Journal of Cancer Therapy
Vol.1 No.3(2010), Article ID:2602,9 pages DOI:10.4236/jct.2010.13018
Green Tea Polyphenols Mediated Apoptosis in Intestinal Epithelial Cells by a Fadd-Dependent Pathway
1Center for Oral Health Research, College of Dentistry, Dallas, USA; 2Department of Internal Medicine, University of Kentucky Medical Center, Lexington, USA.
Email: Helieh.oz@uky.edu
Received May 16th, 2010; revised June 22nd, 2010; accepted July 11th, 2010.
Keywords: Intestinal Epithelial Cells, Green Tea Polyphenols, EGCG, Apoptosis
ABSTRACT
Colorectal cancer is the most common malignant complication in patients with chronic inflammatory bowel disease (IBD). In addition, these patients are at risk for developing painful complications during chemotherapy due to cytotoxic effects of drugs currently in use. Past studies have suggested a protective effect of tea consumption on gastrointestinal (GI) malignancies. Green tea polyphenols (GrTP) inhibited carcinogen-induced GI tumors in rodents and induced apoptosis in various carcinoma cell lines. We hypothesized that GrTP and its polyphenolic compounds regulate apoptosis in the intestinal epithelia. In this study, the effects of GrTP and its polyphenolics on apoptosis was evaluated in intestinal epithelial, IEC-6, cells grown to 85% confluency. GrTP (400-800 mg/ml) induced DNA fragmentation in a dose dependent fashion. Higher concentrations (> 800 mg/ml) induced a mixed apoptosis and cytolysis. Epithelial cells exposed to GrTP and a major polyphenol, EGCG, but not EGC or EC, increased caspase activities in a time and dose dependent manner. The caspase inhibitors rescued cells from GrTP and EGCG-induced cell death. Concomitantly, GrTP resulted in activation of fatty acid synthase (Fas)-associated protein with death domain (FADD) and recruitment to Fas/CD95 domain 30 minutes following treatment. While GrTP also blocked NF-kB activation, an NF-kB inhibitor (MG132) only promoted cytolysis. In conclusion, these data demonstrated GrTP and EGCG induced apoptosis in intestinal epithelia mediated by caspase-8 through a FADD dependent pathway. Future investigation may warrant preventive as well as therapeutic strategies for GrTP in GI malignancy.
1. Introduction
Cancer is known as one of the most prevalent causes of mortality, and colorectal cancer is the most common malignant complication in patients with chronic idiopathic inflammatory bowel disease (IBD) [1]. It is estimated that one in six IBD patients develop colorectal cancer [2], suggesting a profound relationship between the chronic inflammation of IBD and susceptibility to developing colorectal cancer. In addition, these patients are at risk for developing severe gastrointestinal (GI) complications, including diarrhea, and abdominal pain during chemotherapy due to the cytotoxic effects of current anticancer drugs.
In a normal individual, the number of intestinal epithelial cells (IEC) on the villous and crypt are tightly regulated. IEC are generated from stem cells in the crypt and migrate to the villous tip in about 2-3 days, then “sloughed” [3-4], and replaced by new epithelial cells so that the villous height remains constant. This process is mainly regulated through apoptotic pathways.
Evidence indicates that defective apoptosis, impaired intestinal epithelial barrier function, activation of macrophages, and increased production of macrophagederived cytokines (i.e., TNFα) are critical in the inflammatory cascade leading to IBD (5-6). An additional trigger of this process is the host response to the commensal gut microbiota, through innate-defense mechanisms [5-7]. In Crohn’s patients the lamina propria T lymphocytes (LPL) of the intestinal mucosa are chronically activated (6). Defective apoptosis of activated LPL is considered a key pathognomonic mechanism. In fact, increased expression of anti-apoptotic molecules is reported in LPL from these patients [8].
Apoptosis is a gene-directed cellular self-destruction and is recognized by its distinctive morphology, including the fragmentation of DNA into oligomers (180-200 bp) and DNA-ladder formation resulting in later stage activation of endonucleases. Initiation of apoptosis involves extrinsic and intrinsic pathways requiring enzymatic activities of cysteine-aspartate specific proteases “caspases cascades” [9]. The fatty acid synthase (Fas) associated death domain protein (FADD) dependent pathway initiates apoptosis via its death domain which binds to sequence motifs within the prodomain of FLICE/MACH/Caspase- 8 and thereby recruits this apical regulator protease that initiates activation of downstream caspases (e.g., CPP32/ caspase-3).
The balance between generated proapoptotic and anti-apoptotic proteins determines in part, how cells react to apoptotic signaling. Cancerous cells are generally more resistant to apoptotic pathways [9]. Specifically, resistance to cell death (anoikis), triggered by the loss of anchorage to the substratum, has been reported as an essential prerequisite in the proliferation and diffusion of colorectal cancer cells [10].
Various studies suggest a protective effect of tea consumption on GI health and in malignancies. Green tea polyphenols (GrTP) from Camellia sinensis have been reported to inhibit inflammatory responses [11-15] and carcinogen-induced GI tumors in rodent models [16-17]. In addition, GrTP have been shown to inhibit cell growth and are capable of inducing apoptosis in several carcinoma cell lines [18-21]. These polyphenolic compounds are constituted mainly from (-)-epigallocatechin gallate (EGCG) among 4 polyphenolic catechins, (-)-epigallocatechin (EGC), (-)-epicatechin gallate (ECG), and (-)-epicatechin (EC) [11].
However, the protective mechanism(s) of action of GrTP for intestinal cell biology and the pathway(s) involved in green tea-induced apoptosis in these tissues remains unclear. In this study we investigated the effect of GrTP and its major polyphenol, EGCG, on apoptosis and caspase activation in intestinal epithelial cells.
2. Materials and Methods
Materials: Extracted green tea polyphenols (HPLC graded GrTP; > 95% polyphenols) were purchased from LKT Laboratories, Inc (St. Paul, MN). High-performance liquid chromatography analysis of the green tea extracts revealed the percentage composition of the four polyphenols of interest: 35% EC, 4% EGC, 15% EGC, and 38% EGCG. Individual polyphenolic constituent, EC, EGC, EGC, and EGCG (> 98% pure) were purchased from Sigma Chemical (St. Louis, MO). Disposable culture plasticware was purchased from Corning (Corning, NY). Cell culture supplies were obtained from Invitrogen (Carlsbad, CA) and Bio-Whittaker (Walkersville, MD). Material for electrophoresis was purchased from BioRad (Hercules, CA) and the remaining biochemicals were obtained from Sigma Chemical (St. Louis, MO).
