Journal of Cancer Therapy
Vol. 3 No. 4 (2012) , Article ID: 21584 , 9 pages DOI:10.4236/jct.2012.34030
Effect of Genetic Polymorphism of CYP2A6 on Individual Susceptibility to Colorectal Tumors in Japanese Smokers*
![]()
1Laboratory of Drug Metabolism, Graduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo, Japan; 2Department of Molecular-Targeting Cancer Prevention, Kyoto Prefectural University, Kyoto, Japan; 3Department of Public Health, Wakayama Medical University, Wakayama, Japan; 4Showa Pharmaceutical University, Tokyo, Japan.
Email: #hyamazak@ac.shoyaku.ac.jp
Received May 10th, 2012; revised June 12th, 2012; accepted June 29th, 2012
Keywords: P4502A6; Tobacco Smoking; Colorectal Cancer; Colorectal Adenomas; Case-Control Study
ABSTRACT
Tobacco smoking is a risk factor for colorectal cancer and adenomas. To clarify the effect of genetic factors on the risk for tobacco-related colorectal tumors in a Japanese population, we performed a case-control study on 300 patients with two or more tumors and 181 healthy controls; all were genotyped for CYP2A6*4, CYP2A6*7 and CYP2A6*9. Cigarette smoking increased colorectal tumor risk (trend-test P < 0.0000005). Current smokers plus ex-smokers (ever-smokers) with the CYP2A6*4/*4 genotype (whole gene deletion) showed the lowest risk among smokers [odds ratio (OR), 0.17; 95% confidence interval (CI), 0.05 - 0.62 compared to ever-smokers with the wild-type CYP2A6*1/*1]. When the participants were classified into four phenotype groups based on estimated CYP2A6 activity [i.e., normal (*1/*1), intermediate (heterozygotes for the *1 and a variant allele), slow (heterozygotes and homozygotes for variant alleles except for *4/*4) and poor (*4/*4)], the ORs (95% CIs) in ever-smokers of the normal, intermediate, slow and poor groups were 6.75 (2.73 - 16.76), 4.59 (2.10 - 10.06), 3.89 (1.69 - 8.95) and 1.17 (0.31 - 4.40), respectively, compared with never-smokers with normal CYP2A6 activity. The susceptibility to colorectal tumors was dependent on the predicted phenotype among ever-smokers (trend-test P = 0.015), but not among never-smokers (trend-test P = 0.47). Stratifying the subjects with respect to cumulative tobacco exposure and estimated CYP2A6 activity, we found the highest risk of colorectal tumors in subjects with higher CYP2A6 activity and higher cumulative tobacco exposure (trend-test P = 0.000023); the lowest risk was found in subjects with the lowest estimated CYP2A6 activity independent of tobacco exposure (trend-test P = 1.00). These results suggest that the gene-environment interaction (i.e., the CYP2A6-smoking interaction) strongly affects the individual susceptibility to tobacco-related colorectal tumors.
1. Introduction
Colorectal cancer is now one of the most frequent causes of cancer mortality worldwide [1]. The etiology of colorectal cancer is complex and multifactorial, involving both genetic and environmental factors. Numerous case-control and cohort studies have been conducted to investigate the relationship between colorectal cancer and lifestyle factors, including the consumption of red or well-done meat [2], fat [3], and alcohol [4] and cigarette smoking [5]. Cigarette smoking has consistently been identified as a potential risk factor for colorectal adenomas [5], which are recognized as precursor lesions for most cases of colorectal cancer [6]. The association between colorectal cancer and smoking has recently been reported in many studies [5,7,8].
Tobacco smoke contains more than 60 carcinogens, including N-nitrosamines, polycyclic aromatic hydrocarbons (PAHs) and aromatic amines [9]. To exert their carcinogenicity, these carcinogens require metabolic activation, and this activation is mediated mainly by phase I drugmetabolizing enzymes. Among phase I enzymes, the cytochrome P450 (P450 or CYP) family plays a crucial role in this respect. CYP2A6, one of the major members of the P450 family in humans, is involved in the metabolic activation of carcinogens, particularly tobacco-related N-nitrosamines [10-12]. CYP2A6 also catalyzes the elimination of nicotine (this elimination is reported to be a major factor in the maintenance of smoking behavior and tobacco dependence [13]) by metabolizing nicotine to cotinine [14], and cotinine to trans-3’-hydroxy-cotinine [15]. Genetic polymorphisms of CYP2A6 that influence the activity of CYP2A6 have been reported to affect tobacco-related lung cancer risk and smoking behavior [16].
To date, many variants of CYP2A6 have been identified (URL:http://www.imm.ki.se/CYPalleles/ cyp2a6.htm). Among these, the CYP2A6*4 allele, a whole-gene-deletion polymorphism [17-19], is thought to be the most important allele in Asians because it has an allele frequency of up to 20%; however, its frequency is low in Caucasians [19,20]. Subjects homozygous for CYP2A- 6*4 have been reported to show very low capacity to form cotinine from nicotine [21]. The CYP2A6*7 allele causes an amino acid substitution (Ile471Thr) that leads to reduced enzyme activity [20]. The CYP2A6*9 allele, which contains a −48T to G nucleotide substitution in the TATA box of the 5’-flanking region, has been reported to reduce the expression levels of CYP2A6 mRNA and protein in human livers [22]. Both the CYP2A6*7 and CYP2A6*9 alleles are also more common in Asians than in Caucasians [23].
Our recent studies have clarified that the mutant alleles described above reduce the susceptibility to squamous cell carcinoma and small-cell carcinoma, which are known to be types of lung cancer frequently induced by exposure to tobacco smoke [16,24,25]. The reduced risk in those with the CYP2A6*4 allele was also seen in the reduced occurrence of oral cancer in betel/quid chewers with the same allele [26]. However, there is little information on the effects of CYP2A6 polymorphisms on the risks for other smoking-related tumors, including colorectal cancer and adenomas.
In the present study, we found that CYP2A6 polymorphisms, CYP2A6*4, CYP2A6*7 and CYP2A6*9, reduced the tumor risk in combination with exposure to tobacco. We herein provide evidence that CYP2A6 activity is one of the principal factors that determine individual susceptibility to smoking-related colorectal cancer and adenomas.
2. Materials and Methods
2.1. Study Subjects and Data Collection
This study was approved by the ethics committees of the Osaka Medical Center for Cancer and Cardiovascular Diseases, the Osaka Central Hospital and Hokkaido University. All study participants were Japanese and were recruited from June 1993 to September 1997 at the Osaka Medical Center for Cancer and Cardiovascular Diseases. Cases were defined as patients with two or more colorectal tumors removed endoscopically within 3 months before recruitment. The colorectal tumors in this study included both adenomas (mild atypia, moderate atypia or severe atypia) and adenocarcinomas but not advanced or invasive cancer. Patients with one or more adenocarcinomas were assigned to the adenocarcinoma group, and those with adenomas only to the adenoma group. Individuals with a history of intestinal or gastric resection (other than appendectomy), familial adenomatous polyposis, ulcerative colitis, other malignant tumors and severe illness were excluded from the case group. Control subjects were healthy volunteers who required hospitalization for a health checkup. The ages of case and control subjects were defined at the time of the first hospital visit. Signed consent forms and completed questionnaires were collected from all case and control subjects before the collection of blood. The questionnaire covered smoking status (never-, exor current smoker), the total duration of smoking (excluding intermediate periods without smoking) and the average number of cigarettes smoked daily. The number of pack-year was calculated as a measure of cumulative cigarette smoking. One pack-year was defined as smoking 20 cigarettes daily for 1 year. Ex-smokers were defined as subjects with a minimum smoking history of 0.5 pack/day for at least 1 year. Current and ex-smokers were often combined into a group referred to as “eversmokers”.
