- Lucchini, G., Introna, M., Dander, E., Rovelli, A., Balduzzi, A., Bonanomi, S., et al. (2010) Platelet-lysate-expanded mesenchymal stromal cells as a salvage therapy for severe resistant graft-versus-host disease in a pediatric population. Biology of Blood Marrow Transplantation, 16, 1293-1301. doi:10.1016/j.bbmt.2010.03.017
- Sackstein, R., Merzaban, J.S., Cain, D.W., Dagia, N.M., Spencer, J.A., Lin, C.P., et al. (2008) Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nature Medicine, 14, 181-187. doi:10.1038/nm1703
- Bartunek, J., Croissant, J.D., Wijns, W., Gofflot, S., de Lavareille, A., Vanderheyden, M., et al. (2007) Pretreatment of adult bone marrow mesenchymal stem cells with cardiomyogenic growth factors and repair of the chronically infarcted myocardium. American Journal of Physiology Heart Circulatiory Physiology, 292, H1095-1104. doi:10.1152/ajpheart.01009.2005
- Herrmann, J.L., Wang, Y., Abarbanell, A.M., Weil, B.R., Tan, J. and Meldrum, D.R. (2010) Preconditioning mesenchymal stem cells with transforming growth factor-alpha improves mesenchymal stem cell-mediated cardioprotection. Shock, 33, 24-30. doi:10.1097/SHK.0b013e3181b7d137
- Pons, J., Huang, Y., Arakawa-Hoyt, J., Washko, D., Takagawa, J., Ye, J., et al. (2008) VEGF improves survival of mesenchymal stem cells in infarcted hearts. Biochemical Biophysical Research Communication, 376, 419-422. doi:10.1016/j.bbrc.2008.09.003
- Hahn, J.Y., Cho, H.J., Kang, H.J., Kim, T.S., Kim, M.H., Chung, J.H., et al. (2008) Pre-treatment of mesenchymal stem cells with a combination of growth factors enhances gap junction formation, cytoprotective effect on cardiomyocytes, and therapeutic efficacy for myocardial infarction. Journal of the American College of Cardiology, 51, 933-943. doi:10.1016/j.jacc.2007.11.040
- Schmidt, A., Ladage, D, Schinkothe, T., Klausmann, U., Ulrichs, C., Klinz, F.J., et al. (2006) Basic fibroblast growth factor controls migration in human mesenchymal stem cells. Stem Cells, 24, 1750-1758. doi:10.1634/stemcells.2005-0191
- Ponte, A.L., Marais, E., Gallay, N., Langonne, A., Delorme, B., Herault, O., Charbord, P. and Domenech, J. (2007) The in vitro migration capacity of human bone marrow mesenchymal stem cells: Comparison of chemokine and growth factor chemotactic activities. Stem Cells, 25, 1737-1745. doi:10.1634/stemcells.2007-0054
- Sordi, V., Malosio, M.L., Marchesi, F., Mercalli, A., Melzi, R., Giordano, T., et al. (2005) Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood, 106, 419-427. doi:10.1182/blood-2004-09-3507
- Honczarenko, M., Le, Y., Swierkowski, M., Ghiran, I., Glodek, A.M. and Silberstein, L.E. (2006) Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells, 24, 1030- 1041. doi:10.1634/stemcells.2005-0319
- Fox, J.M., Chamberlain, G., Ashton, B.A. and Middleton, J. (2007) Recent advances into the understanding of mesenchymal stem cell trafficking. British Journal of Haematology, 137, 491-502. doi:10.1111/j.1365-2141.2007.06610.x
- Schraufstatter, I.U, DiScipio, R.G., Zhao, M. and Khaldoyanidi, S.K. (2009) C3a and C5a are chemotactic factors for human mesenchymal stem cells, which cause prolonged ERK1/2 phosphorylation. Journal of Immunology, 182, 3827-3836. doi:10.4049/jimmunol.0803055
- Fernandez, H.N., Henson, P.M., Otani, A. and Hugli, T.E. (1978) Chemotactic response to human C3a and C5a anaphylatoxins. I. Evaluation of C3a and C5a leukotaxis in vitro and under simulated conditions in vivo. Journal of Immunology, 120, 109-115.