Cell culture and experimental design: The fetal rat intestinal epithelial cell line, IEC-6, was initially obtained from the American Type Culture Collection (ATCC, Rockville MD). These non-tumorigenic cells are derived originally from intestinal crypt epithelia, and retain their distinct morphological and immunological characteristics through serial culture.
The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM, Bio-Whittaker, Walkersville, MD) supplemented with 10% (v/v) endotoxin-free fetal calf serum, 2 mM glutamine, and 100 U/ml penicillin/100 µg/ml streptomycin, at 37°C in an atmosphere of 10% CO2 and 95% relative humidity. The media was changed every 3 days. Passage consisted of incubating cells in sterile trypsin/EDTA followed by resuspending the cells in fresh medium. The cells were studied between passages 15 and 20. Approximately 1 × 105 cells per well were plated in 6-well plates (Corning, Corning, NY) with 1 ml of media per well or seeded in 100-mm petri dishes with 7 ml of media per petri dish. Cells were used for experiments when they became confluent (85%), usually within 3 to 5 days after plating.
Cell viability assays: Cell viability was assessed using a formazan-based assay [22]. Following treatment of cells in 96 wells at 1 × 104, the incubation medium was removed and replaced with 80 μl of fresh medium. Twenty µl of freshly prepared 3-(4,5-dimethylthiazol2-yl)-2,5 diphenyltetrazolium bromide (MTT, Sigma, St. Louis) solution was added (5 mg/ml stock), and cells were incubated at 37oC for 3 h. Medium was then removed, and 150 μl of DMSO was added and allowed to incubate for 10 min. The degree of formazan production was measured by spectrophotometer at 490 nm. The average absorbance reading was taken from each time point of each plate. Cell survival curves were constructed by plotting cell survival against the various treatments. In addition cells were further examined microscopically using a trypan blue exclusion assay.
DNA fragmentation: DNA from treated cells was extracted and assayed as follow. Cells were collected and lysed with 1.0 ml of lysis buffer [100 mm NaCl, 10 mm Tris (pH 8.0), 25 mm ethylenediaminetetraacetic acid, 0.5% sodium dodecyl sulphate (SDS), 200 μg/ml of DNase free proteinase K (Sigma)] and treated with DNAse free RNase (40 µg/ml) in the presence of 0.1% SDS for 1 h at 37°C. DNA was reextracted with phenol/ chloroform, precipitated with 100% ethanol and resuspended in sterile-distilled water. Ten µg of extracted DNA were loaded into each well and electrophoresed in 1.5% agarose gel (BioRad, Hercules, CA). Gels were stained with ethidium bromide and visualized by ultraviolet fluorescence and photographed with a Polaroid camera system.
Caspase activity assay: Caspase activity was detected by a Quantizyme Assay System (Biomol, Plymouth, PA). The enzyme activities and cleavage of caspase 3 and caspase 8-9 were determined colorimetrically using AcDEVD-pNa (N-acetyl-Asp-Glu-Val-Asp-p-nitroaniline) and Ac-ITED-pNA as substrates respectively and absorbance monitored at 405 nm. The specific caspase-3 (z-DEVD 100 μmol) and caspase-8 and -9 inhibitors (Z-ITETD) were added to the cells immediately before stimulation with GrTP and EGCG. The cells were washed with cold phosphate-buffered saline (PBS) and adherent cells were pelleted by centrifugation at 400 g for 5 min at 4°C. The cell pellet was washed with icecold PBS and resuspended in 100 mmol/l HEPES buffer (pH 7.4) containing protease inhibitors (5 µg/ml aprotinin, 10 µg/ml leupeptin, and 0.5 mmol/l phenyl methyl sulfonyl fluoride (PMSF). The cell suspension was lysed by 3 freeze/thaw cycles, and the cytosolic fraction was obtained by centrifugation at 12,000 g for 20 min at 4°C. Protein concentration was determined using the BCA assay (Sigma, St Louis MO). Cytosolic protein (50 to 100 µg) was combined with 100 µmol/L (2 µl) of the synthetic substrate for caspases-3, -8, and -9 in a total volume of 100 µl of HEPES (100 mmol/l, pH 7.4) containing protease inhibitors. The reaction was conducted for 1 h at 37°C. Cytosolic caspase activities were assayed by measuring the increased absorbance and caspase activity.
Fas/FADD complex analysis: The Fas/CD95 and its associated proteins were immunoprecipitated from cell lysates using mouse anti-Fas antibody and protein G plus agarose. After extensive washes the immuno-complex was denatured, and resolved by 15% SDS-PAGE performed in mini slab gel unit (BioRad, Hercules CA). The proteins were electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked in PBS-Tween buffer [80 mM Na2HPO4, 10 mM NaCl, and 0.05% Tween-20 (pH 7.5)] containing 10% nonfat milk. To identify FADD the membrane was incubated for 2 h with specific anti-FADD antibody. To ensure equal loading of samples the membrane was re-probed with mouse anti-Fas antibody. Anti-FADD and anti-Fas antibodies were visualized by an enhanced chemiluminescence blotting kit (ECL, Amersham, Bioscience, Pittsburgh, PA). Band intensity was quantified by the absolute integrated optical intensity.
Statistical analysis: Results are expressed as mean + SD unless otherwise stated. Data were evaluated using ANOVA, followed by appropriate post hoc test using GraphPad Instat version 3 for Windows (GraphPad Software, San Diego, CA). Statistical significance was set at p < 0.05.