2.2. Genotyping of CYP2A6
The CYP2A6*4 allele genotyped in this study was the CYP2A6*4A variant, which is identical to CYP2A6*4C [20]. The genotyping of CYP2A6*4 was based on polymerase chain reaction (PCR)/restriction fragment length polymorphism and was performed by a previously described method [20] with minor modifications. Briefly, a novel forward primer named 2A6-B6 (5’-CCT CAT CAC ACA CAA CTT CCT C-3’) and a reverse primer named 2A6-UTRAS1 (5’-TGT AAA ATG GGC ATG AAC GCC C-3’) [20] were used to amplify the common regions of CYP2A6*1 and CYP2A6*4. The reaction mixture contained 1 × LA PCR Buffer II, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.2 μM 2A6-B6, 0.2 μM 2A6-UTRAS1, 0.5 U of LA Taq DNA polymerase (TaKaRa BIO, Shiga, Japan), and approximately 50 ng of genomic DNA in a final volume of 25 μL. PCR was carried out under the following conditions: initial denaturation at 94˚C for 3 min; followed by 30 cycles of denaturation at 94˚C for 30 s, annealing at 64˚C for 30 s and extension at 72˚C for 1.5 min; and subsequently a final extension at 72˚C for 3 min. The PCR products consisted of 1358-bp fragments from the CYP2A6*1 allele and 1356-bp fragments from the CYP2A6*4 allele; these fragments were digested with Eco81 I. Fragments of 824 bp and 728 bp were derived from the CYP2A6*1 and CYP2A6*4 alleles, respectively. The CYP2A6*7 allele was genotyped using a two-step allele-specific-PCR method [20] in which the PCR products described above were used as templates.
For CYP2A6*9 genotyping, a new method based on allele-specific-PCR was developed for this study. In the first PCR, a 440-bp fragment from the 5’-flanking region of exon 1 of CYP2A6 was amplified using the primers 2A6 up-0.1 kb and 2A6 ex1R, as described previously [22]. In the second PCR, 2A6 TATA (5’-TCC CTC TTT TTC AGG CAG TAT-3’) and 2A6 TAGA (5’-TCC CTC TTT TTC AGG CAG TAG-3’) were employed as forward primers. The reaction mixture (25 μL) contained 1 μL of the first PCR products; 1 × PCR Buffer II, 2.0 mM MgCl2, 0.2 mM dNTPs, 0.2 μM 2A6 TATA or 0.2 μM 2A6 TAGA, 0.2 μM 2A6 ex1R; and 0.5 U of AmpliTaq DNA polymerase (Applied Biosystems, Foster City, CA). Amplification was performed by initial denaturation at 94˚C for 1 min; followed by 20 cycles of denaturation at 94˚C for 30 s, annealing at 64˚C for 30 s and extension at 72˚C for 45 s; and subsequently a final extension at 72˚C for 1 min. An aliquot (5 µL) of second PCR products (387 bp) was analyzed by electrophoresis with a 1% agarose gel. Alleles that were not classified as CYP2A6*4, CYP2A6*7 or CYP2A6*9 were assigned to the wild-type CYP2A6*1 allele.
In the analysis of putative CYP2A6 phenotypes, subjects were tentatively assigned to four groups based on their CYP2A6 genotypes according to the definitions of Fujieda et al. [16] : the putative “normal” phenotype group, CYP2A6*1/*1; the “intermediate” group, those heterozygous for the CYP2A6*1 allele (CYP2A6*1/*4, *1/*7 and *1/*9); the “slow” group, those heterozygous or homozygous for variant alleles except for those homozygous for the CYP2A6*4 allele (CYP2A6*4/*7, *4/*9, *7/*7, *7/*9 and *9/*9); and the “poor” group, CYP2A6*4/*4.
2.3. Data Analysis
Differences in the distribution of relevant characteristics between cases and controls were evaluated using t tests and χ2 tests. Compliance with Hardy-Weinberg equilibrium among controls was examined using the χ2 test. Associations between susceptibility to colorectal tumors and smoking-related indicators or the CYP2A6 genotypes/phenotypes were assessed using odds ratios (ORs). ORs and 95% confidence intervals (CIs) were calculated from logistic regression models to adjust for age and sex. When an association between smoking and colorectal tumor risk was detected, the never-smoker group was defined as the reference. The tertile values of continuous smoking variables (smoking duration, daily cigarette consumption and pack-year smoked) among ever-smokers in the controls were used as cutoff points. To investigate the impact of CYP2A6 genotypes and the putative CYP2A6 phenotypes on colorectal tumor risk, the ORs among the neverand ever-smokers were estimated by comparing with the reference group of the never-smokers with the CYP2A6*1/*1 genotype and the putative normal phenotype, respectively. Subsequently, among ever-smokers, the effects of the CYP2A6 genotypes and putative CYP2A6 phenotypes on the risk were assessed, defining eversmokers with the CYP2A6*1/*1 genotype and the normal phenotype, respectively, as the reference groups. More detailed investigations were conducted in association with the combination of putative CYP2A6 phenotypes and cigarette smoking. The median values of smoking duration, daily cigarette consumption and pack-year smoked were used as the cutoff points.
To investigate whether the estimated risks were dependent on increased smoking variables, trend test was performed by assigning ordinal scores as continuous variables in the logistic regression models. When the median or tertile values of the smoking indicators among the control group were used as the cutoff points, P values for the trend were estimated with the ordinal scores 1 - 3 assigned to the three levels of smoking exposure (1 for never-smokers, 2 for light smokers and 3 for heavy smokers). When the trends of associations between putative CYP2A6 phenotypes and the risks were tested, ordinal scores of 1 - 4 were assigned to the normal, intermediate, slow and poor groups, respectively. All tests of statistical significance were two-sided. A P value of 0.05 was considered the threshold of significance. All statistical analyses were carried out by using the statistical software SAS, version 5.0 (SAS Institute, Inc., Cary, NC).
