Journal of Modern Physics
Vol.5 No.4(2014), Article ID:43093,8 pages DOI:10.4236/jmp.2014.54023
Molecular Dynamics Simulations of the DNA-CNT Interaction Process: Hybrid Quantum Chemistry Potential and Classical Trajectory Approach
1Tajik Technical University Named after M.S. Osimi, Dushanbe, Tajikistan
2Moscow State University of Technology “STANKIN”, Moscow, Russia3Institute of Nuclear Physics, Tashkent, Uzbekistan4Laboratory of Radiation Biology, JINR, Dubna, Moscow Region, Russia5Dubna International University, Dubna, Moscow Region, RussiaEmail: mirzo85@inbox.ru, dushanov@jinr.ru, mirzo@jinr.ru
Copyright © 2014 Mirzoaziz A. Khusenov et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Mirzoaziz A. Khusenov et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received December 13, 2013; revised January 11, 2014; accepted February 9, 2014
Keywords: Molecular Dynamics; Carbon Nanotube; DNA Molecule; Drug Delivery; DNA-CNT Interaction
ABSTRACT
In this work the quantum chemistry Tersoff potential in combination with classical trajectory calculations was used to investigate the interaction of the DNA molecule with a carbon nanotube (CNT). The so-called hybrid approach—the classical and quantum-chemical modeling, where the force fields and interaction between particles are based on a definite (but not unique) description method, has been outlined in some detail. In such approach the molecules are described as a set of spheres and springs, thereby the spheres imitate classical particles and the spring the interaction force fields provided by quantum chemistry laws. The Tersoff potential in hybrid molecular dynamics (MD) simulations correctly describes the nature of covalent bonding. The aim of the present work was to estimate the dynamical and structural behavior of the DNA-CNT system at ambient temperature conditions. The dynamical configurations were built up for the DNA molecule interacting with the CNT. The analysis of generated МD configurations for the DNA-CNT complex was carried out. For the DNA-CNT system the observations reveal an encapsulation-like behavior of the DNA chain inside the CNT chain. The discussions were made on possible use of the DNA-CNT complex as a candidate material in drug delivery and related systems.
1. Introduction
The novel computational approaches allow treating important problems in material fabrication and biomedical applications. Powerful modern computer-based molecular simulation methods became traditional tools in the industry of new materials and drugs. Herewith, the methods of computer molecular simulation involve conventional (classical based approach) or hybrid molecular dynamics, Monte-Carlo and ab initio quantum chemistry, and so on. Computer MD simulations of large molecular systems have quickly shown their power with the invention of modern supercomputers, as a powerful technique which allows one to investigate various physical or biological processes at the atomic/molecular level. X-ray or neutron measurements of physical, chemical, and biological structures inevitably involve the further use of computer models based on molecular and atomic simulation techniques [1-5].
Molecular simulation (conventional and hybrid MD) is based on classical Newtonian physics, modeling the particle interaction in molecules via the force fields defined in advance—empirically or calculated by other methods. Computer molecular simulation is a set of molecular simulation and quantum chemistry methods, or hybrids of these two kinds of methods exhibiting new possibilities.
The methods of computer molecular simulation (conventional MD, hybrid MD or MC (Monte Carlo)), which were first proposed more than 50 years ago, have been rapidly developing in the last 5 - 10 years with the invention of modern computing specialized clusters and supercomputers. In a MD study, the molecular systems are modeled deterministically by the integration of classical equations of motions; in MC, stochastically—with various ensembles. The MD methods are capable of modeling atomic molecular systems of up to thousands and millions of particles and simulating many system parameters and environmental configurations. MD simulation allows one to predict efficiently the ensemble properties and behavior, such as {P-V-T} relations, phase equilibrium, transport properties, structures of synthetic and biological macromolecules, docking of one molecule against another, etc. [1-8].
A scheme shown below (see Table 1) illustrates an example of hybrid MD approach used for a description of hydrogen molecule H2 (two protons, (a, b) and two electrons, i = 1, 2; r12 and rab; r is interatomic distance).
Another example of a similar approach in quantum mechanics is an adiabatic (Born-Zommerfeldt’s) approximation (see Table 2).
Thus, in so-called hybrid approach—the classical and quantum-chemical MD modeling—the molecules are described as a set of spheres and springs, where the force fields and interaction between particles are based on a definite (but not unique) description method and technique. In such technique the classical spheres and springs would imitate the interaction force fields which are actually provided by quantum chemistry laws (Figure 1). It is worth noting that for the development of multiscale models for complex chemical systems the Nobel Prize in Chemistry 2013 was awarded jointly to Martin Karplus, Michael Levitt and Arieh Warshel (Ref: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/2013). So far, creating models of molecules as balls and sticks laid foundation for the powerful techniques and computer programs that are used to understand and predict protein structure conformations and dynamics, chemical reactions and related important processes [9-20].
Computational quantum chemistry research—ab initio, density functional theories (DFT), and others—in contrast to the conventional molecular simulations, is based on quantum physics. The computational quantum chemistry methods were first applied to the electronic structure of atoms or molecules, which yielded wave functions or

Table 1. A scheme illustrating hybrid classical molecular dynamics (MD) and quantum chemistry molecular dynamics (qMD) approach.

Table 2. A scheme illustrating adiabatic (Born-Zommerfeldt’s) approximation in hybrid classical MD and quantum chemisstry qMD approach.
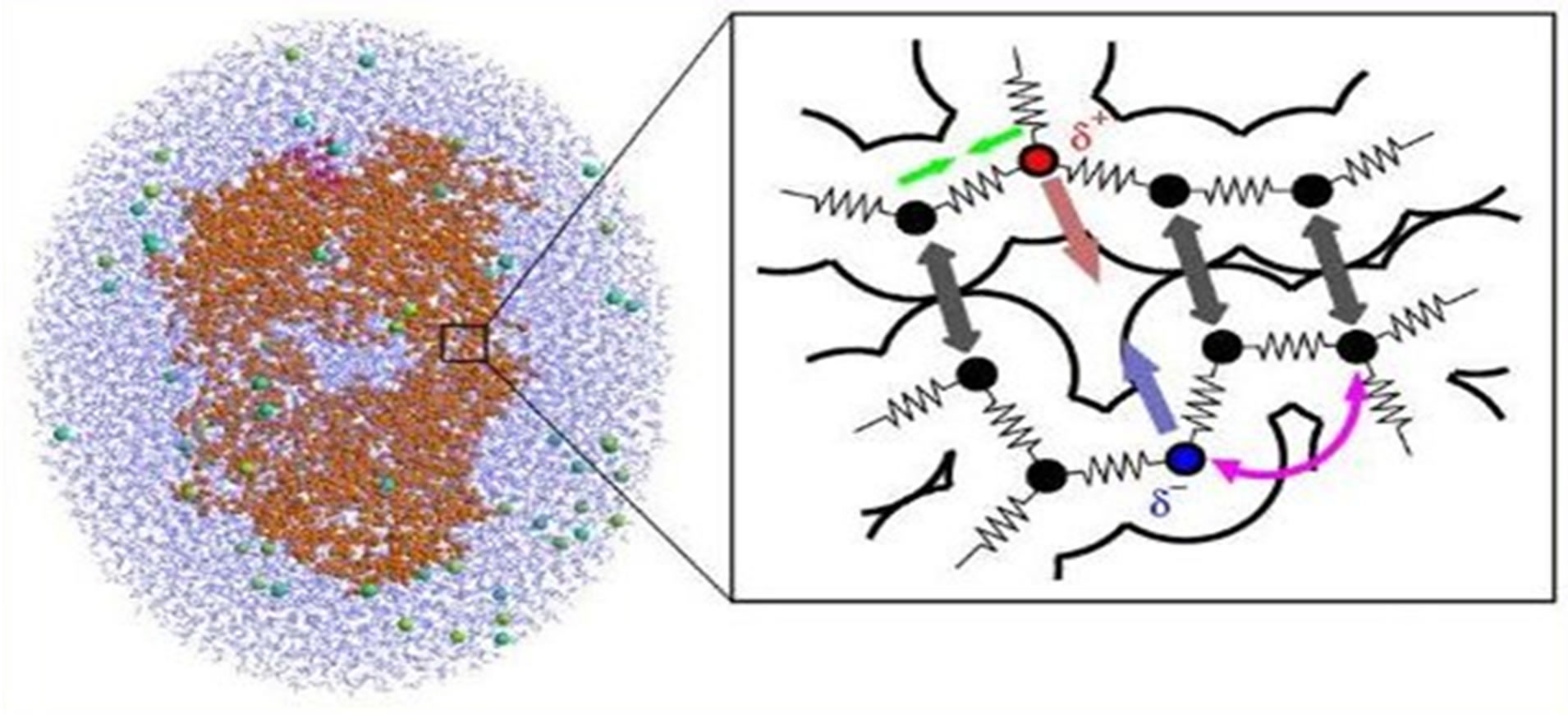
Figure 1. Molecules are described as a set of classical spheres & springs, which imitate the interaction force field provided by quantum chemistry laws. (Ref: a public domain of the World Wide Web).