3. Results
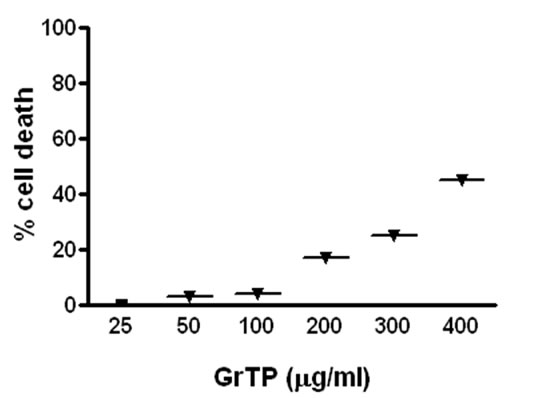
Intestinal cells were treated with different concentrations of GrTP (100-400 µg/ml) or EGCG and EGC (10-500 µM) for a period of 24 h. Cell viability and death were determined with trypan blue exclusion and confirmed with MTT assay. While lower doses of GrTP had no significant effect on the viable cells, higher concentrations caused cell death in a dose dependent manner. GrTP induced (43 + 0.5) cell death at a high dose of 400 µg/ml (Figure 1(a) which correlated with DNA fragmentation and ladder formation in these cells (Figure 1(b)). There was no significant differences (p > 0.05) detected between untreated control cells and those treated with lower doses of GrTP (100-200 µg/ml). GrTP at higher concentrations (400-800 µg/ml) induced DNA fragmentation in a dose responsive fashion, although, higher concentrations (> 800 µg/ml) of GrTP promoted a mixed portrait of apoptosis and cytolysis (data not shown). Accordingly,
 (a)
(a)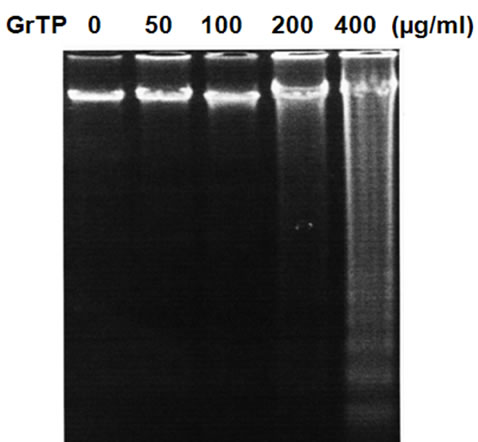 (b)
(b)
Figure 1. Intestinal Epithelial Cells (IEC-6) were treated with different concentrations of Green tea polyphenols (GrTP) for 24 h. (a) GrTP induced cell death in IEC-6 cells. The data is expressed as mean + SD, n = 3; (b) GrTP induced DNA fragmentation in IEC-6. Cells were treated with indicated doses of GrTP for 24 h.
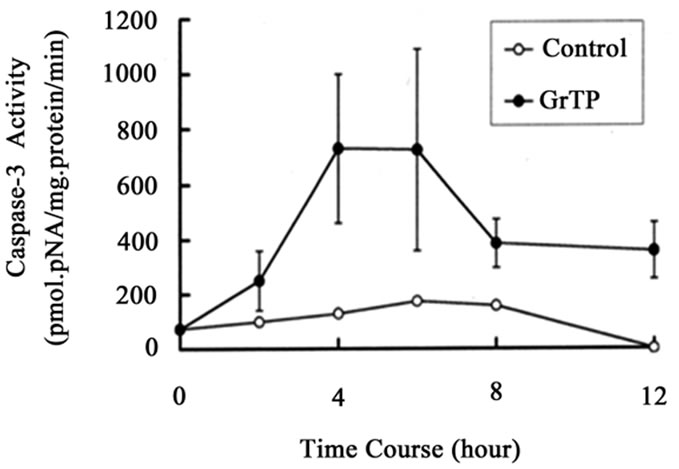
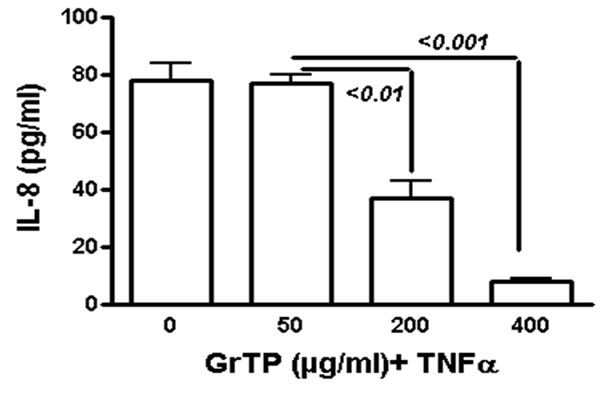
GrTP upregulated caspase-3 activity that was detected within 2 h (p < 0.01) and increased after 4 h (p < 0.001) of intestinal epithelial cell’ exposure (Figure 2(a)). GrTP also increased caspase-8 activity in a similar fashion with a sharp peek after 4 h (p < 0.001) of exposure (Figure 2(b)). GrTP (Figure 3(a)) and EGCG (data not shown) blocked TNFα induced IL-8 release from intestinal epithelial cells in a dose dependent fashion (p < 0.001). TNFα by itself (25 ng/ml) did not induce DNA fragmentation in intestinal epithelial cells, nor did it synergize with GrTP in this outcome (Figure 3(b)).
Although, GrTP decreased NF-kB activation in intestinal epithelial cells, the NF-kB inhibitor (MG132) promoted only cytolysis and with no induction of apoptotic events (Figure 4). In contrast, treatment of cells with specific translational inhibitors, staurosporine and actinomycin D (50 ng/ml), provoked DNA fragmentation of intestinal epithelia is similar to high doses of GrTP (400 µg/ml) (Figure 4). In addition, epithelial cells were pretreated with actinomycin D, staurosporine or sham control (saline) for 30 min and then TNFa (25 ng/ml) or PBS was added for 15 h. TNFα treatment alone or in combina-
 (a)
(a)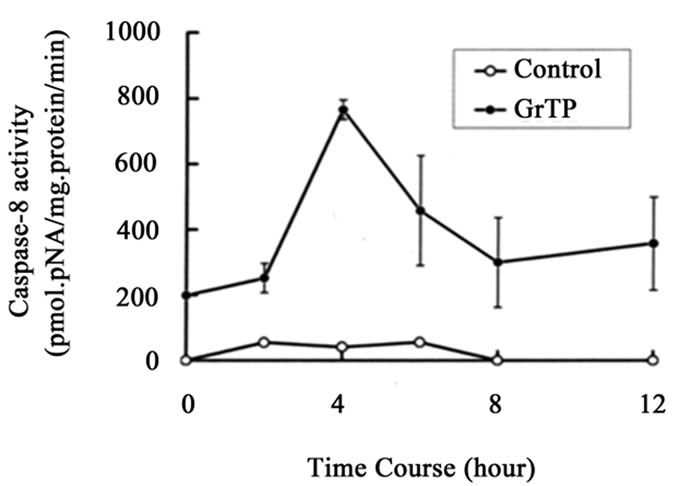 (b)
(b)
Figure 2. (a) Activation of caspase-3 and (b) caspase-8. IEC- 6 cells were treated with 400 µg/ml of GrTP for the indicated time. Caspase activities were significantly increased following exposure of cells to GrTP after 4 h (p < 0.001) into the incubation. Data is expressed as mean + SD, (n = 3).