3. Results
The characteristics and smoking-related indicators of the colorectal tumor patients and healthy controls are summarized in Table 1. The mean ages of the case and control groups were significantly different (P = 0.0048). There more men than women in both case and control groups; however, the gender distributions were not significantly different between the two groups (χ2 = 0.59, P = 0.44). More ever-smokers (i.e., ex-smokers and current smokers combined) were present in case group than in the control group (72.3% versus 51.9%; χ2 = 20.6, P = 0.0000058). Ever-smokers in the case group had significantly longer durations of smoking (P = 0.0013) and higher values of pack-year smoked (P = 0.000080) than ever-smokers in the control group. No significant difference was found in the daily cigarette consumption between cases and controls (P = 0.080).
Table 2 shows the association between the various smoking-related indicators (smoking status, smoking years, daily cigarette consumption and pack-year smoked) and the risk for colorectal tumors. Ever having smoked cigarettes was associated with a significantly increased risk for colorectal tumors (OR, 2.95; 95% CI, 1.85 - 4.70). In particular, current smokers were at the highest risk (OR, 3.16; 95% CI, 1.90 - 5.26). The association with smoking was also examined in terms of smoking years, daily cigarette consumption and packyear. The highest exposure levels were associated with the highest
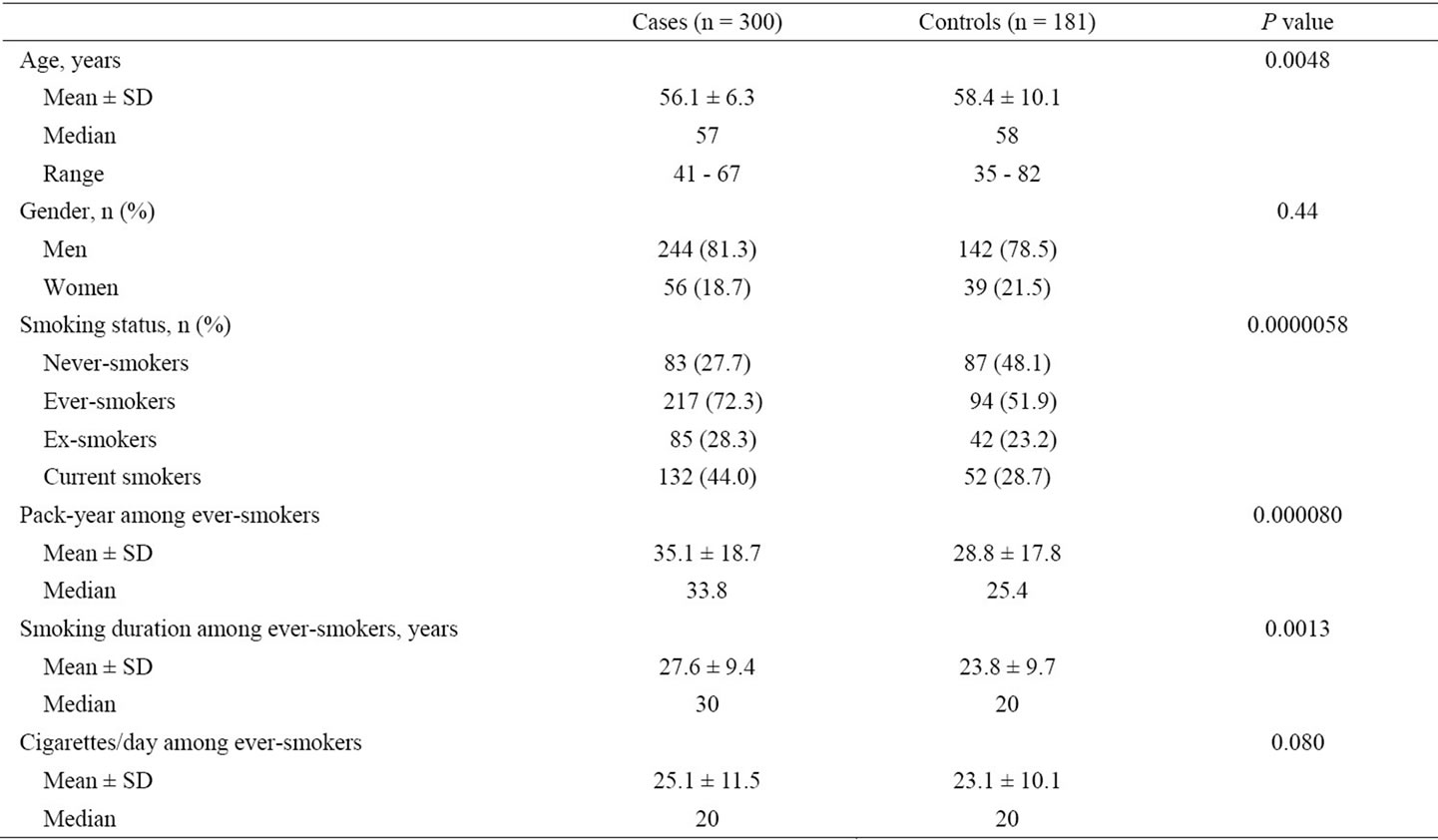
Table 1. Characteristics and smoking-related indicators of colorectal tumor patients and healthy controls enrolled in the present study.
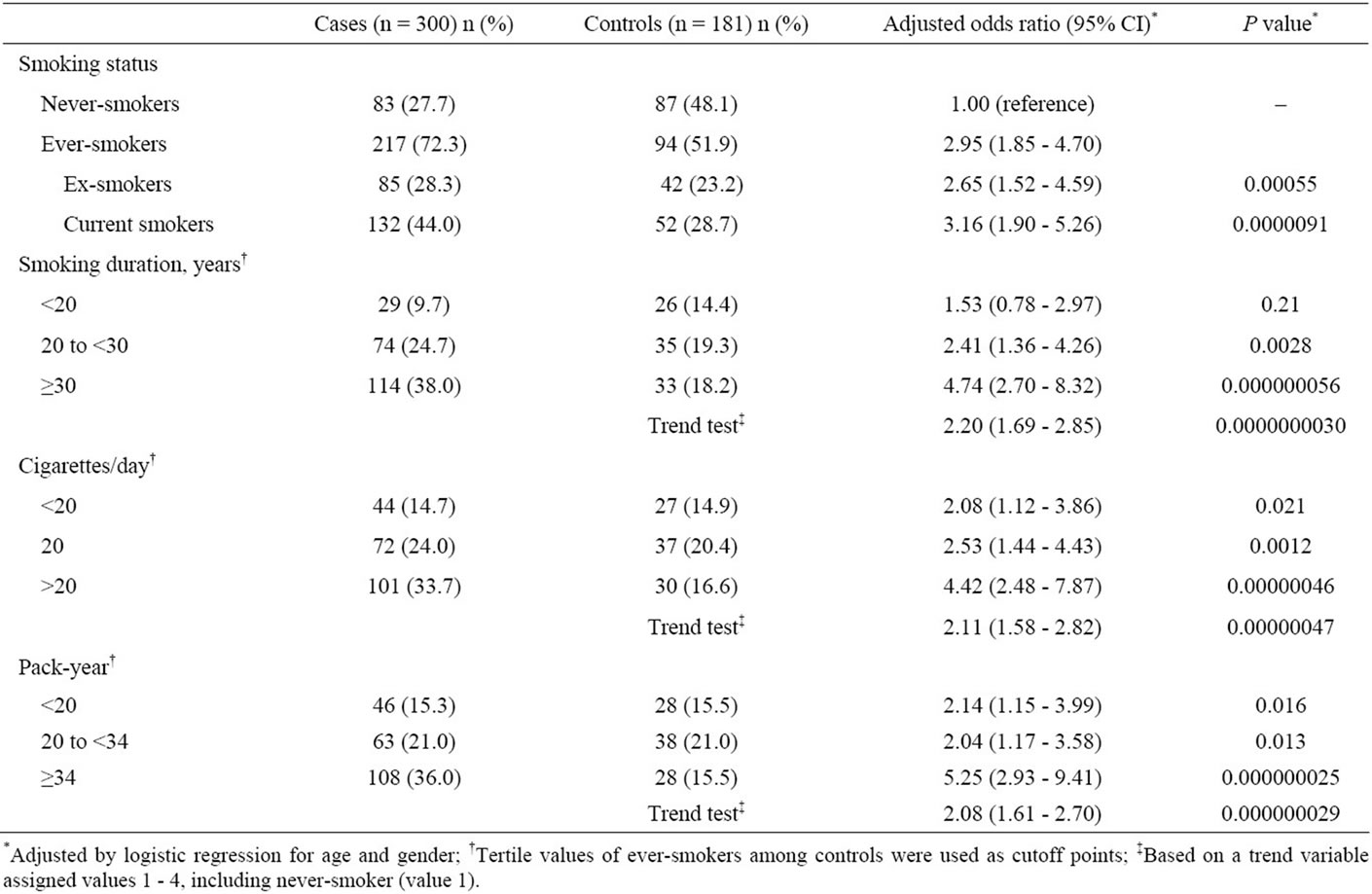
Table 2. Association between cigarette smoking and risk of colorectal tumors.