a probability density functional describing the electron states. The quantum chemistry methods provide greater accuracy but are restricted to a smaller molecular size because of their complexity and CPU costs. Quantum chemistry simulation is essential when chemical bonds are formed or broken. It is also used when force parameters are unknown or not applicable. The DFT methods are well established and used with increasing accuracy; the high-level wave function methods with large atomic orbital basis sets currently remain standard. As results of our quantum chemistry studies, we got force—field data, which enabled us to calculate the thermochemical, kinetic, and optical properties, NMR shifts, etc.
Thus, hybrid classical MD and quantum chemistry qMD facilitates a powerful multi-scale computational scheme (see Table 3).
In this work we have employed the quantum chemistry Tersoff potential in combination with classical trajectory calculations to investigate the interaction of the DNA molecule with a CNT. Tersoff potential is efficiently used for MD simulation of systems that contain carbon, silica, germanium, etc. alloys. In carbon nanotubes (CNTs) we have chemical bonding is hybridization sp2 (as graphite), which is stronger than sp3 bond (of diamond). The nature of chemical bonding in CNTs is described by quantum chemistry, through the process of orbital hybridization. The Tersoff potential in hybrid MD simulations correctly describes the nature of covalent bonding. It’s good for simulating systems that contain carbon, silica, germanium and alloys of these elements. The peculiarity of Tersoff potential is that it allows the breaking and formation of chemical bonds. That is associated with hybridization process. Tersoff potential is pair wise potential, but coefficient in attractive term depends on local environment. Thus, Tersoff potential possesses a many body nature. It is also worth noting that CNTs exhibit a unique electrical and chemical properties for organic materials, they possess a great interest for the material research and electronic applications. Depending on their chemical structure, CNTs can be used as an alternative to organic or
Table 3. A scheme illustrating multi-scale hybrid classical MD and quantum chemisstry qMD approach.
inorganic semiconductors as well as conductors. The chemical bonding of nanotubes is composed entirely of sp2 bonds, similar to those of graphite. This bonding structure, which is stronger than the sp3 bonds found in diamonds, provides the molecules with their unique strength. Nanotubes naturally align themselves into “ropes” held together by Van der Waals forces. The nature of the bonding of a nanotube is described by quantum chemistry, specifically, orbital hybridization [21,22].
The physical and molecular mechanism of the interaction of DNA as the genetic material with CNT represents a great interest in today biophysics and biochemistry. Understanding the DNA-CNT interaction mechanism has crucially important for the purposes of a drug delivery in bio-medicine and nanotechnology applications. Along with the DNA-NP (nanoparticle) objects the DNA-CNT system represents a great interest in today biomedicine applications due to diagnostic and treatment of oncology diseases. Cancer, in which cells grow and divide abnormally, is one of the primary diseases with regard to how it responds to CNT drug delivery. Representing a revolutionarily potential for the biochemistry and medicine the use of CNTs in drug delivery has based on the enhancing of sufficient solubility and allowing of efficient tumor targeting. These aspects prevent CNTs from being cytotoxic and altering the function of immune cells. For today, cancer therapy involves surgery, radiation therapy, and chemotherapy. For example, recent experimental and simulation studies involve the interaction of DNA with highly localized high power beams and various nanoparticles (Ag, Au, etc.). These studies are aimed on targeted cancer therapy through the injection of metal microor nanoparticles into the tumor tissue with consequent local microwave or laser heating. Due to their good heat conductivities of NPs (Ag, Au, and so on) the experiments reveal that the only tumor cells to destroy, remaining normal cells undamaged. Nevertheless, such kind treatment methods are usually painful and kill normal cells in addition to producing adverse side effects. CNTs as drug delivery vehicles have shown a potential interest due to a targeting of specific cancer cells with a lower dosage rather than conventional drugs have [9-22].
The aim of this study is to perform the MD simulations to investigate the dynamical and structural behavior of the DNA-CNT model at ambient temperature conditions. The structural radial distribution functions and the dynamical configurations have built up for the DNA molecule interacting with the CNT. For the DNA-CNT system we have to observe an encapsulation-like behavior of the DNA chain inside the carbon nanotube to penetrate deep into. The discussions have made for possible use of the DNA-CNT formation as a candidate in drug delivery and related systems.