 (a)
(a)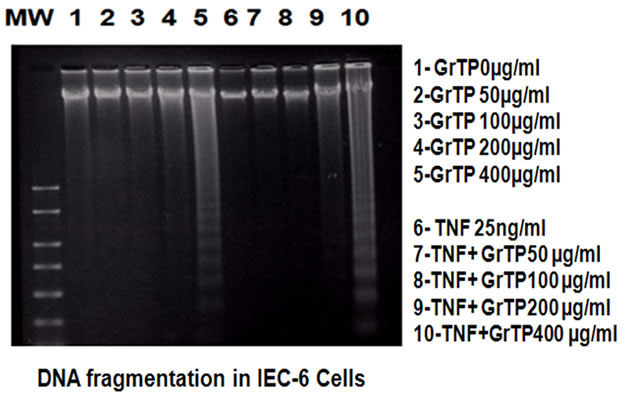 (b)
(b)
Figure 3. GrTP prevented NF-kB activation and IL-8 release mediated by TNFα stimulation of IEC-6 cells. (a) shows a dose dependent response of GrTP treatment; (b) demonstrates inhibition of DNA degradation.
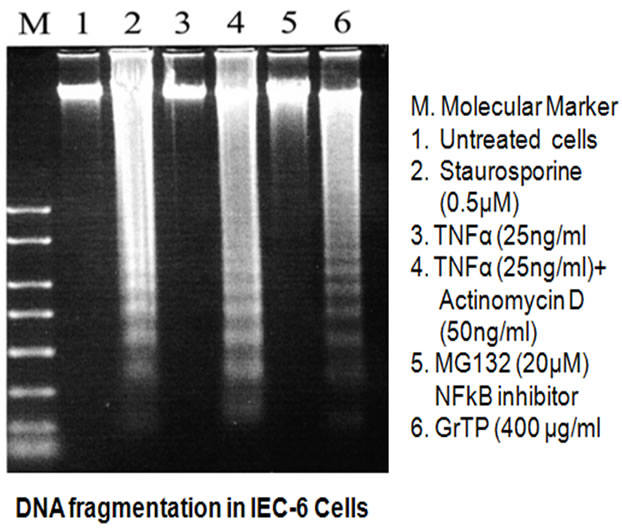
Figure 4. Induction of DNA fragmentation in IEC-6 cells. Cells were treated with indicated stimuli for 24 h. (M) denotes molecular weight marker, (1) Untreated cells; (2) Staurosprine (0.5 µM); (3) TNFα (25 ng/ml); (4) TNFα (25 ng/ml) + Actinomycin D (50 ng/ml); (5) MG132 (20 µM); (6) GrTP (400 µg/ml).
tion with these translational inhibitors did not provoke nor synergize in eliciting DNA laddering.
Concomitantly, GrTP activated FADD and recruited the Fas/CD95 domain in a time dependent manner evidenced 30 minutes after cell treatment. This increased at 1 h, as shown by Western blot analysis (Figure 5) demonstrating activation of death pathways through a FADD dependent pathway.
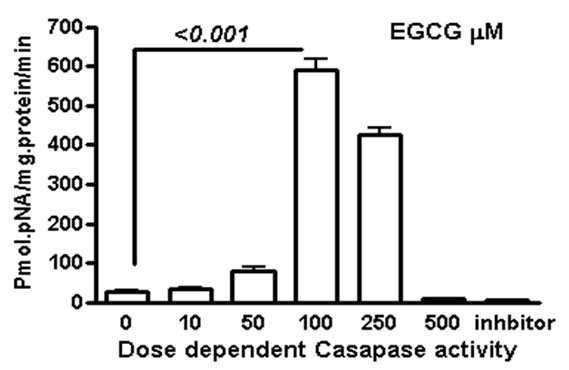
We measured apoptosis induced by different concentrations of various catechins in GrTP. The major catechin component of GrTP is EGCG (approx. 40%). Amongst polyphenols, EGCG at 100 µM caused activation of caspase-3 and caspase-8 in a time dependent manner which sharply increased after 8 h of treatment (Figure 6). Minimal caspase activation was induced by other polyphenolic constituents (EC or EGC data not shown). Lower doses of EGCG (10-50 µM) did not promote caspase activation. The caspase activity was prevented by specific (caspase-3) and general (caspase-8 and -9) caspase inhibitors leading to cell survival rescued after treatment which mimicked untreated normal intestinal epithelia.
4. Discussion
We investigated the efficacy of GrTP and its polyphenolic constituents to regulate apoptosis in fetal rat intestinal epithelial cells. While GrTP and EGCG in lower doses had no significant effect on cell viability, higher concentrations provoked apoptosis in a dose and time dependent manner which correlated with caspase (3,8 and 9) activation.
Under normal circumstances, the gut immune system is in a constant balance between inflammatory and apoptotic states, as epithelial cells and activated T lymphocytes are rapidly and efficiently eliminated via apoptosis. However, IBD patients develop a dysfunction in apoptosis [23-25], that can contribute an increased incidence of colorectal carcinoma, and chemotherapy cytotoxicity of the gut epithelia. Apoptosis can be initiated from cell cycle activation [26-27], cytokine production [28], infective agents [29], or as an adverse response to drugs [30-31]. Based on the agonist or initiating milieu, apoptosis is accompanied by the activation of various cells
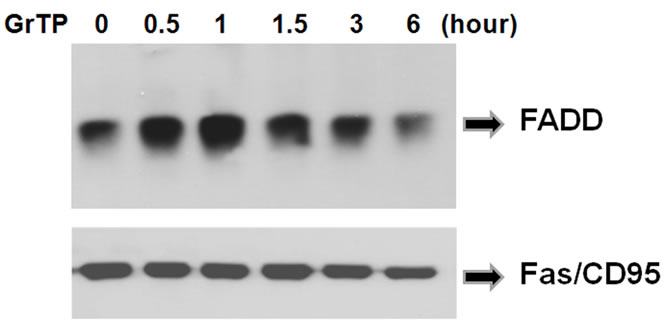
Figure 5. GrTP induced recruitment of FADD to Fas/CD95. IEC-6 cells were treated with 400 µg/ml of GTP for the indicated time. The Fas/CD95 and its associated proteins were immuno-precipetated using anti-Fas antibody. The presence of FADD was identified by Western immunoblotting. Equal loading of samples was verified by re-probing the membrane with anti-Fas antibody.