- Norgauer, J., Dobos, G., Kownatzki, E., Dahinden, C., Burger, R., Kupper, R., et al. (1993) Complement fragment C3a stimulates Ca2+ influx in neutrophils via a pertussis toxin sensitive G protein. European Journal of Biochemistry, 217, 289-294. doi:10.1111/j.1432-1033.1993.tb18245.x
- Daffern, P.J., Pfeifer, P.H., Ember, J.A. and Hugli, T.E. (1995) C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation is secondary to eosinophil activation. Journal of Experimental Medicine, 181, 2119-2127. doi:10.1084/jem.181.6.2119
- Zwirner, J., Gotze, O., Moser, A., Sieber, A., Begemann, G., Kapp, A., et al. (1997) Bloodand skin-derived monocytes/macrophages respond to C3a but not C3a (desArg) with a transient release of calcium via a pertussis toxin sensitive pathway. European Journal of Immunology, 27, 2317-2322. doi:10.1002/eji.1830270928
- Elsner, J., Oppermann, M., Czech, W. and Kapp, A. (1994) C3a activates the respiratory burst in human polymorphonuclear neutrophilic leukocytes via pertussis toxin-sensitive G-proteins. Blood, 83, 3324-3331.
- Klos, A., Bank, S., Gietz, C., Bautsch, W., Köhl, J., Burg, M. and Kretzschmar, T. (1992) C3a receptor on dibutyryl-cAMP-differentiated U937 cells and human neutronphils: The human C3a receptor characterized by functional responses and 125I-C3a binding. Biochemistry, 31, 11274-11282. doi:10.1021/bi00161a003
- DiScipio, R.G., Daffern, P.J., Jagels, M.A., Broide, D.H. and Sriramarao, P. (1999) A comparison of C3a and C5amediated stable adhesion of rolling eosinophils in postcapillary venules and transendothial migration in vitro and in vivo. Journal of Immunology, 162, 1127-1136.
- Guo, R.F. and Ward, P.A. (2005) Role of C5a in inflammatory responses. Annual Review of Immunology, 23, 821- 852. doi:10.1146/annurev.immunol.23.021704.115835
- Schraufstatter, I.U., Trieu, K., Sikora, L., Sriramarao, P. and DiScipio R. (2002) Complement C3a and C5a induce different signal transduction cascades in endothelial cells. Journal of Immunology, 169, 2102-2110.
- Venkatesha, R.T., Berla Thangam, E., Zaidi, A.K. and Ali, H. (2005) Distinct regulation of C3a-induced MCP- 1/CCL2 and RANTES/CCL5 production in human mast cells by extracellular signal regulated kinase and PI3 kinase. Molecular Immunology, 42, 581-587. doi:10.1016/j.molimm.2004.09.009
- Chao, T.S., Ember, J.A., Wang, M., Bayon, Y., Hugli, T.E. and Ye, R.D. (1999) Role of the second extracellular loop of human C3a receptor in agonist binding and receptor function. Journal of Biological Chemistry, 274, 9721- 9728. doi:10.1074/jbc.274.14.9721
- Lee, D.K., Lanca, A.J., Cheng, R., Nguyen, T., Ji, X.D., Gobeil, F., et al. (2004) Agonist-independent nuclear localization of the apelin, angiotensin AT1, and bradykinin B2 receptors. Journal of Biological Chemistry, 279, 7901- 7908. doi:10.1074/jbc.M306377200
- Lu, D., Yang, H. and Raizada, M.K. (1996) Angiotensin II regulation of neuromodulation: Downstream signaling mechanism from activation of mitogen-activated protein kinase. Journal of Cell Biology, 135, 1609-1617. doi:10.1083/jcb.135.6.1609
- Brunet, A., Roux, D., Lenormand, P., Dowd, S., Keyse, S. and Pouyssegur, J. (1999) Nuclear translocation of p42/ p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO Journal, 18, 664-674. doi:10.1093/emboj/18.3.664
- Goetzl, E.J. (2007) Diverse pathways for nuclear signaling by G protein-coupled receptors and their ligands. FASEB Journal, 21, 638-642. doi:10.1096/fj.06-6624hyp
- Hsu, M.H., Wang, M., Browning, D.D., Mukaida, N. and Ye, R.D. (1999) NF-κB activation is required for C5a-induced interleukin-8 gene expression in mononuclear cells. Blood, 93, 3241-3249.