2. Materials and Methods
A configuration snapshot of the DNA-CNT model is shown in Figure 2. The DNA strand was consisted of 1260 atoms and its structure was taken from the DL_ POLY database. Thereby specifying the DNA interaction potential, it contained 21 bonds, 1358 constraints, 2442 angles and 3525 dihedrals. The CNT (carbon nanotube) consists of 800 carbon atoms in a nanotube 41.7 angstrom in length (see Figure 3). In the description of the physical properties of CNT we employ the Tersoff potential. The Tersoff potential [23-33] is a special example of a density-dependent potential, which reproduces the properties of covalent bonding in systems containing carbon, silicon, germanium, etc., and alloys of these elements. A special feature of the potential is that it allows bond breaking and associated changes in bond hybridi-
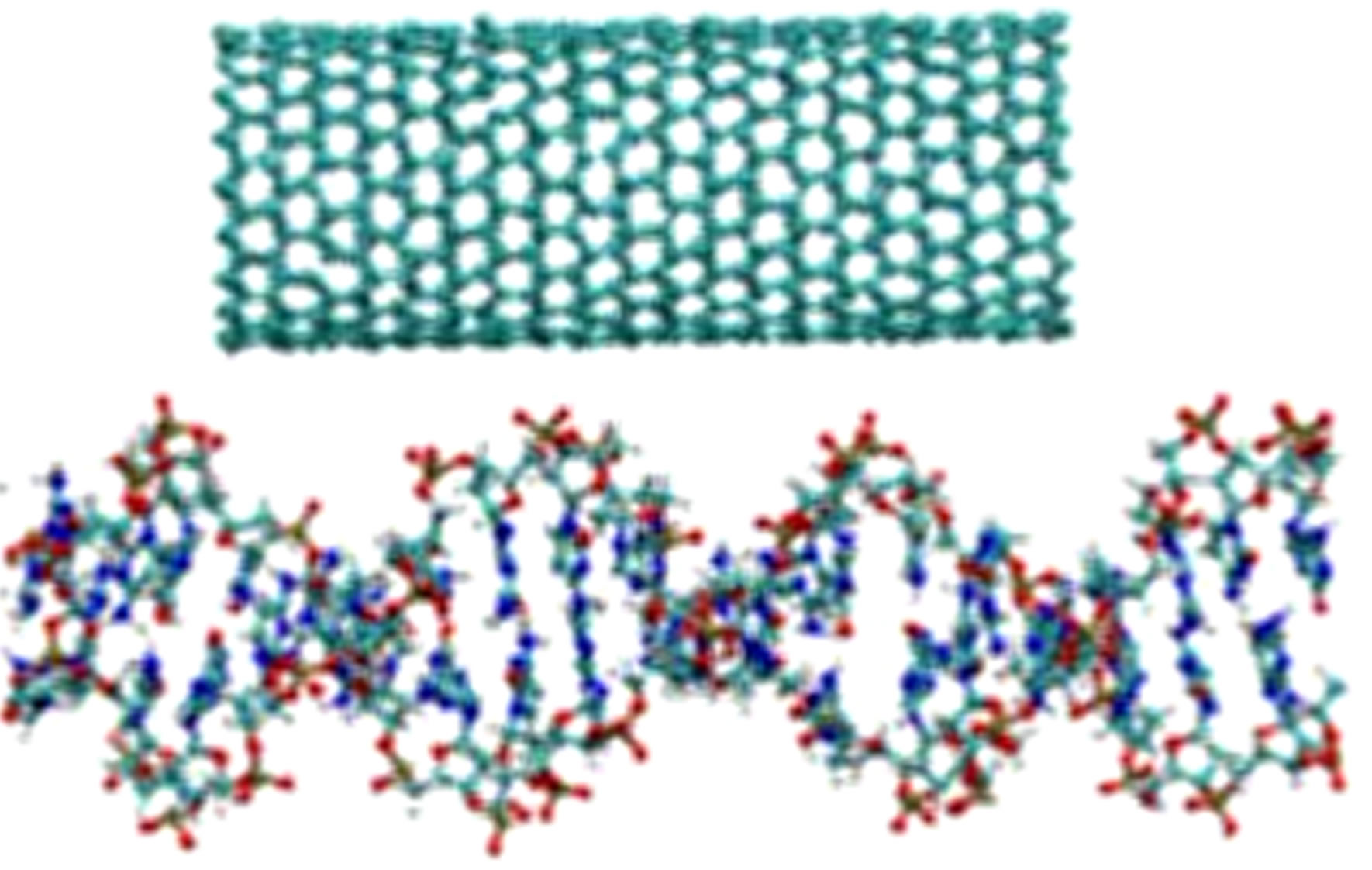
Figure 2. A configuration snapshot of the CNT (top) and DNA (bottom).


Figure 3. The CNT structural presentation (side and top views).
zation. The energy is modelled as a sum of pair-like interactions where, however, the coefficient of the attractive term in the pair-like potential (which plays the role of a bond order) depends on the local environment giving a many-body potential.
The Tersoff potential has 11 atomic and 2 bi-atomic parameters and looks like:
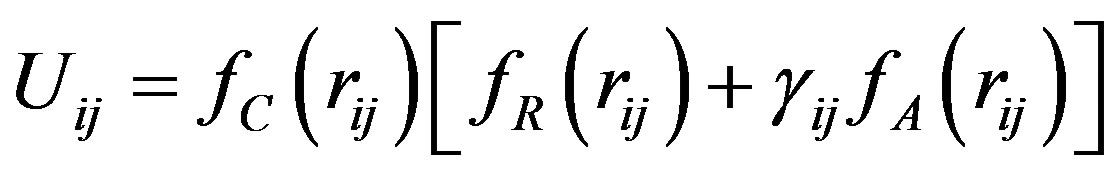 , (1)
, (1)
where the potential parameters have the following forms:
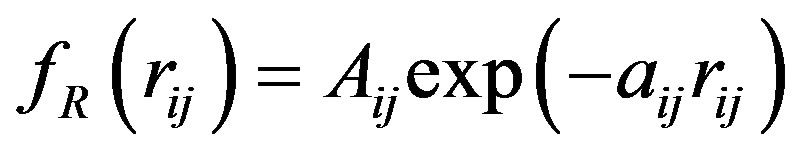 ,
, 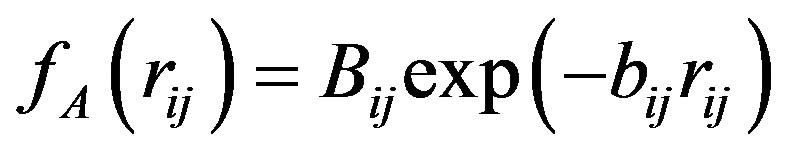 ,
,
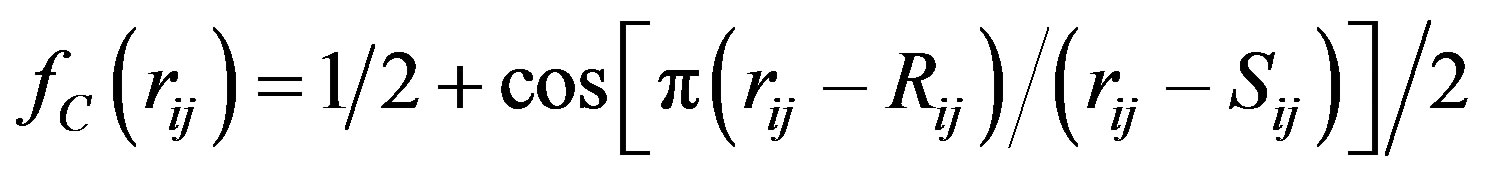 ,
,
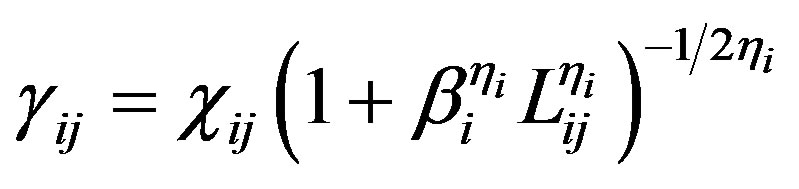 ,
, 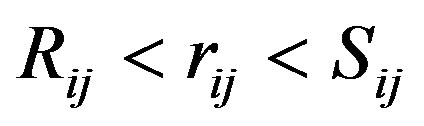 ,
,
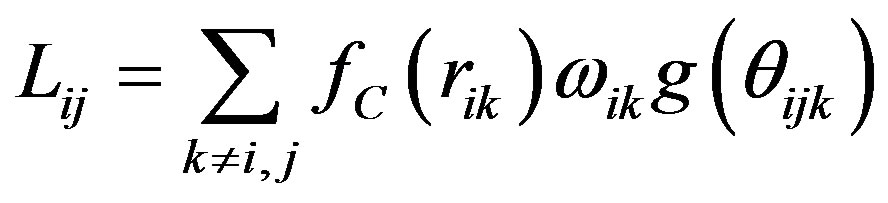 ,