 (a)
(a)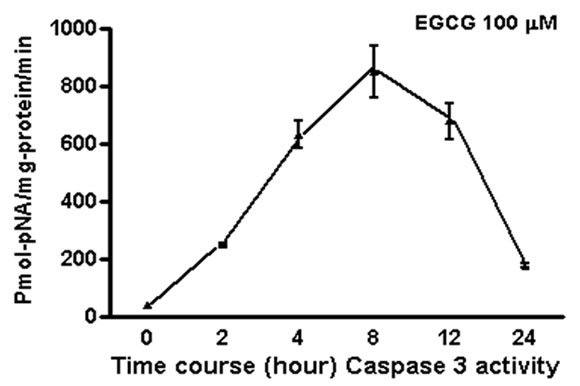 (b)
(b)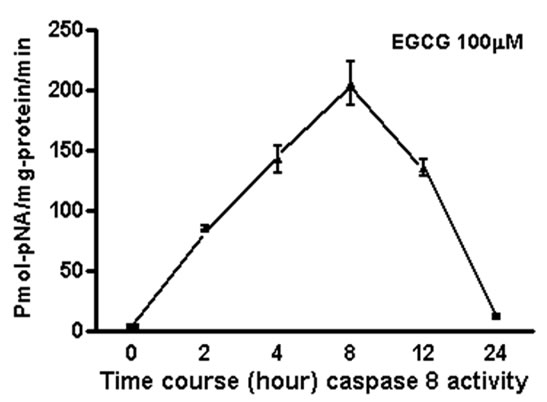 (c)
(c)
Figure 6. EGCG induces caspase activation in IEC-6 cells. To provide dose and time dependent response relationship, cells were treated with different concentrations of EGCG. (a) or 100 µM/ml of EGCG for the indicated time, and caspase’ activity was measured (b-c). EGCG at 10-50 µM had no caspase activity (p > 0.05). Caspase inhibitor rescued the cells from EGCG apoptotic effects of EGCG activated -3; (b) and to a lesser extend caspase-8; (c) in treated cells with a time depended response. Data expressed as mean + SD (n = 3). Also, caspase inhibitors rescued the cells from EGCG apoptotic effects.
death pathways such as caspases, altered gene expression, mitochondrial dysfunction and consumption of ATP with DNA repair [32]. Apoptosis is crucial in maintenance of intestinal epithelial integrity and regulated by numerous factors including NF-kB activity [33]. It is speculated that excessive secretion of cytokines including IL-6 in the gut of Crohn’s disease patients, upregulates expression of antiapoptotic molecules and impairs mucosal homeostasis [34]. The transcription inhibitor and chemotherapeutic agent, actinomycin D, elicits apoptosis in cell types, by anchoring into (purine–pyrimidine) DNA-base pairs via intercalation and inhibiting mRNA biosynthesis [35]. Early studies demonstrated that actinomycin D enhanced the rate of apoptosis in the intestinal epithelial crypts in mice [36] and the apoptotic bodies were either extruded into the lumen or phagocytized and degraded by adjacent cells with essentially different ultrastructure than necrosis induced in the center of tumors [36]. The distinctive morphology of apoptotic cells included, chromatin condensation, cytoplasmic shrinkage, and membrane-bound apoptotic bodies and genomic breakdown. In this study high dose GrTP provoked fragmentation of the intestinal epithelial genome into oligomers similar to actinomycin D and staurosporine.
TNFα a pleiotropic cytokine is involved in regulation of proinflammatory gene expression [37-38], cytotoxicity, apoptosis and pathogenesis in IBD patients [5,39] and in IBD-models [13,40]. Therapeutic modalities using monoclonal antibodies to neutralizing or reducing TNFα result in reduction in mucosal inflammatory response [5,41]. GrTP significantly lowered TNFα production, cyclooxygenases and B-cell lymphoma (BCL)-2 upregulation, and restored glutathione resources, which protected against acetaminophen induced hepatotoxicity [12]. TNFa treatment alone or combined with specific translational inhibitors actinomycin D and staurosporine did not provoke nor synergize in eliciting DNA laddering in these epithelia cells. The present data also extends these findings by confirming that, TNFα stimulation, by itself, is not a major factor inducing apoptosis in intestinal epithelia [42-43]. Also, caspase activation is required in these cells for the progression of apoptosis [44-45]. EGCG treatment of cells resulted in a significant increase in protein expression and activation of caspases and induced proteolytic cleavage of NF-kB/p65 subunit, leading to the loss of transactivation domains, and apoptosis [46]. In addition, GrTP and EGCG-mediated apoptosis was significantly blocked by caspase inhibitors.
Murine intestinal epithelial cells (IEC-18) exposed to oxidants, peroxynitrite and H2O2, and pretreated with low dose green tea were protected against necrotic cell death [31]. Dysregulation of intracellular redox balance with depletion of antioxidant reserves is associated with the activation of transcription factors like NF-kB, and genes associated with apoptosis (TNFα), proliferation and inflammation [12,28,33,47]. Indeed, dietary antioxidants including green tea [46,48] inhibited gene expression associated with inflammation, and immune activation, and protected the gut from pro-apoptotic oxidant stress at levels distinct from the scavenging of the oxidant signal alone. Taken together, these studies demonstrate that GrTP and EGCG-mediated activation of caspases is critical, at least in part, for inhibition of NF-kB and subsequent apoptosis.
The 2 principal signaling pathways that mediate apoptosis are associated with the secondary generation of ROS [49], and mitochondrial dysfunction in cells, which trigger mitochondria-dependent apoptosis (intrinsic pathway) [10]. Alternatively, over-expression of antioxidants such as intracellular superoxide dismutase reduced induction of cell death [50]. The extrinsic pathway (Fas/ FasL) initiates recruitment of FADD to the death receptors (TNF receptor, Fas/CD95) and causes autocatalytic activation of caspases leading to apoptosis [10]. Colonic cancer tissues show alterations in the CD95 (Fas/FasL) as tumors progress from local to metastatic disease. Fas antigen inhibits apoptosis by increasing Fas-mediated proliferation and cell survival in local colonic tumor growth, and coexpression of FasL may be involved in metastatic nature of these neoplastic tumors [10].
GrTP have been shown to inhibit carcinogenesis and tumorigenesis on different organs in models. Green tea is speculated to modulate the signaling pathways leading to transformation, and apoptosis of preneoplastic and neoplastic cells, as well as inhibition of tumor invasion and angiogenesis [16]. For instance, EGCG exerts growth inhibitory effects on a number of human tumor cells [20-21,49,51] including colorectal cancer (Caco-2) cells [18]. The anti-proliferative effect of EGCG in Caco-2 cells is possibly modulated by c-fos and c-myc gene expression [18]. Based on these findings, EGCG may be a candidate for consideration as treatment option for several human carcinomas.
GrTP induced DNA fragmentation in IEC-6 cells in a dose and time dependent manner. At doses above 200 µg/ml GrTP activated caspase-3 and caspase-8 in these cells that peaked 4 h following exposure consistent with increased recruitment of FADD to Fas/CD95 and formation of Fas/FADD complex. Overall GrTP and EGCG may be useful in the chemoprevention of carcinomas with overexpression of Fas signaling. Tumor cells with deleted caspase-3 gene treated with GrTP did not undergo apoptosis, suggesting GrTP-induce apoptosis is a mitochondria-targeted and, caspase-3 executed mechanism in the tumor cells [52]. In addition Fas/Fas ligand deletion was associated with increased levels of IFNγ, TNFα and Fas expression and significant reduction in mouse intestinal epithelial damage and apoptosis [53].