risk, as shown by the groups smoking for ≥30 years (OR, 4.74; 95% CI, 2.70 - 8.32), smoking >20 cigarettes/day (OR, 4.42; 95% CI, 2.48 - 7.87) and having smoked ≥34 pack-years (OR, 5.25; 95% CI, 2.93 - 9.41). Elevated risks for colorectal tumors were signifycantly associated with increased smoking years, daily cigarette consumption and pack-years (trend-test P = 0.00047, 0.015 and 0.0026, respectively), even when the tests for trend excluded never-smokers.
To confirm the effects of CYP2A6 genotypes on the risk for colorectal tumors in everand never-smokers, all case and control subjects were genotyped for CYP2A6*1, *4, *7 and *9 (Table 3). The distribution of CYP2A6 genotypes among controls were in Hardy-Weinberg equilibrium in ever-smokers (χ2 = 2.81, P = 0.97) and never-smokers (χ2 = 5.74, P = 0.77). This fact indicates that control subjects were sufficiently random and representative in both groups. Among never-smokers, no positive association was seen between CYP2A6 genotype and the risk for colorectal tumors. Compared to never-smokers with CYP2A6*1/*1 as a reference group, estimated risks among ever-smokers were significantly increased in CYP2A6*1/*1, *1/*4, *1/*7, *1/*9, *4/*7, *4/*9 and *7/*9 genotypes (e.g., CYP2A6*1/*1: OR, 6.85; 95% CI, 2.76 - 17.01). To investigate the impact of CYP2A6 genetic polymorphisms among smokers only, ever-smokers with the CYP2A6*1/*1 genotype were defined as the reference group. This analysis revealed that ever-smokers with the CYP2A6*4/*4 genotype had a significantly reduced risk (OR, 0.17; 95% CI, 0.05 - 0.62). The ORs among ever-smokers with other genotypes tended to be lower than 1, but these ORs were not significant.
To further investigate the effects of CYP2A6 genetic polymorphisms on the tumor risk in smokers, we classified all study subjects into four groups (normal, intermediate, slow and poor metabolizers) according to putative CYP2A6 phenotypes predicted from the CYP2A6 genotypes, as previously defined by Fujieda et al. ADDIN EN.CITE ADDIN EN.CITE.DATA [16] . Among never-smokers, the putative CYP2A6 phenotypes did not affect the risk for colorectal tumors (trend-test P = 0.47, see Table 4). However, compared with CYP2A6-normal never-smokers, the ORs in predicted normal, intermediate and slow phenotype groups among ever-smokers were significantly increased to 6.75 (95% CI, 2.73 - 16.76), 4.59 (95% CI, 2.10 - 10.06) and 3.89 (95% CI, 1.69 - 8.95), respectively. However, compared with CYP2A6- normal ever-smokers, the estimated risk in CYP2A6-poor ever-smokers was significantly low (OR, 0.17; 95% CI, 0.05 - 0.62). These results clearly indicate that the risk of colorectal tumors in smokers is decreased by polymorphisms that reduce the activity of CYP2A6 (trend-test P = 0.015).
Table 4 shows the effects of putative CYP2A6 phenotypes on the risk of colorectal tumors with stratification by smoking-related indicators. The ORs among exsmokers and current smokers relative to CYP2A6-normal never-smokers were significantly high, except for these in CYP2A6-poor groups. With respect to pack-years smoked, the highest OR was seen in groups with the putative normal phenotype with >25 pack-years smoked (OR, 10.03; 95% CI, 3.55 - 28.34) (see Table 4). In subjects with at least some CYP2A6 activity (i.e., all except
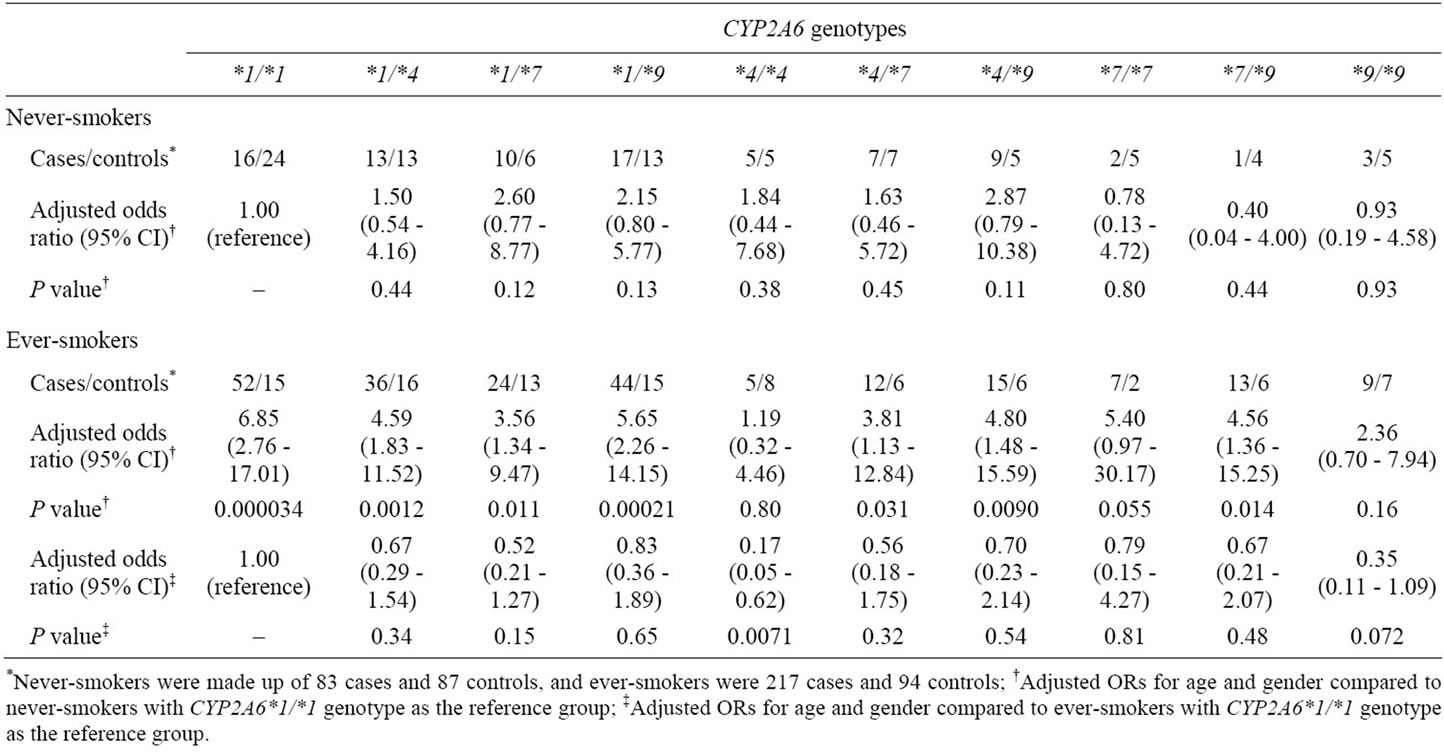
Table 3. Effect of CYP2A6 genotypes on risk of colorectal tumors in neverand ever-smokers.