, 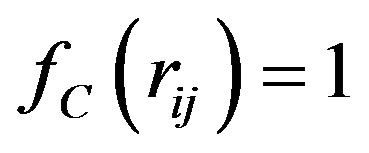 , for
, for 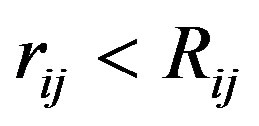
and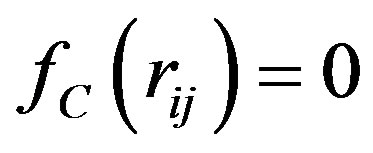 , for
, for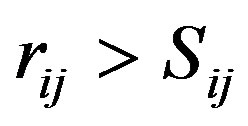 ,
, 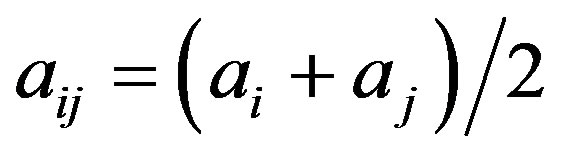 ,
,
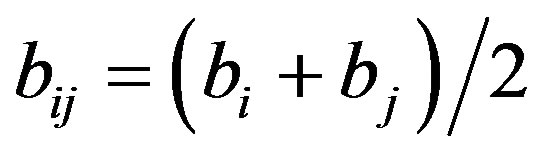 ,
,
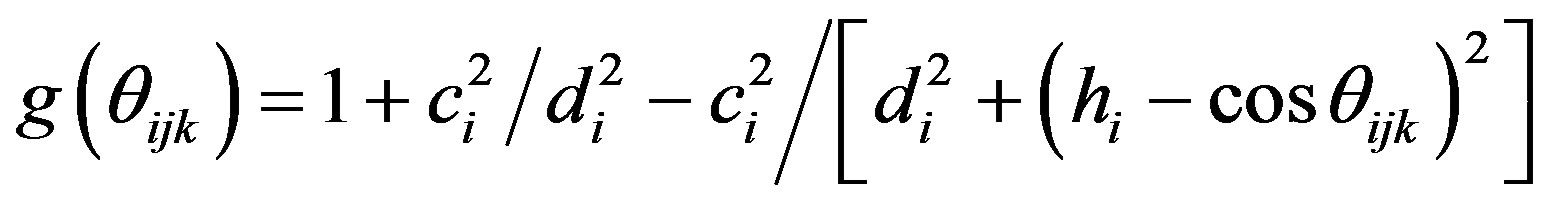 ,
,
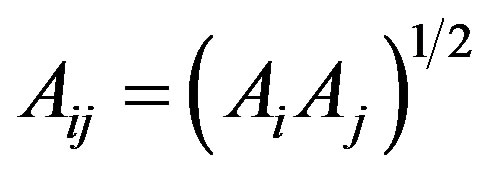 ,
, 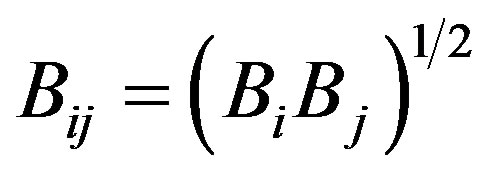 ,
, 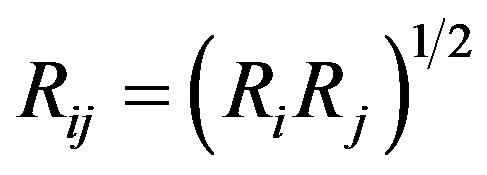 ,
,
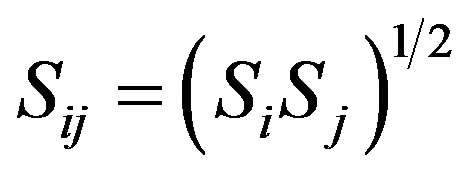 .
.
We have accepted the following values: cii = 1, cij = cji, wii = 1, wij = wji.
We simulated several DNA-CNT model systems using molecular dynamics (MD) simulation method. The MD simulation has been performed on the basis of the DL_POLY general-purpose code [32,33]. The elementary MD cell was a parallelipipped with a volume of V = (130, 130, 65) Å3. The integration algorithm was Berendsen NPT ensemble with the termostat and barostat relaxation times 2.0 ps.
For the CNT we used the Tersoff potential parameters of the DL_POLY software data base [24-26,33,34]: A = 1393.6, a = 3.4879, B = 346.74, b = 2.2119, R = 1.8, S = 2.1, b = 1.5724 × 10−7, h = 0.72751, c = 38049, d = 4.3484, h = −0.57058. The position of the CNT was fixed up using tethering potential as rigid body.
For the Van der Waals interactions between the DNA and CNT we used the Lennard-Jones (LJ), for the DNA used 12-6 potential:
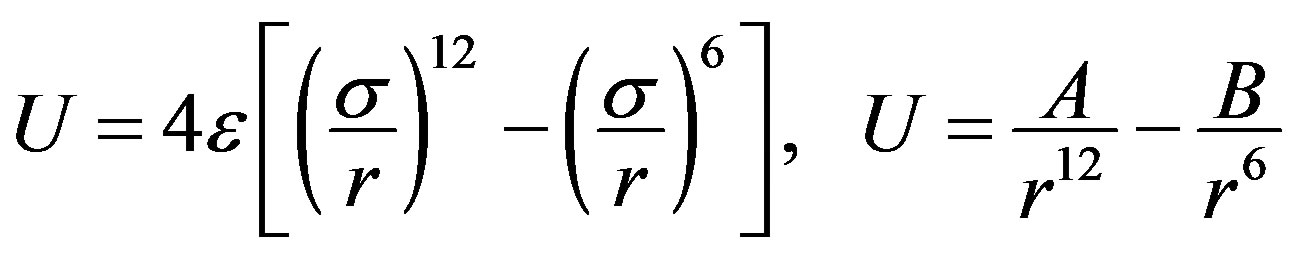 . (2)
. (2)
All potential and parameters are shown in Tables 4-6, where C, O, N and P stands for carbon, oxygen, nitrogen and sulphur atoms of DNA and CC denotes the carbon
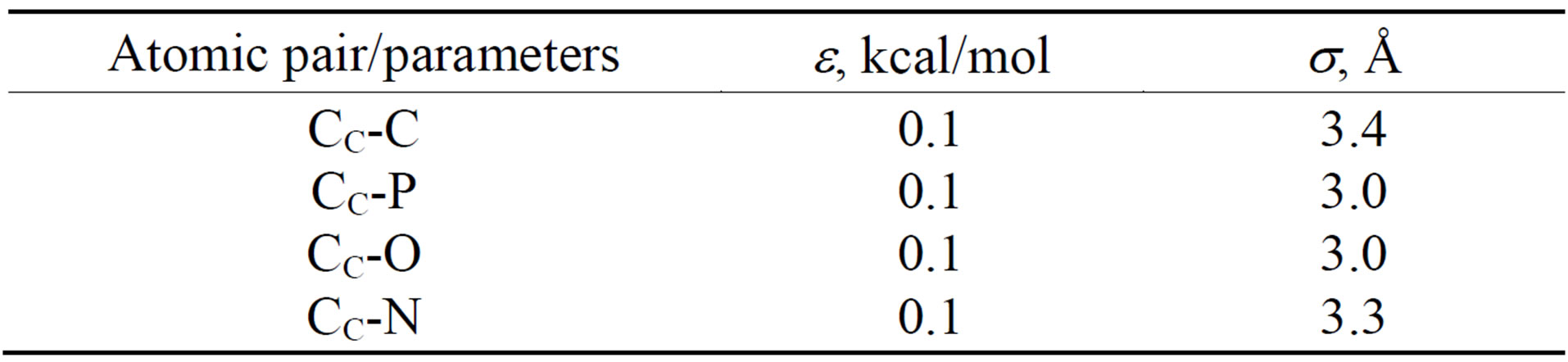
Table 4. The LJ potential parameters of the DNA-CNT model.