Our previous investigations demonstrated that GrTP and EGCG in low concentrations blocked NF-kB activation and translocation in intestinal epithelial cells [11]. The NF-kB inhibitor, MG132, promoted only cytolysis with no induction of apoptotic events, indicating that GrTP may act by different mechanism(s) than solely inhibiting NF-kB activation. At low concentrations GrTP acts as an antioxidant and inhibits NF-kB activation [11- 14], while in higher doses it provoked apoptosis, as documented in this study. The anti-tumor efficacy of several chemotherapeutic agents is correlated with their ability to induce apoptosis [54,55]. Some of these regimens are partially effective in tumor cells being particularly resistant to radiotherapy and/or chemotherapy [55]. These data support that apoptosis induced by GrTP is caspasedependent and involves both extrinsic and intrinsic pathways. EGCG has been reported (800 mg/day) to be safe with minor side effect in a preclinical trial [56]. Therefore, a safer and effective apoptotic agent represents an attractive approach for the development of potential anticancer agents and, GrTP may serve as a candidate for future translational cancer studies and preventive or therapeutic modality for GI malignancy.
5. Acknowledgements
This research was supported by the National Institutes of Health grants NCCAM-AT1490 and NIDCR DE019177 (H. Oz). A portion of this study was presented as a poster at the DDW 2010 in New Orleans.
REFERENCES
- W. Goessling and R. J. Mayer, “Systemic Treatment of Patients who Have Colorectal Cancer and Inflammatory Bowel Disease,” Gastroenterology Clinics of North America, Vol. 35, No. 3, 2006, pp. 713-727.
- J. Suchy, E. Kłujszo-Grabowska, J. Kładny, C. Cybulski, D. Wokołorczyk, J. Szymańska-Pasternak, G. Kurzawski, R. J. Scott and J. Lubiński, “Inflammatory Response Gene Polymorphisms and their Relationship with Colorectal Cancer Risk,” BMC Cancer, Vol. 23, No. 8, 2008, p. 112.
- J. H. Hendry and C. S. Potten, “Cryptogenic Cells and Proliferative Cells in Intestinal Epithelium,” International Journal of Radiation Biology & Related Studies in Physics, Chemistry & Medicine, Vol. 25, No. 4, 1974, pp. 583- 588.
- J. A. Hermos, M. Mathan and J. S. Trier, “DNA Synthesis and Proliferation by Villous Epithelial Cells in Fetal Rats,” Journal of Cell Biology, Vol. 50, No. 1, 1971, pp. 255- 258.
- H. S. Oz and J. L. Ebersole, “Application of Prodrugs to Inflammatory Diseases of the Gut,” Molecules, Vol. 13, No. 2, 2008, pp. 452-474.
- W. Strober, I. J. Fuss and R. S. Blumberg, “The Immunology of Mucosal Models of Inflammation,” Annual Review of Immunology, Vol. 20, No. 1, 2002, pp. 495-549.
- H. S. Oz, J. Zhong and W. de Villiers, “The Pattern Recognition Scavenger Receptors, SR-A & CD36, Have Additive Roles in the Development of DSS-Induced Colitis in Mice,” Digestive Diseases Sciences, Vol. 54, No. 3, 2009, pp. 2247-2252.
- J. Doering, B. Begue, M. J. Lentze, F. Rieux-Laucat, O. Goulet, J. Schmitz, N. Cerf-Bensussan and F. M. Ruemmele, “Induction of T Lymphocyte Apoptosis by Sulphasalazine in Patients with Crohn’s Disease,” Gut, Vol. 53, No. 11, 2004, pp. 1632-1638.
- I. Mercier, M. Vuolo, J. F. Jasmin, C. M. Medina, M. Williams, J. M. Mariadason, H. Qian, X. Xue, R. G. Pestell, M. P. Lisanti and R. N. Kitsis, “ARC (Apoptosis Repressor with Caspase Recruitment Domain) is a Novel Marker of Human Colon Cancer,” Cell Cycle, Vol. 7, No. 11, 2008, pp. 1640-1647.
- H. Li, G. Ray, B. H. Yoo, M. Erdogan and K. V. Rosen, “Down-Regulation of Death-Associated Protein Kinase-2 is Required for Beta-Catenin-Induced Anoikis Resistance of Malignant Epithelial Cells,” Journal of Biological Chemistry, Vol. 284, No. 4, 2009, pp. 2012-2022.
- F. Yang, H. S. Oz, S. Barve, W. J. de Villiers, C. J. McClain and G. W. Varilek, “The Green Tea Polyphenol (-)-Epigallocatechin-3-Gallate Blocks Nuclear FactorKappa B Activation by Inhibiting Ikappa B Kinase Activity in the Intestinal Epithelial Cell Line IEC-6,” Molecular Pharmacology, Vol. 60, No. 3, 2001, pp. 528-533.
- H. S. Oz, C. J. McClain, H. T. Nagasaw, M. B. Ray, W. S. J. de Villiers and T. S. Chen, “Diverse Antioxidants Protect against Acetaminophen Hepatotoxicity,” Journal of Biochemical and Molecular Toxicology, Vol. 18, No. 6, 2004, pp. 361-368.
- G. W. Varilek, F. Yang, E. Y. Lee, W. J. de Villiers, J. Zhong, H. S. Oz and C. J. McClain, “Green Tea Polyphenol Extract Attenuates Inflammation in Interleukin- 2-Deficient Mice, a Model of Autoimmunity,” Journal of Nutrition, Vol. 131, No. 7, 2001, pp. 2034-2039.
- H. S. Oz, T. Chen, C. McClain and W. de Villiers, “Antioxidants a Novel Therapy in a Murine Model of Colitis,” Journal of Nutritional Biochemistry, Vol. 16, No. 5, 2005, pp. 297-304.
- H. S. Oz and T. S. Chen, “Green-Tea Polyphenols Downregulate Cyclooxygenase and Bcl-2 Activity in Acetaminophen-Induced Hepatotoxicity,” Digestive Diseases Sciences, Vol. 53, No. 11, 2008, pp. 2980-2988.
- C. S. Yang, P. Maliakal and X. Meng, “Inhibition of Carcinogenesis by Tea,” Annual Review of Pharmacology and Toxicology, Vol. 42, No. 1, 2002, pp. 25-54.