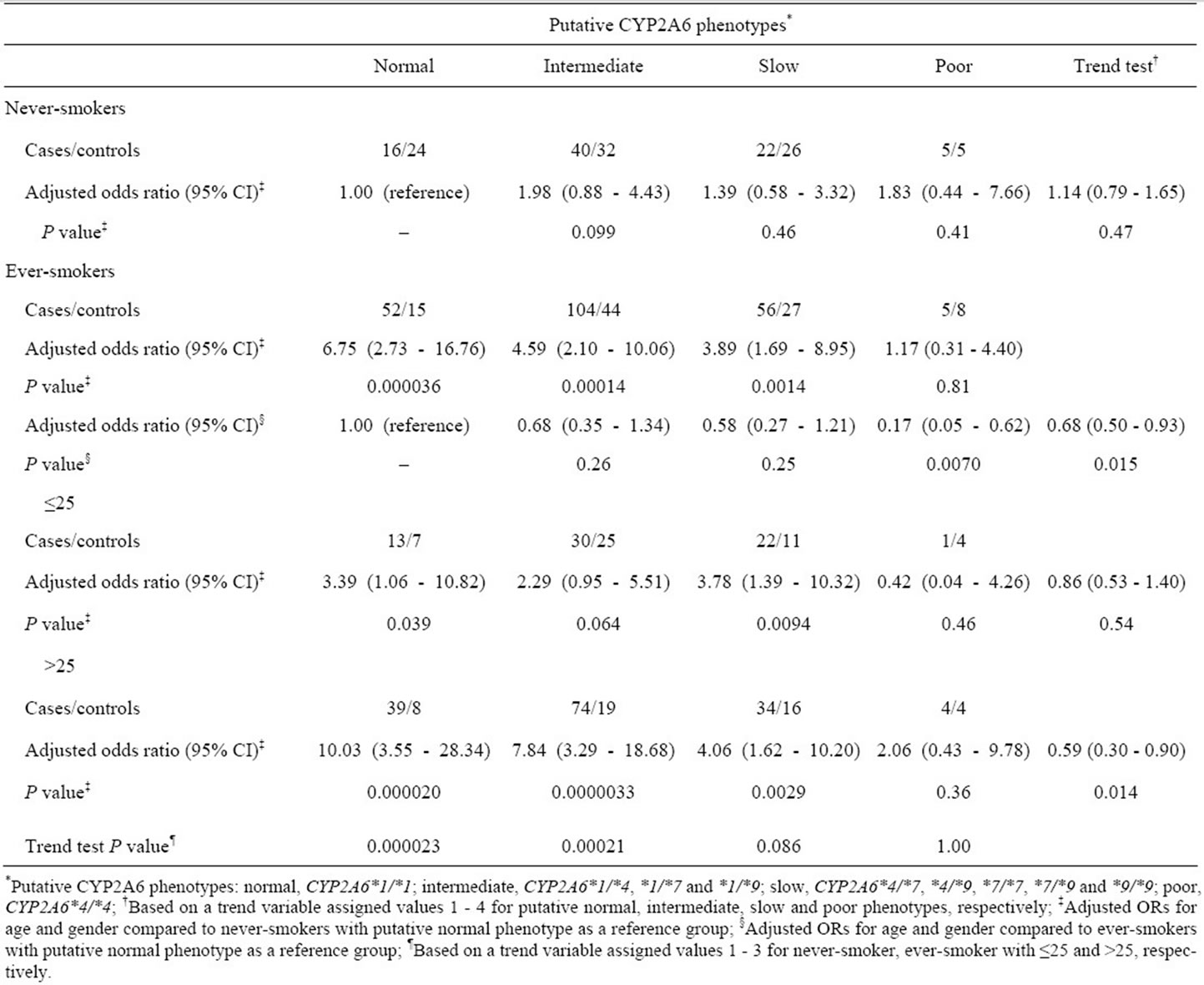
Table 4. Effects of putative CYP2A6 phenotypes on risk of colorectal tumors in never-smokers and ever-smokers.
those with CYP2A6*4/*4), the estimated risks for colorectal tumors were elevated with increased pack-years smoked (putative normal phenotype group, trend-test P = 0.000023; intermediate, P = 0.00021; and slow, P = 0.086). The ORs in subjects assigned to the putative normal or intermediate group were significantly dose dependent with regard to daily cigarette consumption and smoking duration (data not shown). In the CYP2A6-poor group, there was no evidence that the estimated risk was dependent on smoking-related indicators (pack-years, trend-test P = 1.00). Individuals who had smoked for >25 pack-years was strongly affected by the CYP2A6 genotype (trend-test P = 0.014). In contrast, no significant association between the susceptibility to colorectal tumors and putative CYP2A6 phenotypes was observed in light smokers or never-smokers (trend-test P = 0.54 or P = 0.47, respectively). Thus, the combination of CYP2A6 genetic polymorphism and cigarette smoking was clearly associated with the risk for colorectal tumors.