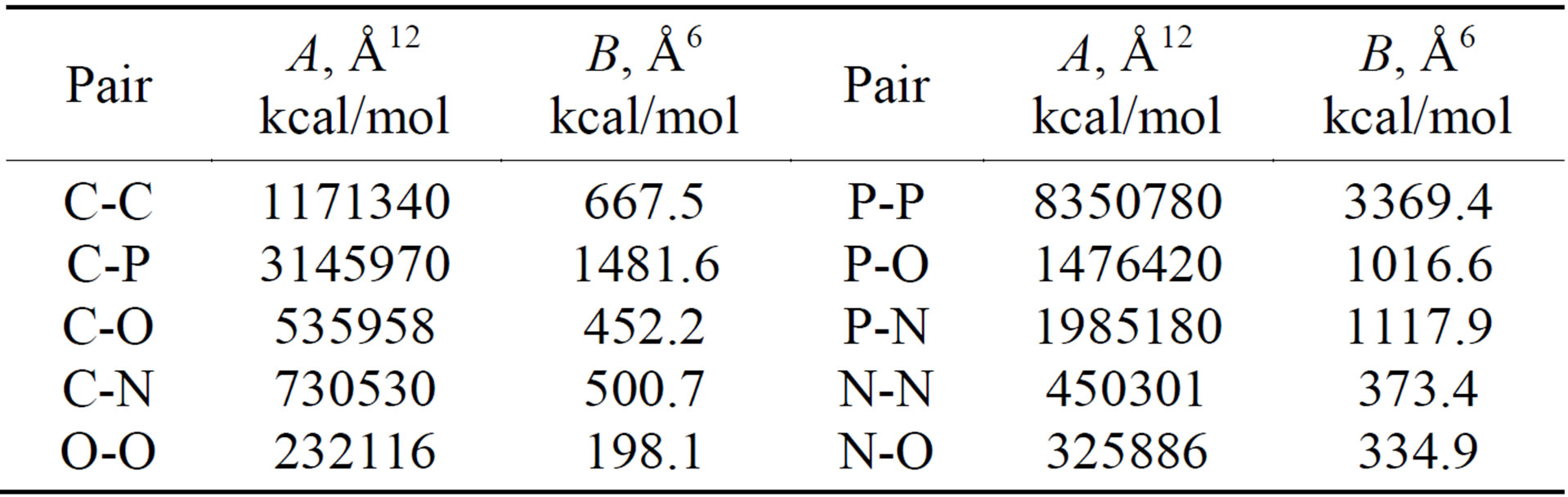
Table 5. The 12-6 potential parameters of the DNA model.
atom of CNT.
3. Results and Discussions
Multiple configurations were generated for the DNACNT model with the MD simulation parameters described above. The DNA insertion into the CNT were relaxed at the temperatures around of T = 310 K. The output MD data were analyzed and visualized using different software facilities (VMD [35], DL_POLY utilities [32,33], etc.).
3.1. MD Simulated Structural DNA-CNT Configurations
In Figure 4 several sequential snapshots are shown to illustrate the DNA-CNT interaction process. In Figure 4 the top (left pictures) and side (right pictures) views of the DNA-CNT system along with the positions of the arbitrarily chosen DNA (P601 and P631) and CNT (CC1630) atoms are presented. From the stage of a close DNA-CNT contact formation, the DNA chain to start its penetration deeper into the CNT region. This is a fully spontaneous process; the only nature of the atomic interactions has governing the DNA-CNT complex formation. The penetration process is like as an initial stage of DNA encapsulation inside of a CNT. It is worth noting that this process is rather slow; for these MD simulations all processes