- T. Ohishi, Y. Kishimoto, N. Miura, G. Shiota, T. Kohri, Y. Hara, J. Hasegawa and M. Isemura, “Synergistic Effects of (-)-Epigallocatechin Gallate with Sulindac against Colon Carcinogenesis of Rats Treated with Azoxymethane,” Cancer Letters, Vol. 177, No. 1, 2002, pp. 49-56.
- Z. P. Chen, J. B. Schell, C. T. Ho and K. Y. Chen, “Green Tea Epigallocatechin Gallate Shows a Pronounced Growth Inhibitory Effect on Cancerous Cells but not on their Normal Counterparts,” Cancer Letters, Vol. 129, No. 2, 1998, pp. 173-179.
- M. Isemura, K. Saeki, T. Kimura, S. Hayakawa, T. Minami and M. Sazuka, “Tea Catechins and Related Polyphenols as Anti-Cancer Agents,” Biofactors, Vol. 13, No. 1-4, 2000, pp. 81-85.
- P. P. Wu, S. C. Kuo, W. W. Huang, J. S. Yang, K. C. Lai, H. J. Chen, K. L. Lin, Y. J. Chiu, L. J. Huang and J. G. Chung, “(-)-Epigallocatechin Gallate Induced Apoptosis in Human Adrenal Cancer NCI-H295 Cells through Caspase-Dependent and Caspase-Independent Pathway,” Anticancer Research, Vol. 29, No. 4, 2009, pp. 1435-1442.
- A. Basu and S. Haldar, “Combinatorial Effect of Epigallocatechin-3-Gallate and TRAIL on Pancreatic Cancer Cell Death,” International Journal of Oncology, Vol. 34, No. 1, 2009, pp. 281-286.
- P. R. Twentyman and M. Luscombe, “A Study of Some Variables in a Tetrazolium Dye (MTT) Based Assay for Cell Growth and Chemosensitivity,” British Journal of Cancer, Vol. 56, No. 3, 1994, pp. 279-285.
- C. Fiocchi, “Inflammatory Bowel Disease: Etiology and Pathogenesis,” Gastroenterology, Vol. 115, No. 1, 1998, pp. 182-205.
- D. K. Podolsky, “Inflammatory Bowel Disease,” The New England Journal of Medicine, Vol. 347, No. 6, 2002, pp. 417-429.
- A. D. Levine, “Apoptosis: Implications for Inflammatory Bowel Disease,” Inflammatory Bowel Disease, Vol. 6, No. 3, 2000, pp. 191-205.
- D. Jourd'heuil, Z. Morise, E. M. Conner, J. Kurose and M. B. Grisham, “Oxidant-Regulation of Gene Expression in the Chronically Inflamed Intestine,” The Keio Journal of Medicine, Vol. 46, No. 1, 1997, pp. 10-15.
- J. Marks-Honczalik, S. C. Chu and J. Moss, “Cytokine-Mediated Transcriptional Induction of Human Inducible Nitric Oxide Synthase Gene Requires both Activator Protein-1 and Nuclear Factor κB Binding Sites,” Journal of Biological Chemistry, Vol. 273, No. 35, 1998, pp. 22201-22208.
- M. F. Neurath, C. Becker and K. Barbulescu, “Role of NF-Kappa B in Immune and Inflammatory Responses in the Gut,” Gut, Vol. 43, No. 6, 1998, pp. 856-860.
- J. Shibata, H. Goto, T. Arisawa, Y. Niwa, T. Hayakawa, A. Nakayama and N. Nori, “Regulation of Tumor Necrosis factor (TNF)-induced Apoptosis by Soluble TNF-Receptors in Helicobacter pylori Infections,” Gut, Vol. 45, No. 1, 1999, pp. 24-31.
- M. J. Miller, F. M. Angeles, B. K. Reuter, P. Bobrowski and M. Sandoval, “Dietary Antioxidants Protect Gut Epithelial Cells from Oxidant-Induced Apoptosis,” BMC Complementary and Alternative Medicine, Vol. 1, No. 1, 2001, p. 11.
- S. Fiorucci, E. Antonelli, L. Santucci, O. Morelli, M. Miglietti, B. Federici, R. Mannucci, P. del Soldato and A. Morelli, “Gastrointestinal Safety of Nitric Oxide-Derived Aspirin is Related to Inhibition of ICE-Like Cysteine Proteases in Rats,” Gastroenterology, Vol. 116, No. 5, 1999, pp. 1089-1106.
- C. Jobin and R. B. Sartour, “The IκB/NF-κB System - A Key Determinant of Mucosal Inflammation and Protection,” American Journal of Physiology, Vol. 278, No. 3, 2000, pp. C451-C462.
- T. Zou, J. N. Rao, X. Guo, L. Liu, H. M. Zhang, E. D. Strauch, B. L. Bass and J. Y. Wang, “NF-KappaB-Mediated IAP Expression Induces Resistance of Intestinal Epithelial Cells to Apoptosis after Polyamine Depletion,” American Journal of Physiology and Cell Physiology, Vol. 286, No. 5, 2004, pp. C1009-C1018.
- R. Atreya and M. F. Neurath, “New Therapeutic Strategies for Treatment of Inflammatory Bowel Disease,” Mucosal Immunology, Vol. 1, No. 3, 2008, pp. 175-182.
- D. Shim, H. Y. Kang, B. W. Jeon, S. S. Kang, S. I. Chang, H. Y. Kim, “Protein Kinase B Inhibits Apoptosis Induced by Actinomycin D in ECV304 Cells through Phosphorylation of Caspase 8,” Archives of Biochemistry and Biophysics, Vol. 425, No. 2, 2004, pp. 214-220.
- J. Searle, T. A. Lawson, P. J. Abbott, B. Harmon, J. F. Kerr, “An Electron-Microscope Study of the Mode of Cell Death Induced by Cancer-Chemotherapeutic Agents in Populations of Proliferating Normal and Neoplastic Cells,” Journal of Pathology, Vol. 116, No. 3, 1975, pp. 129-138.
- H. S. Oz, T. Chen and M. Neuman, “Methionine Deficiency and Liver Injury in a Dietary NASH Model,” Digestive Diseases Sciences, Vol. 53, No. 3, 2008, pp. 767-776.
- H. S. Oz, H. Im, T. Chen, W. de Villiers and C. McClain, “Glutathione Enhancing Agents Protect against Steatohepatitis in a Model,” Journal of Biochemical and Molecular Toxicology, Vol. 20, No. 1, 2006, pp. 39-47.
- M. Neuman, “Immune Dysfunction in Inflammatory Bowel Disease,” Trans-Research, Vol. 149, No. 4, 2007, pp. 173-186.