4. Discussion
Enzymes such as N-acetyltransferase [27-29], CYP1A1 [30,31], microsomal epoxide hydrolase [29,31-33], sulfotransferase [29] and glutathione S-transferase [28-30] are known to activate tobacco-related carcinogens (mainly PAHs and aromatic amines). Thus, a close association between genetic polymorphisms of these enzymes and susceptibility to colorectal cancer or adenomas is to be expected. However, few studies have provided definitive evidence that colorectal tumor risk is influenced by genetic polymorphisms of these enzymes, despite positive associations between the risk and smoking. The results of this current study are the first to show a clear association between CYP2A6 genotypes and the risk of colorectal cancer and adenomas. Supporting this association, we previously reported that CYP2A6 polymorphism is one of the principal determinants of the risks of tobacco-related lung cancer and betel/quid-related oral cancer [16,24-26].
Nowell et al. [34] reported a relationship between colorectal cancer risk and the urinary metabolite ratio of caffeine, which probably reflects the phenotype of CYP- 2A6, although genetic variants were not analyzed. They demonstrated that subjects with high or medium putative CYP2A6 activity had an increased risk for colorectal cancer (high: OR, 2.9; 95% CI, 1.6 - 5.0 and medium: OR, 2.0; 95% CI, 1.0 - 3.7) compared with subjects with low activity. Their data strongly supported our concept that CYP2A6 could be one of the most important factors contributing to colorectal cancer risk. In contrast, Sachse et al. [35] reported that, in a Caucasian population, no significant association was seen between CYP2A6 inactive alleles, particularly CYP2A6*2, and colorectal cancer risk (OR, 0.51; 95% CI, 0.28 - 1.06). This discrepancy between our results and theirs may be explained by several possibilities. First, ethnic differences exist in the frequencies of CYP2A6 variants; the frequency of inactive alleles is lower in Caucasian than in Japanese populations [23] . The frequencies of the CYP2A6*4, CYP2A6- *7 and CYP2A6*9 alleles among controls in this study were 19.8%, 16.9% and 20.7%, respectively. These allele frequencies were similar to those previously observed in a large-scale Japanese population (χ2 = 0.93, P = 0.82) [16] (in that study CYP2A6*10 and CYP2A6*11 alleles were classified as CYP2A6*7 and CYP2A6*1, respectively) indicating that no selection bias was evident in our study population. In Caucasians, the frequencies of the CYP2A6*4, CYP2A6*7 and CYP2A6*9 alleles are reported to be 0.5%, 0.0% and 5.2%, respectively [23]. As a result, the statistical power is likely insufficient in the report by Sachse et al. [35]. Second, they analyzed the samples without distinguishing smokers from nonsmokers [35]. We previously proposed that the positive relationship between cancer risk and CYP2A6 polymorphism can be detected only in smokers [16,20,25,26]. In accordance with our earlier studies, we found that CYP2A6 polymorphisms altered the susceptibility to colorectal tumors in smokers but not in non-smokers (Table 3). Finally, most of the case subjects in the present study had colorectal adenomas, and most adenocarcinomas in the present study were cancer in adenomas; those with advanced or invasive cancer were excluded. The adenoma-carcinoma sequence is now widely accepted as a central pathway of carcinogenesis. Adenomas are recognized as the precursor lesions for the majority of cases of colorectal cancer [6]. The main environmental risk factors for colorectal adenomas are meat and fat consumption [36] and cigarette smoking [5], which are similar to those for colorectal cancer. However, it has been reported that the risk for colorectal adenomas was more strongly increased by cigarette smoking than was the risk for colorectal cancer. The effects of CYP2A6 genetic polymorphism on the susceptibility to colorectal adenomas are thought to be clearer than those on the susceptibility to colorectal cancer.
We propose possible mechanisms for the correlation between CYP2A6 genotypes and the susceptibility to smoking-related colorectal cancer. CYP2A6 is reportedly responsible for the metabolic activation of tobacco-related N-nitrosamines, including N-nitrosodiethylamine, N-nitrosonorntine, 4-(methylnitrosa mino)-1-(3-pyridyl)-1-butanone, N-nitroso-piperidine and N-nitrosopyrrolidine [11,12]. DNA adducts with these N-nitrosamines have been detected in human colonic tissues [37]. In addition, when human colon microsomes were incubated with known carcinogen methyl-n-pentylnitrosamine, the addition of coumarin, a typical substrate of CYP2A6, inhibited methyln-pentylnitrosamine metabolite formation [38] . These results suggest that CYP2A6 is expressed in human colon. We recently clarified that CYP2A6 mRNA and protein were over-expressed in colorectal adenocarcinoma. Kumarakulasingham et al. [39] examined CYP2A6 protein expression in normal colon and colorectal cancer tissues. Considering these lines of evidence together with our results, it seems reasonable to assume that CYP2A6 expressed in colon tissues is a factor critical for colorectal tumorigenesis.
In conclusion, the findings of this current study indicate that CYP2A6 genetic polymorphisms alter the susceptibility to colorectal tumors in Japanese smokers, particularly in individuals highly exposed to tobacco smoke.
5. Acknowledgements
This work was supported in part by a Grant-in-Aid for Cancer Research from the Ministry of Health, Labour and Welfare of Japan and an SRF Grant for Biomedical Research in Japan.
REFERENCES
- P. Pisani, D. M. Parkin, F. Bray and J. Ferlay, “Estimates of the Worldwide Mortality from 25 Cancers in 1990,” International Journal of Cancer, Vol. 83, No. 1, 1999, pp. 18-29. doi:10.1002/(SICI)1097-0215(19990924)83:1<18::AID-IJC5>3.0.CO;2-M
- M. S. Sandhu, I. R. White and K. McPherson, “Systematic Review of the Prospective Cohort Studies on Meat Consumption and Colorectal Cancer Risk: A Meta-Analytical Approach,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 10, No. 5, 2001, pp. 439-446.
- G. R. Howe, K. J. Aronson, E. Benito, R. Castelleto, J. Cornee, S. Duffy, R. P. Gallagher, J. M. Iscovich, J. Deng-ao, R. Kaaks, et al., “The Relationship between Dietary Fat Intake and Risk of Colorectal Cancer: Evidence from the Combined Analysis of 13 Case-Control Studies,” Cancer Causes and Control, Vol. 8, No. 2, 1997, pp. 215-228. doi:10.1023/A:1018476414781
- E. Giovannucci, “Alcohol, One-Carbon Metabolism, and Colorectal Cancer: Recent Insights from Molecular Studies,” Journal of Nutrition, Vol. 134, No. 9, 2004, pp. 2475S- 2481S.
- E. Giovannucci, “An Updated Review of the Epidemiological Evidence that Cigarette Smoking Increases Risk of Colorectal Cancer,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 10, No. 7, 2001, pp. 725-731.
- B. Morson, “President’s Address. The Polyp-Cancer Sequence in the Large Bowel,” Proceedings of the Royal Society of Medicine, Vol. 67, No. 6, 1974, pp. 451-457.