are happening in vacuum. Obviously, the inclusion of water into consideration would accelerate the DNA encapsulation into the CNT. Therefore, CNTs have been proved to be an excellent transport candidate for many biomolecules, proteins or drugs [15-18].
3.2. The DNA-CNT Encapsulation
In Figure 5 a distance diagram is presented for the DNACNT close contact interaction. The positional changes of two distances, DNA (P601, P631)-CNT(C1630), illustrates a DNA penetration behavior deeper into a CNT
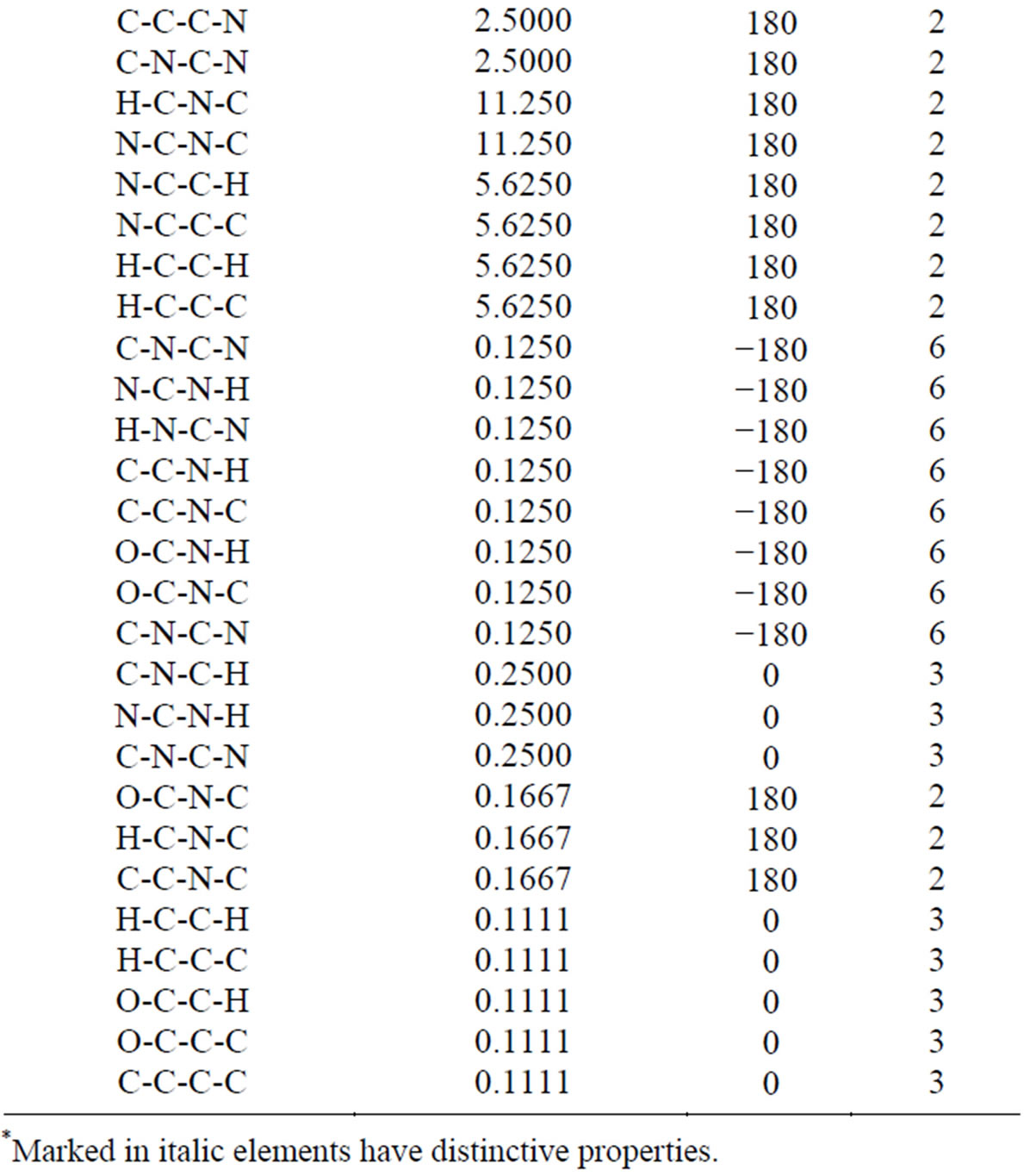
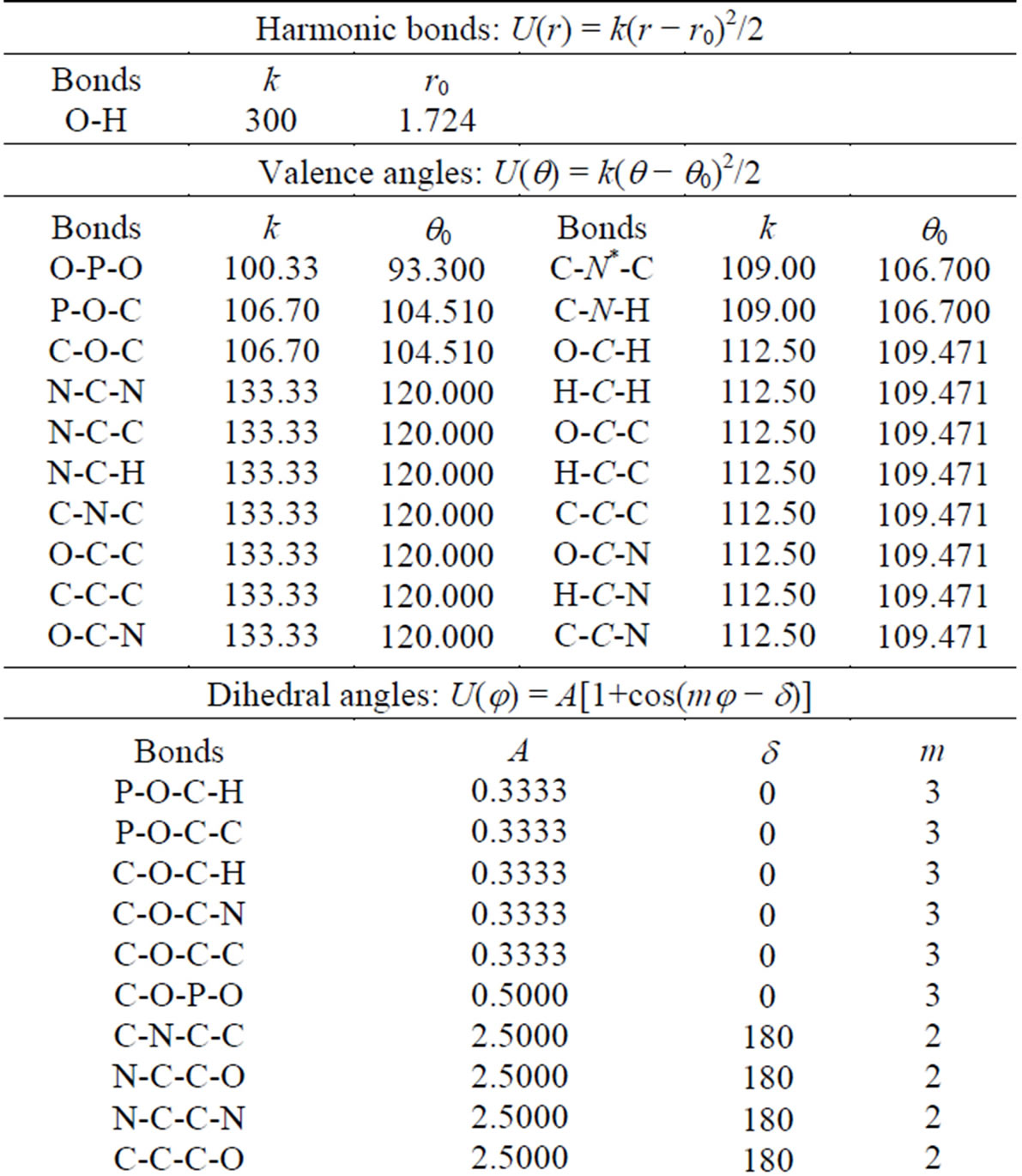
Table 6. The intermolecular potential parameters of the DNA model.
*Marked in italic elements have distinctive properties.
region. Some fluctuations of the distance between DNA and CNT take place, indicating the self-adjustment of the DNA positions before it begins to enter into the CNT. This process has seen to be quite similar with the encapsulation process, as indicated by other authors on biomolecule-CNT interactions [15-22]. Obviously, the Van der Waals and electrostatics interactions used in our simula
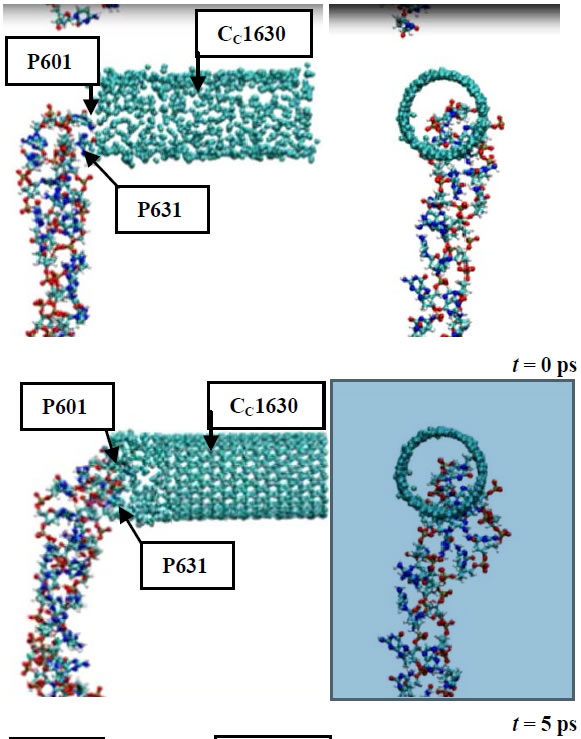
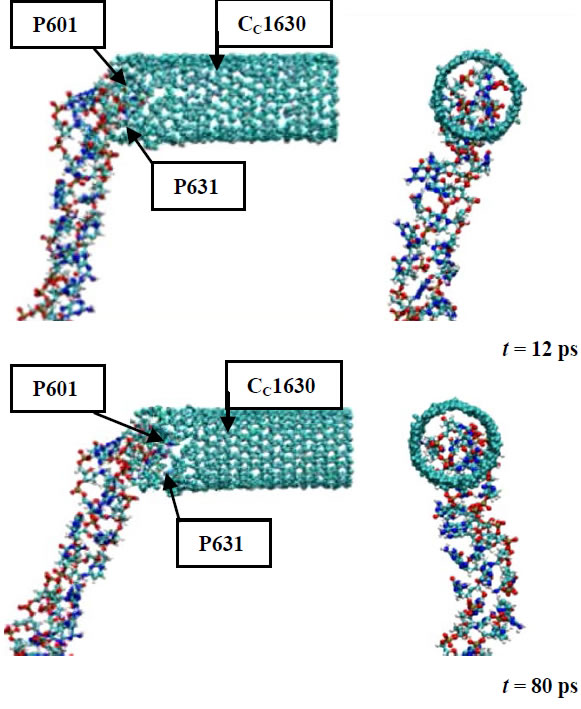
Figure 4. The DNA-CNT dynamics from their contact to penetrtion.