- Huang, S., Chen, L.Y., Zuraw, B.L., Ye, R.D. and Pan, Z.K. (2001) Chemoattractant-stimulated NF-κB activation is dependent on the jow molecular weight GTPase rhoA. Journal of Biological Chemistry, 276, 40977-40981. doi:10.1074/jbc.M105242200
- van den Berk, L.C.J., Jansen, B.J.H., Siebers-Vermeulen, K.G.C., Roelofs, H., Figdor, C.G., Adema, G.J., et al. (2010) Mesenchymal stem cells respond to TNF but do not produce TNF. Journal of Leukocyte Biology, 87, 283- 289. doi:10.1189/jlb.0709467
- Li, Z., Wei, H., Deng, L., Cong, X. and Chen, X. (2010) Expression and secretion of interleukin-1β, tumour necrosis factor-α and interleukin-10 by hypoxiaand serumdeprivation-stimulated mesenchymal stem cells. FEBS Journal, 277, 3688-3698. doi:10.1111/j.1742-4658.2010.07770.x
- Caunt, C.J., Finch, A.R., Sedgley, K.R. and McArdle, C.A. (2006) Seven-transmembrane receptor signalling and ERK compartmentalization. Trends in Endocrinology and Metabolism, 17, 276-283. doi:10.1016/j.tem.2006.07.008
- Sasaki, T., Irie-Sasaki, J., Jones, R.G., Oliveira-dos-Santos, A.J., Stanford, W.L., Bolon, B., et al. (2000) Function of PI3K in thymocyte development, T cell activation, and neutrophil migration. Science, 287, 1040-1046. doi:10.1126/science.287.5455.1040
- Yasumoto, K., Okamoto, S., Mukaida, N., Murakami, S., Mai, M. and Matsushima, K. (1992) Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. Journal of Biological Chemistry, 267, 22506-22511.
- Bobrovnikova-Marjon, E.V., Marjon, P.L., Barbash, O., Vander Jagt, D.L. and Abcouwer, S.F. (2004) Expression of angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 is highly responsive to ambient glutamine availability: Role of nuclear factor-kappaB and activating protein-1. Cancer Research, 64, 4858-4869. doi:10.1158/0008-5472.CAN-04-0682
- Fang, H.Y., Hughes, R., Murdoch, C., Coffelt, S.B., Biswas, S.K., Harris, A.L., et al. (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood, 114, 844-859. doi:10.1182/blood-2008-12-195941
- Sparmann, A. and Bar-Sagi, D. (2004) Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell, 6, 447-458. doi:10.1016/j.ccr.2004.09.028
- Rak, J., Mitsuhashi, Y., Sheehan, C., Tamir, A., ViloriaPetit, A., Filmus, J., et al. (2000) Oncogenes and tumor angiogenesis: Differential modes of vascular endothelial growth factor up-regulation in ras-transformed epithelial cells and fibroblasts. Cancer Research, 60, 490-498.
- Zhao, M., Discipio, R.G., Wimmer, A.G. and Schraufstatter, I.U. (2006) Regulation of CXCR4-mediated nuclear translocation of ERK1/2. Molecular Pharmacology, 69, 66-75.
- Maekawa, M., Ishizaki, T., Boku, S., Watanabe, N., Fujita, A., Iwamatsu, A., et al. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science, 285, 895-899. doi:10.1126/science.285.5429.895
- Posern, G., Sotiropoulos, A. and Treisman, R. (2002) Mutant actins demonstrate a role for unpolymerized actin in control of transcription by serum response factor. Molecular Biology of the Cell, 13, 4167-4178. doi:10.1091/mbc.02-05-0068
- Beqaj, S., Jakkaraju, S., Mattingly, R.R., Pan, D. and Schuger, L. (2002) High RhoA activity maintains the undifferentiated mesenchymal cell phenotype, whereas RhoA down-regulation by laminin-2 induces smooth muscle myogenesis. Journal of Cell Biology, 156, 893-903. doi:10.1083/jcb.200107049
- Marais, R., Wynne, J. and Treisman, R. (1993) The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell, 73, 381-393. doi:10.1016/0092-8674(93)90237-K
- Gayer, C.P., Craig, D.H., Flanigan, T.L., Reed, T.D., Cress, D.E. and Basson, M.D. (2010) ERK regulates strain-induced migration and proliferation from different subcellular locations. Journal of Cellular Biochemistry, 109, 711-725.