- H. S. Oz, M. Ray, T. Chen and C. McClain, “Efficacy of a TGF-β2 Containing Nutritional Support Formula in a Murine Model of IBD,” Journal of American College of Nutrition, Vol. 23, No. 3, 2004, pp. 220-226.
- L. M. Gaetke, H. S. Oz, R. Frederich and C. McClain, “Anti-TNF-α Antibody Normalizes Serum Leptin in IL-2 Deficient Mice,” Journal of American College of Nutrition, Vol. 22, No. 5, 2003, pp. 415-420.
- K. Wright, G. Kolios, J. Westwick and S. G. Ward, “Cytokine-Induced Apoptosis in Epithelial HT-29 Cells is Independent of Nitric Oxide Formation,” Journal of Biological Chemistry, Vol. 274, No. 24, 1999, pp. 17193- 17201.
- Q. Chang and B. L. Tepperman, “The Role of Protein Kinase C Isozymes in TNF-Alpha-Induced Cytotoxicity to a Rat Intestinal Epithelial Cell Line,” American Journal of Physiology Gastrointest Liver Physiology, Vol. 280, No. 4, 2001, pp. G572-G583.
- G. S. Salvesen and V. M. Dixit, “Caspases: Intracellular Signaling by Proteolysis,” Cell, Vol. 91, No. 4, 1997, pp. 443-446.
- M. Krajewska, H. G. Wang, S. Krajewski, J. M. Zapata, A. Shabaik, R. Gascoyne and J. C. Reed, “Immunohistochemical Analysis of in Vivo Patterns of Expression of CPP32 (Caspase-3), a Cell Death Protease,” Cancer Research, Vol. 57, No. 8, 1997, pp. 1605-1613.
- S. Gupta, K. Hastak, F. Afaq, N. Ahmad and H. Mukhtar, “Essential Role of Caspases in Epigallocatechin-3-Gallate-Mediated Inhibition of Nuclear Factor KappaB and Induction of Apoptosis,” Oncogene, Vol. 23, No. 14, 2004, pp. 2507-2516.
- J. Li, C. Y. Huang, R. L. Zheng, K. R. Cui and J. F. Li, “Hydrogen Peroxide Induces Apoptosis in Human Hepatoma Cells and Alters Cell Redox Status,” Cell Biology International, Vol. 24, No. 1, 2000, pp. 9-23.
- M. Nihal, H. Ahsan, I. A. Siddiqui, H. Mukhtar, N. Ahmad and G. S. Wood, “(-)-Epigallocatechin-3-Gallate (EGCG) Sensitizes Melanoma Cells to Interferon Induced Growth Inhibition in a Mouse Model of Human Melanoma,” Cell Cycle, Vol. 8, No. 13, 2009, pp. 2057-2063.
- T. Bohler, J. Waiser, H. Hepburn, J. Gaedeke, C. Lehmann, P. Hambach, K. Budde and H. H. Neumayer, “TNF-Alpha and IL-1 Alpha Induce Apoptosis in Subconfluent Rat Mesangial Cells. Evidence for the Involvement of Hydrogen Peroxide and Lipid Peroxidation as Second Messengers,” Cytokine, Vol. 12, No. 7, 2000, pp. 986-991.
- K. Kahlos, Y. Soini, P. Paako, M. Saily, K. Linnainmaa, V. L. Kinnula, “Proliferation, Apoptosis, and Manganese Superoxide Dismutase in Malignant Mesothelioma,” International Journal of Cancer, Vol. 88, No. 11, 2000, pp. 37-43.
- M. H. Pan, C. C. Lin, J. K. Lin and W. J. Chen, “Tea Polyphenol (-)-Epigallocatechin 3-Gallate Suppresses Heregulin-Beta1-Induced Fatty Acid Synthase Expression in Human Breast Cancer Cells by Inhibiting Phosphatidylinositol 3-Kinase/Akt and Mitogen-Activated Protein Kinase Cascade Signaling,” Journal of Agricultural and Food Chemistry, Vol. 55, No. 13, 2007, pp. 5030-5037.
- S. Hsu, J. Lewis, B. Singh, P. Schoenlein, T. Osaki, M. Athar, A. G. Porter and G. Schuster, “Green Tea Polyphenol Targets the Mitochondria in Tumor Cells Inducing Caspase 3-Dependent Apoptosis,” Anticancer Research, Vol. 23, No. 2B, 2003, pp. 1533-1539.
- B. S. Mantej, R. Borojevic, S. Basak, E. Ho, P. Zhou and K. Croitoru, “IL-10 Protects Mouse Intestinal Epithelial Cells from Fas-Induced Apoptosis via Modulating Fas Expression and Altering Caspase-8 and FLIP Expression,” American Journal of Physiology Gastrointest Liver Physiology, Vol. 291, No. 5, 2006, pp. G820-G829.
- R. W. Johnstone, A. A. Ruefli and S. W. Lowe, “Apoptosis: A Link Review between Cancer Genetics and Chemotherapy,” Cell, Vol. 108, No. 2, 2002, pp. 153-164.
- F. Vandermeers, P. Hubert, P. Delvenne, C. Mascaux, B. Grigoriu, A. Burny, A. Scherpereel and L. Willems, “Valproate, in Combination with Pemetrexed and Cisplatin, Provides Additional Efficacy to the Treatment of Malignant Mesothelioma,” Clinical Cancer Research, Vol. 15, No. 8, 2009, pp. 2818-2828.
- H. H. Chow, Y. Cai, I. A. Hakim, J. A. Crowell, F. Shahi, C. A. Brooks, R. T. Dorr, Y. Hara and D. S. Alberts, “Pharmacokinetics and Safety of Green Tea Polyphenols after Multiple-Dose Administration of Epigallocatechin Gallate and Polyphenon E in Healthy Individuals,” Clinical Cancer Research, Vol. 9, No. 9, 2003, pp. 3312- 3319.
Abbreviations
Caspases, Cysteine-aspartate specific proteases; DISC, death inducing signalling complex; EC, (-)-epicatechin; EG, (-)-epigallocatechin; EGC, (-)-epigallocatechin; ECG, (-)-epicatechin gallate; EGCG, (-)-epigallocatechin gallate; Fas, Fatty acid snthase; FasL, Fas Ligand; FADD, Fas associated death domain; FLIP, FADD-like inhibitory protein; GI, gastrointestinal; GrTP, Green tea polyphenols; IL, interleukin; IBD, Inflammatory bowel disease; IEC, Intestinal epithelial cells; LPL, lamina propria T lymphocytes; NF-kB, Transcription nuclear factor kB; PBL, peripheral blood lymphocytes; ROS, reactive oxygen species; TNFα, Tumor necrosis factor.