- M. L. Slattery, J. D. Potter, G. D. Friedman, K. N. Ma and S. Edwards, “Tobacco Use and Colon Cancer,” International Journal of Cancer, Vol. 70, No. 3, 1997, pp. 259-264. doi:10.1002/(SICI)1097-0215(19970127)70:3<259::AID-IJC2>3.0.CO;2-W
- A. Chao, M. J. Thun, E. J. Jacobs, S. J. Henley, C. Rodriguez and E. E. Calle, “Cigarette Smoking and Colorectal Cancer Mortality in the Cancer Prevention Study II,” Journal of the National Cancer Institute, Vol. 92, No. 23, 2000, pp. 1888-1896. doi:10.1093/jnci/92.23.1888
- D. Hoffmann and I. Hoffmann, “The Changing Cigarette, 1950-1995,” Journal of Toxicology and Environmental Health, Vol. 50, No. 4, 1997, pp. 307-364. doi:10.1080/009841097160393
- C. L. Crespi, B. W. Penman, J. A. Leakey, M. P. Arlotto, A. Stark, A. Parkinson, T. Turner, D. T. Steimel, K. Rudo, R. L. Davies, et al., “Human Cytochrome P450IIA3: cDNA Sequence, Role of the Enzyme in the Metabolic Activation of Promutagens, Comparison to Nitrosamine Activation by Human Cytochrome P450IIE1,” Carcinogenesis, Vol. 11, No. 8, 1990, pp. 1293-1300. doi:10.1093/carcin/11.8.1293
- H. Yamazaki, Y. Inui, C. H. Yun, F. P. Guengerich and T. Shimada, “Cytochrome P450 2E1 and 2A6 Enzymes as Major Catalysts for Metabolic Activation of N-Nitrosodialkylamines and Tobacco-Related Nitrosamines in Human Liver Microsomes,” Carcinogenesis, Vol. 13, No. 10, 1992, pp. 1789-1794. doi:10.1093/carcin/13.10.1789
- H. Kushida, K. Fujita, A. Suzuki, M. Yamada, T. Endo, T. Nohmi and T. Kamataki, “Metabolic Activation of N-Alkylnitrosamines in Genetically Engineered Salmonella typhimurium Expressing CYP2E1 or CYP2A6 Together with Human NADPH-Cytochrome P450 Reductase,” Carcinogenesis, Vol. 21, No. 6, 2000, pp. 1227-1232. doi:10.1093/carcin/21.6.1227
- J. E. Henningfield, K. Miyasato and D. R. Jasinski, “Abuse Liability and Pharmacodynamic Characteristics of Intravenous and Inhaled Nicotine,” Journal of Pharmacology and Experimental Therapeutics, Vol. 234, No. 1, 1985, pp. 1-12.
- M. Nakajima, T. Yamamoto, K. Nunoya, T. Yokoi, K. Nagashima, K. Inoue, Y. Funae, N. Shimada, T. Kamataki and Y. Kuroiwa, “Role of Human Cytochrome P4502A6 in C-Oxidation of Nicotine,” Drug Metabolism and Disposition, Vol. 24, No. 11, 1996, pp. 1212-1217.
- M. Nakajima, T. Yamamoto, K. Nunoya, T. Yokoi, K. Nagashima, K. Inoue, Y. Funae, N. Shimada, T. Kamataki and Y. Kuroiwa, “Characterization of CYP2A6 Involved in 3’-Hydroxylation of Cotinine in Human Liver Microsomes,” Journal of Pharmacology and Experimental Therapeutics, Vol. 277, No. 2, 1996, pp. 1010-1015.
- M. Fujieda, H. Yamazaki, T. Saito, K. Kiyotani, M. A. Gyamfi, M. Sakurai, H. Dosaka-Akita, Y. Sawamura, J. Yokota, H. Kunitoh, et al., “Evaluation of CYP2A6 Genetic Polymorphisms as Determinants of Smoking Behavior and Tobacco-Related Lung Cancer Risk in Male Japanese Smokers,” Carcinogenesis, Vol. 25, No. 12, 2004, pp. 2451-2458. doi:10.1093/carcin/bgh258
- K. Nunoya, T. Yokoi, Y. Takahashi, K. Kimura, M. Kinoshita and T. Kamataki, “Homologous Unequal Cross-Over within the Human CYP2A Gene Cluster as a Mechanism for the Deletion of the Entire CYP2A6 Gene Associated with the Poor Metabolizer Phenotype,” The Journal of Biochemistry, Vol. 126, No. 2, 1999, pp. 402-407. |doi:10.1093/oxfordjournals.jbchem.a022464
- K. I. Nunoya, T. Yokoi, K. Kimura, T. Kainuma, K. Satoh, M. Kinoshita and T. Kamataki, “A New CYP2A6 Gene Deletion Responsible for the in Vivo Polymorphic Metabolism of (+)-cis-3,5-dimethyl-2-(3-pyridyl)thiazolidin-4-one Hydrochloride in Humans,” Journal of Pharmacology and Experimental Therapeutics, Vol. 289, No. 1, 1999, pp. 437- 442.
- M. Oscarson, R. A. McLellan, H. Gullsten, Q. Y. Yue, M. A. Lang, M. L. Bernal, B. Sinues, A. Hirvonen, H. Raunio, O. Pelkonen, et al., “Characterisation and PCR-Based Detection of a CYP2A6 Gene Deletion Found at a High Frequency in a Chinese Population,” FEBS Letters, Vol. 448, No. 1, 1999, pp. 105-110. doi:10.1016/S0014-5793(99)00359-2
- N. Ariyoshi, Y. Takahashi, M. Miyamoto, Y. Umetsu, S. Daigo, T. Tateishi, S. Kobayashi, Y. Mizorogi, M. A. Loriot, I. Stucker, et al., “Structural Characterization of a New Variant of the CYP2A6 Gene (CYP2A6*1B) Apparently Diagnosed as Heterozygotes of CYP2A6*1A and CYP2A6*4C,” Pharmacogenetics, Vol. 10, No. 8, 2000, pp. 687-693. doi:10.1097/00008571-200011000-00003
- M. Nakajima, J. T. Kwon, N. Tanaka, T. Zenta, Y. Yamamoto, H. Yamamoto, H. Yamazaki, T. Yamamoto, Y. Kuroiwa and T. Yokoi, “Relationship between Interindividual Differences in Nicotine Metabolism and CYP2A6 Genetic Polymorphism in Humans,” Clinical Pharmacology & Therapeutics, Vol. 69, No. 1, 2001, pp. 72-78. doi:10.1067/mcp.2001.112688
- K. Kiyotani, H. Yamazaki, M. Fujieda, S. Iwano, K. Matsumura, S. Satarug, P. Ujjin, T. Shimada, F. P. Guengerich, A. Parkinson, et al., “Decreased Coumarin 7-hydroxylase Activities and CYP2A6 Expression Levels in Humans Caused by Genetic Polymorphism in CYP2A6 Promoter Region (CYP2A6*9),” Pharmacogenetics, Vol. 13, No. 11, 2003, pp. 689-695. doi:10.1097/00008571-200311000-00005
- M. A. Gyamfi, M. Fujieda, K. Kiyotani, H. Yamazaki and T. Kamataki, “High Prevalence of Cytochrome P450 2A- 6*1A Alleles in a Black African Population of Ghana,” European Journal of Clinical Pharmacology, Vol. 60, No. 12, 2005, pp. 855-857. doi:10.1007/s00228-004-0854-9
- M. Miyamoto, Y. Umetsu, H. Dosaka-Akita, Y. Sawamura, J. Yokota, H. Kunitoh, N. Nemoto, K. Sato, N. Ariyoshi and T. Kamataki, “CYP2A6 Gene Deletion Reduces Susceptibility to Lung Cancer,” Biochemical and Biophysical Research Communications, Vol. 261, No. 3, 1999, pp. 658-660. doi:10.1006/bbrc.1999.1089
- N. Ariyoshi, M. Miyamoto, Y. Umetsu, H. Kunitoh, H. Dosaka-Akita, Y. Sawamura, J. Yokota, N. Nemoto, K. Sato and T. Kamataki, “Genetic Polymorphism of CYP2A6 Gene and Tobacco-Induced Lung Cancer Risk in Male Smokers,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 11, No. 9, 2002, pp. 890-894.