- Zhao, Y., Lv, M., Lin, H.S., Hong, Y., Yang, F.C., Sun, Y.L., et al. (2012) ROCK1 induces ERK nuclear translocation in PDGF-BB-stimulated migration of rat vascular smooth muscle cells. IUBMB Life, 64, 194-202. doi:10.1002/iub.598
- McBeath, R., Pirone, D.M., Nelson, C.M., Bhadriraju, K. and Chen, C.S. (2004) Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Developmental Cell, 6, 483-495. doi:10.1016/S1534-5807(04)00075-9
- Meriane, M., Duhamel, S., Lejeune, L., Galipeau, J. and Annabi, B. (2006) Cooperation of matrix metalloproteinases with the RhoA/Rho kinase and mitogen-activated protein kinase kinase-1/extracellular signal-regulated kinase signaling pathways is required for the sphingosine- 1-phosphate-induced mobilization of marrow-derived stromal cells. Stem Cells, 24, 2557-2565. doi:10.1634/stemcells.2006-0209
- Pappu, R., Schwab, S.R., Cornelissen, I., Pereira, J.P., Regard, J.B., Xu, Y., et al. (2007) Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science, 316, 295-298. doi:10.1126/science.1139221
- Bokisch, V.A. and Muller-Eberhard, H.J. (1970) Anaphylatoxin inactivator of human plasma: Its isolation and characterization as a carboxypeptidase. Journal of Clnical Investigation, 49, 2427-2436. doi:10.1172/JCI106462
- Hart, M.J., Jiang, X., Kozasa, T., Roscoe, W., Singer, W.D., Gilman, A.G., et al. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by galpha13. Science, 280, 2112-2114. doi:10.1126/science.280.5372.2112
- Prockop, D.J. (2009) Repair of tissues by adult stem/ progenitor cells (MSCs): Controversies, myths, and changing paradigms. Molecular Therapy, 17, 939-946. doi:10.1038/mt.2009.62
- Crisan, M., Yap, S., Casteilla, L., Chen, C.W., Corselli, M., Park, T.S., et al. (2008) A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell, 11, 301-313. doi:10.1016/j.stem.2008.07.003
- Hung, S.C., Pochampally, R.R., Chen, S.C., Hsu, S.C. and Prockop, D.J. (2007) Angiogenic effects of human multipotent stromal cell conditioned medium activate the PI3K-Akt pathway in hypoxic endothelial cells to inhibit apoptosis, increase survival, and stimulate angiogenesis. Stem Cells, 25, 2363-2370. doi:10.1634/stemcells.2006-0686
- Boomsma, R.A. and Geenen, D.L. (2012) Mesenchymal stem cells secrete multiple cytokines that promote angiogenesis and have contrasting effects on chemotaxis and apoptosis. PLoS ONE, 7, e35685. doi:10.1371/journal.pone.0035685