tion technique are both acceleration factors in the DNACND encapsulation process. It is well known that during the DNA insertion into CNT their interaction energies decrease.
The continuous DNA encapsulation into CNT suggests a practical aspect of using such kind systems in an innovation technology, in material design for medicine in the
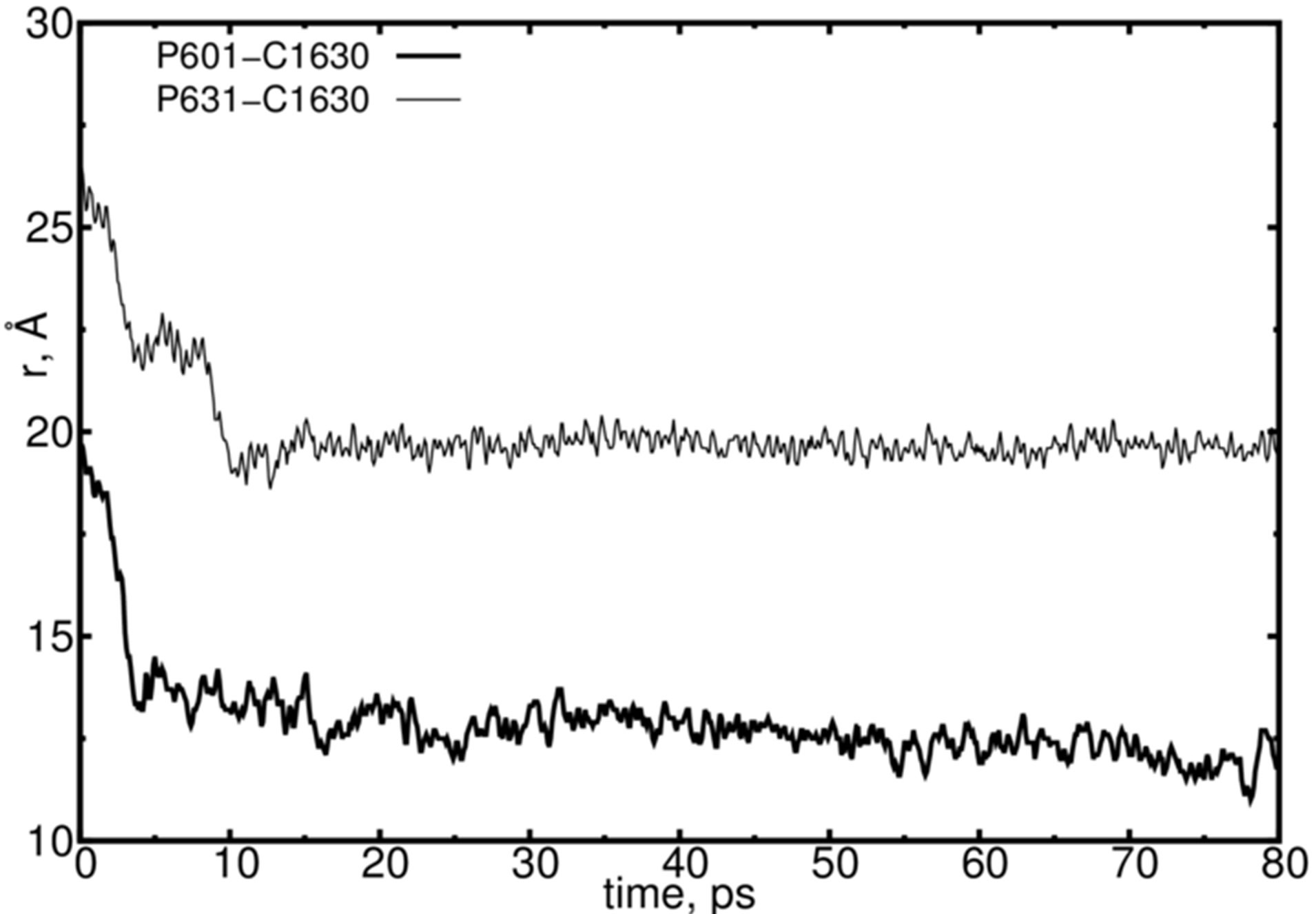
Figure 5. The DNA-CNT penetration dynamics from their contact. The positions of the arbitrarily chosen DNA (P601 and P631) and CNT (C1630) atoms are presented.
future. Say, in manufacturing practical CNT-based devices for the delivery of genes, inhibitors, drugs and so on [15-22,34-38].
4. Conclusion
The quantum chemistry potentials in combination with classical trajectory calculations have to be an efficient approach in today computer molecular modeling. A lot of the physical, chemical and biological systems were under investigation in modern research using this kind hybrid approach. In the hybrid approach—the classical and quantum-chemical MD modeling, the molecules are described as a set of spheres and springs where the spheres imitate classical particles and the spring—the interaction force fields governed by quantum chemistry laws. In this work the interaction of DNA molecule with a carbon nanotube (CNT) was studied using Tersoff potential. The dynamical configurations were built up for the DNACNT model at ambient temperatures. The observations reveal an encapsulation-like behavior of the DNA chain inside the CNT. A possible use of the DNA-CNT complex as a candidate material in drug delivery and related systems was discussed.
Acknowledgements
This work has been performed as part of collaboration between JINR (Russia), RIKEN (Japan), and Keio University (Japan). This work was jointly supported by the JSPS (Japan Society for the Promotion of Science) and the RFBR (Russian Foundation for Basic Research); Grant No. 13-04-92100. The MD simulations have been performed using computer software, hardware facilities, and cluster machines at the CICC (JINR), RICC (RIKEN), and the Yasuoka Laboratory of Keio University (Japan).