- Z. Topcu, I. Chiba, M. Fujieda, T. Shibata, N. Ariyoshi, H. Yamazaki, F. Sevgican, M. Muthumala, H. Kobayashi and T. Kamataki, “CYP2A6 Gene Deletion Reduces Oral Cancer Risk in Betel Quid Chewers in Sri Lanka,” Carcinogenesis, Vol. 23, No. 4, 2002, pp. 595-598. doi:10.1093/carcin/23.4.595
- J. D. Potter, J. Bigler, L. Fosdick, R. M. Bostick, E. Kampman, C. Chen, T. A. Louis and P. Grambsch, “Colorectal Adenomatous and Hyperplastic Polyps: Smoking and NAcetyltransferase 2 Polymorphisms,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 8, No. 1, 1999, pp. 69-75.
- O. L. van der Hel, H. B. Bueno de Mesquita, M. Roest, B. Slothouber, C. van Gils, P. A. van Noord, D. E. Grobbee and P. H. Peeters, “No Modifying Effect of NAT1, GSTM1, and GSTT1 on the Relation between Smoking and Colorectal Cancer Risk,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 12, No. 7, 2003, pp. 681-682.
- E. W. Tiemersma, A. Bunschoten, F. J. Kok, H. Glatt, S. Y. de Boer and E. Kampman, “Effect of SULT1A1 and NAT2 Genetic Polymorphism on the Association between Cigarette Smoking and Colorectal Adenomas,” International Journal of Cancer, Vol. 108, No. 1, 2004, pp. 97-103. doi:10.1002/ijc.11533
- H. Inoue, C. Kiyohara, T. Marugame, S. Shinomiya, E. Tsuji, K. Handa, H. Hayabuchi, K. Onuma, H. Hamada, H. Koga, et al., “Cigarette smoking, CYP1A1 Mspl and GSTM1 Genotypes, and Colorectal Adenomas,” Cancer Research, Vol. 60, No. 14, 2000, pp. 3749-3752.
- L. Hou, N. Chatterjee, W. Y. Huang, A. Baccarelli, S. Yadavalli, M. Yeager, R. S. Bresalier, S. J. Chanock, N. E. Caporaso, B. T. Ji, et al., “CYP1A1 Val462 and NQO1 Ser187 Polymorphisms, Cigarette Use, and Risk for Colorectal Adenoma,” Carcinogenesis, Vol. 26, No. 6, 2005, pp. 1122- 1128. doi:10.1093/carcin/bgi054
- C. M. Ulrich, J. Bigler, J. A. Whitton, R. Bostick, L. Fosdick and J. D. Potter, “Epoxide Hydrolase Tyr113His Polymorphism Is Associated with Elevated Risk of Colorectal Polyps in the Presence of Smoking and High Meat Intake,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 10, No. 8, 2001, pp. 875-882.
- G. J. Tranah, E. Giovannucci, J. Ma, C. Fuchs, S. E. Hankinson and D. J. Hunter, “Epoxide Hydrolase Polymorphisms, Cigarette Smoking and Risk of Colorectal Adenoma in the Nurses’ Health Study and the Health Professionals Follow-Up Study,” Carcinogenesis, Vol. 25, No. 7, 2004, pp. 1211-1218. doi:10.1093/carcin/bgh126
- S. Nowell, C. Sweeney, G. Hammons, F. F. Kadlubar and N. P. Lang, “CYP2A6 Activity Determined by Caffeine Phenotyping: Association with Colorectal Cancer Risk,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 11, No. 4, 2002, pp. 377-383.
- C. Sachse, G. Smith, M. J. Wilkie, J. H. Barrett, R. Waxman, F. Sullivan, D. Forman, D. T. Bishop and C. R. Wolf, “A Pharmacogenetic Study to Investigate the Role of Dietary Carcinogens in the Etiology of Colorectal Cancer,” Carcinogenesis, Vol. 23, No. 11, 2002, pp. 1839-1849. doi:10.1093/carcin/23.11.1839
- R. W. Haile, J. S. Witte, M. P. Longnecker, N. ProbstHensch, M. J. Chen, J. Harper, H. D. Frankl and E. R. Lee, “A Sigmoidoscopy-Based Case-Control Study of Polyps: Macronutrients, Fiber and Meat Consumption,” International Journal of Cancer, Vol. 73, No. 4, 1997, pp. 497-502. doi:10.1002/(SICI)1097-0215(19971114)73:4<497::AID-IJC7>3.0.CO;2-V
- H. Autrup, C. C. Harris and B. F. Trump, “Metabolism of Acyclic and Cyclic N-Nitrosamines by Cultured Human Colon,” Proceedings of the Society for Experimental Biology and Medicine, Vol. 159, No. 1, 1978, pp. 111-115.
- S. C. Chen, L. Zhou, X. Ding and S. S. Mirvish, “Depentylation of the Rat Esophageal Carcinogen, Methyl-nPentylnitrosamine, by Microsomes from Various Human and Rat Tissues and by Cytochrome P450 2A3,” Drug Metabolism and Disposition, Vol. 29, No. 9, 2001, pp. 1221-1228.
- M. Kumarakulasingham, P. H. Rooney, S. R. Dundas, C. Telfer, W. T. Melvin, S. Curran and G. I. Murray, “Cytochrome p450 Profile of Colorectal Cancer: Identification of Markers of Prognosis,” Clinical Cancer Research, Vol. 11, No. 10, 2005, pp. 3758-3765. doi:10.1158/1078-0432.CCR-04-1848
Abbreviations
PAHs: polycyclic aromatic hydrocarbons;
CYP: cytochrome P450;
OR: odds ratio;
CI: confidence interval.
NOTES
*There are non-financial competing interests in this study.
#Corresponding author.