- [61] Monsinjon, T., Gasque, P., Chan, P., Ischenko, A., Brady, J.J. and Fontaine, M. (2003) Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. FASEB Journal, 17, 1003-1014. doi:10.1096/fj.02-0737com
- [62] Klinz, F.J., Schmidt A., Schinköthe, T., Arnhold, S., Desai, B., Popken, F., et al. (2005) Phospho-eNOS Ser-114 in human mesenchymal stem cells: Constitutive phosphorylation, nuclear localization and upregulation during mitosis. European Journal of Cell Biology, 8, 809-818. doi:10.1016/j.ejcb.2005.06.003
- [63] Rahpeymai, Y., Hietala, M.A., Wilhelmsson, U., Fotheringham, A., Davies, I., Nilsson, A.K., et al. (2006) Complement: A novel factor in basal and ischemia-induced neurogenesis. The EMBO Journal, 25, 1364-1374. doi:10.1038/sj.emboj.7601004
- [64] Perez-Terzic, C., Behfar, A., Mery, A., Van Deursen, J.M.A., Terzic, A. and Puceat, M. (2003) Structural adaptation of the nuclear pore complex in stem cell-derived cardiomyocytes. Circulation Research, 92, 444-452. doi:10.1161/01.RES.0000059415.25070.54
- [65] Ratajczak, J., Reca, R., Kucia, M., Majka, M., Allendorf, D.J., Baran, J.T., et al. (2004) Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood, 103, 2071- 2078. doi:10.1182/blood-2003-06-2099
SUPPLEMENTARY INFORMATION
List of Abbreviations
C3aR: C3a Receptor; C5aR: C5a Receptor, DAPI 4,6- diamino-2-phenylindole; Elk: Ets-like transcription factor; EMSA Electrophoretic Mobility Shift Assay; ERK: Extracellular Signal Regulated Kinase; GPCR: G-Protein Coupled Receptor; HIF-1: Hypoxia Induced Factor; HUVEC: Human umbilical vein endothelial cell; MAPK: Mitogen activated Protein Kinase; MSC: Mesenchymal Stem Cells; MSK: Mitogen and Stress Activated Protein Kinase; NFkB: Nuclear factor kappa-B; PI-3K: Phosphoinositol-3 kinase; PMSF: Phenyl Methyl Sulfonyl Fluoride; PMA: Phorbol Myristate Acid; S1P: Sphingosine-1- phosphate; SRF: Serum Response Factor: TCF: Ternary Complex Factors; VEGF: Vascular Endothelial Growth Factor.
List of RT-PCR Primers
Actin forward : 5’-GAGCTGCCCGATGGCCAGGTCATCA CC-3’,
Actin reverse : 5’-TTAGAAGCATTTGCGGTGGAC GATGGA-3’,
CXCL-8 forward : 5’-GGATCCGCTAAAGAACTTAGATGTCAGT-3’,
CXCL-8 reverse : 5’-TGAATTCTAGCCCTCTTCAAAAACTT-3’,
bFGF forward : 5’-CCCGCCTTGCCCGAGGATGGC -3’,
bFGF reverse : 5’-TCAGCTCTTAGCAGACATTGGAAGAAA-3’,
VEGF forward : 5’-GCACCCATGGCAGAAGGAGGAGGG-3’,
VEGF reverse: 5’-TTACCGCCTCGGCTTGTCACATCT-3’,
TGFβ1 forward : 5’-AACACACTGCAAGTGGACATCAACGGG-3’,
TGFβ1 reverse: 5’-TCAGCTGCACTTGCAGGAGCGCAGA-3’
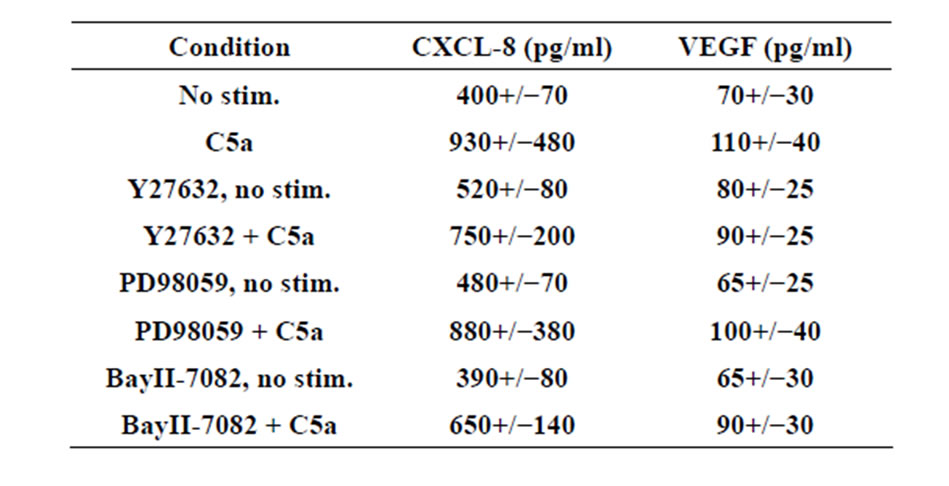
Table 1.Supplementary.