REFERENCES
- D. S. D. Larsson, Y. Wang and D. van der Spoel, Biochemistry, Vol. 48, 2009, pp. 1006-1015. http://dx.doi.org/10.1021/bi801952f
- E. G. Marklund, D. S. D. Larsson, D. van der Spoel, A. Patriksson and C. Caleman, Physical Chemistry Chemical Physics, Vol. 11, 2009, pp. 8069-8078. http://dx.doi.org/10.1039/b903846a
- R. Friemann, D. S. D. Larsson, Y. Wang and D. van der Spoel, Journal of the American Chemical Society, Vol. 131, 2009, pp. 16606-16607. http://dx.doi.org/10.1021/ja902962y
- D. van der Spoel, E. Marklund, D. S. D. Larsson and C. Caleman, Macromolecular Bioscience, Vol. 11, 2011, pp. 50-59. http://dx.doi.org/10.1002/mabi.201000291
- Kh. Kholmurodov, “Molecular Simulation Studies in Material and Biological Sciences,” Nova Science Publishers Ltd., 2007, 196 p.
- Kh. Kholmurodov, “Molecular Simulation in Material and Biological Research,” Nova Science Publishers Ltd., 2009, 155 p.
- Kh. Kholmurodov, “Molecular Dynamics of Nanobistructures,” Nova Science Publishers Ltd., 2011, 210 p.
- Kh. Kholmurodov, “Models in Bioscience and Materials Research: Molecular Dynamics and Related Techniques,” Book of International Workshop MSSMBS’12, Nova Science Publishers Ltd., 2013, 208 p.
- J. Santa Lucia Jr. and D. Hicks, Annual Review of Biophysics and Biomolecular Structure, Vol. 33, 2004, pp. 415-440. http://dx.doi.org/10.1146/annurev.biophys.32.110601.141800
- K. J. Breslauert, R. Franks, H. Blockers and L. A. Markyt, Proceedings of the National Academy of Sciences of the United States of America, Vol. 83, 1986, pp. 3746-3750. http://dx.doi.org/10.1073/pnas.83.11.3746
- V. Freyre-Fonseca, et al., Toxicology Letters, Vol. 202, 2011, pp. 111-119. http://dx.doi.org/10.1016/j.toxlet.2011.01.025
- B. Trouiller, R. Reliene, A. Westbrook, P. Solaimani and R. H. Schiestl, Cancer Research, Vol. 69, 2009, pp. 8784- 8789. http://dx.doi.org/10.1158/0008-5472.CAN-09-2496
- Q. Saquib, A. A. Al-Khedhairy, M. A. Siddiqui, F. M. Abou-Tarboush, A. Azam and J. Musarrat, Toxicology in Vitro, Vol. 26, 2012, pp. 351-361. http://dx.doi.org/10.1016/j.tiv.2011.12.011
- R. Dunford, A. Salinaro, L. Cai, N. Serpone, S. Horikoshi, H. Hidaka and J. Knowland, FEBS Letters, Vol. 418, 1997, pp. 87-90. http://dx.doi.org/10.1016/S0014-5793(97)01356-2
- C. Srinivasan, Current Science, Vol. 94, 2008, pp. 300- 301.
- T. A. Hilder and J. M. Hill, Current Applied Physics, Vol. 8, 2008, pp. 258-261. http://dx.doi.org/10.1016/j.cap.2007.10.011
- T. A. Hilder and J. M. Hill, Micro & Nano Letters, Vol. 3, 2008, pp. 41-49. http://dx.doi.org/10.1049/mnl:20080008
- Q. Chen, Q. Wang, Y. C. Liu, T. Wu, Y. Kang, J. D. Moore and K. G. Gubbins, Journal of Chemical Physics, Vol. 131, 2009, Article ID: 015101.
- I. Ali, D. Marenduzzo and J. M. Yeomans, Physical Review Letters, Vol. 96, 2006, Article ID: 208102. http://dx.doi.org/10.1103/PhysRevLett.96.208102
- I. Ali, D. Marenduzzo and J. M. Yeomans, Biophysical Journal, Vol. 94, 2008, pp. 4159-4164. http://dx.doi.org/10.1529/biophysj.107.111963
- J. V. Veetil and K. Ye, Biotechnology Progress, Vol. 23, 2007, pp. 517-531. http://dx.doi.org/10.1021/bp0602395
- S. Dhar, Z. Liu, J. Thomale, H. J. Dai and S. J. Lippard, Journal of the American Chemical Society, Vol. 130, 2008, pp. 11467-11476. http://dx.doi.org/10.1021/ja803036e
- J. Tersoff, Physical Review B, Vol. 39, 1989, pp. 5566- 5568. http://dx.doi.org/10.1103/PhysRevB.39.5566
- Kh. Kholmurodov, G. Aru and K. Yasuoka, Natural Science, Vol. 2, 2010, pp. 902-910. http://dx.doi.org/10.4236/ns.2010.28111
- Kh. Kholmurodov, et al., Chemical Physics, Vol. 402, 2012, pp. 41-47. http://dx.doi.org/10.1016/j.chemphys.2012.04.002
- K. A. Affholter, S. J. Henderson, G. D. Wignall, G. J. Bunick, R. E. Haufler and R. N. Compton, The Journal of Chemical Physics, Vol. 99, 1993, pp. 9224-9229. http://dx.doi.org/10.1063/1.465538
- Y. B. Melnichenko, G. D. Wignall, R. N. Compton and G. Bakale, The Journal of Chemical Physics, Vol. 111, 1999, pp. 4724-4728. http://dx.doi.org/10.1063/1.479234
- H. E. Smorenburg, R. M. Crevecoeur, I. M. de Schepper and L. A. de Graaf, Physical Review E, Vol. 52, 1995, pp. 2742-2752. http://dx.doi.org/10.1103/PhysRevE.52.2742
- F. Migliardo, V. Magazù and M. Migliardo, Journal of Molecular Liquids, Vol. 10, 2004, pp. 3-6. http://dx.doi.org/10.1016/j.molliq.2003.08.010
- T. Tomiyama, S. Uchiyama and H. Shinohara, Chemical Physics Letters, Vol. 264, 1997, pp. 143-148. http://dx.doi.org/10.1016/S0009-2614(96)01290-0
- A. D. Bokare and A. Patnaik, Journal of Chemical Physics, Vol. 119, 2003, pp. 4529-4538. http://dx.doi.org/10.1063/1.1594177
- W. Smith and T. R. Forester, Journal of Molecular Graphics, Vol. 14, 1996, pp. 136-141. http://dx.doi.org/10.1016/S0263-7855(96)00043-4
- W. Smith, T. R. Forester and I. T. Todorov, “The DL_POLY 2 User Manual,” STFC Daresbury Laboratory Daresbury, Warrington WA4 4AD Cheshire, UK, Version 2.19, 2008.
- Y. Kang, Y. C. Liu, Q. Wang, T. Wu and W. J. Guan, Biomaterials, Vol. 30, 2009, pp. 2807-2815. http://dx.doi.org/10.1016/j.biomaterials.2009.01.024
- Y. H. Xie and A. K. Soh, Materials Letters, Vol. 59, 2005, pp. 971-975. http://dx.doi.org/10.1016/j.matlet.2004.10.079
- D. S. D. Larsson, L. Liljas and D. van der Spoel, PLOS Computational Biology, Vol. 8, 2012, Article ID: e100 2502. http://dx.doi.org/10.1371/journal.pcbi.1002502
- B.-D. Chen, C.-L. Yang, M.-S. Wang and X.-G. Ma, Chinese Physics B, Vol. 21, 2012, Article ID: 083103.
- W. Humphrey, A. Dalke and K. Schulten, Journal of Molecular Graphics, Vol. 14, 1996, pp. 33-38. http://dx.doi.org/10.1016/0263-7855(96)00018-5