Advances in Bioscience and Biotechnology
Vol. 4 No. 2 (2013) , Article ID: 28402 , 11 pages DOI:10.4236/abb.2013.42023
Evaluation of a novel oral iron chelator 1-(N-acetyl-6-aminohexyl)-3-hydroxypyridin-4-one (CM1) for treatment of iron overload in mice
![]()
1Department of Biochemistry, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand
2College of Medicine and Public Health, Ubon Ratchathani University, Ubon Ratchathani, Thailand
3Institute of Research and Development, Government Pharmaceutical Organization, Ministry of Public Health, Bangkok, Thailand
4Thalassemia Research Center, Institute of Molecular Bioscience, Mahidol University Salaya Campus, Nakornpathom, Thailand
Email: ssrichai@med.cmu.ac.th, u4305084@hotmail.com, chada@health.moph.go.th, *grsfc@mahidol.ac.th
Received 9 January 2013; revised 9 February 2013; accepted 16 February 2013
Keywords: Thalassemia; Iron Overload; Iron Chelator; Hydroxypyridinone; Lipid Peroxidation; Malondialdehyde
ABSTRACT
Desferrioxamine (DFO), deferiprone (DFP) and deferasirox (DFX) are promising effective iron chelators for the treatment of iron overload in (-thalassemia patients; nonetheless, their side effects have also been reported. 3-Hydroxypyridinone derivatives are being developed as a safer new chelator and in combined chelation therapy. We evaluated the iron-chelating activity of 1-(N-acetyl-6-aminohexyl)-3-hydroxypyridin-4-one (CM1) in iron-loaded C57BL6 mice. The feeding of a ferrocene-supplemented diet (Fe diet) to mice resulted in iron overload, detectable plasma nontransferrin-bound iron (NTBI) and labile plasma iron (LPI), and increases of red cell membrane iron, plasma malondialdehyde (MDA) and excessive tissue iron deposits. Like DFP, the CM1 lowered the levels of the membrane non-heme iron, the NTBI and LPI (p < 0.05) and the MDA after 3 months of treatment. Administration of the Fe diet and the Fe diet along with the chelators did not change the morphology of the liver and heart. Numerous iron accumulations were observed in the liver and spleen tissues of the Fe dietfed mice, whereas the CM1 reduced such iron deposition. Thus, 1-(N-acetyl-6-aminohexyl)-3-hydroxypyridin-4-one (CM1) can be considered a candidate bidentate oral iron chelator and is effective in the removal of toxic irons in blood compartment and tissues. The effectiveness and toxicity of the CM1 need to be investigated extensively in thalassemia mice and patients with iron overload.
1. INTRODUCTION
In iron overload, excessive amounts of iron catalyzes the production of reactive oxygen species (ROS) via HaberWeiss and Fenton reactions, and in turn the ROS can damage cells and tissues of the heart, liver, pancreas and endocrine glands, leading to dysfunctions of these vital organs [1]. Non-transferrin bound iron (NTBI) and labile plasma iron (LPI) appearing in the plasma of thalassemia patients with iron overload play a major role in many pathological conditions [2,3]. These two toxic forms of the iron seem to be translocated across cell membrane irregularly; nonetheless, they are susceptible to chelation outside the cells [4]. Intracellular labile iron pool (LIP) is the other source of chelatable and redox-active iron, comprising transitory Fe(II) and Fe(III) forms and serves as a crossroad of the cellular iron metabolism [4,5]. Iron chelation therapy aims to prevent the accumulation of toxic iron and eliminate the excess iron in transfusion-dependent b-thalassemia patients. Reduction of the redox-active irons requires aggressive chelation therapy. Levels of the NTBI, LPI and LIP have been proposed for the assessment of iron overload and evaluation of the efficiency in chelation therapy [6].
Nowadays, only desferrioxamine (DFO), deferiprone (DFP) and deferasirox (DFX) are promising iron chelators used for treatment in b-thalassemia patients with iron overload. Unfortunately, most patients develop adverse effects such as local irritation, skin redness and mild pain at the applied sites, as well as poor compliance treatment from DFO [7,8]. Other side effects involve GI disturbances, arthopathy, zinc deficiency, agranulocytosis and thrombocytopenia from DFP [9]; as well as renal toxicity and Fanconi syndrome in DFX treatment [10]. Chelation therapy has been successful in the treatment of iron overload in patients with transfusional thalassemia, sickle cell disease, myelodysplatic syndrome and other hematologic disorders [11]. Effective iron chelation can result in a net negative iron balance, lowered incidence of hepatic fibrosis, a reduction of cardiac mortality, and a striking improvement in survival and quality of life in such patients [12-14]. In addition, several candidate compounds such as 2,3-dihydroxybenzoic acid (DHB), diethylene triamine pentaacetic acid (DTPA), N,N’-bis(2- hydroxybenzyl)-ethylenediamine diacetic acid (HBED), pyridoxal isonicotinoyl hydrazone (PIH), 3-hydroxy-4- pyridinones derivative, curcumin and green tea catechins have been evaluated for their potential iron-chelating activity in iron overload conditions [15-25].
To ensure the safety of clinical trial in human, animal models are commonly used to evaluate efficacy, pharmacokinetics, drug metabolism and relative toxicity of different chelators [26]. Recently, we have reported our new bidentate iron chelator, N-[6-(3-hydroxy-2-methyl- 4-oxo-4H-pyridin-1-yl)-hexyl]-acetamide (CM1), which is more lipophilc than DFP and can bind iron efficiently [27]. The compound was able to chelate plasma NTBI, but was not toxic to peripheral blood mononuclear cells (PBMC), cardiomyocytes and hepatocytes in vitro. Importantly, CM1 had reduction efficiency of LIP in ironloaded hepatocyte as well as in myocyte cultures [28]. In the present study, we investigated a potential role of our new chelator, CM1 in the treatment of iron-loaded mice.
2. MATERIALS AND METHODS
2.1. Animal Models
Wild type (WT, muβth+/+) mice (strain C57BL/6) aged between 6 - 10 weeks and having a body weight 24 - 30 grams were kindly supplied by the Thalassemia Research Center, Institute of Molecular Biosciences, Mahidol University, Salaya Campus [29,30]. The animals were housed in polyethylene cages and maintained in a clean air-conditioned room under the controlled conditions of 12-h day/12-h night cycle at 23˚C ± 1˚C and at 40% - 70% humidity. The study protocol has been approved by the Animal Ethical Committee of Medical Faculty, Chiang Mai University, Thailand) (Reference Number- 3/2554).
2.2. Iron Loading and Chelation Treatment
Iron overload was induced by feeding the mice a normal chow diet (N diet) and the N diet supplemented with 0.2% (w/w) ferrocene (Fe diet) for 90 days [31]. The iron-loaded mice were randomly subdivided into 4 groups. The study group was fed with the Fe diet along with treatments for another 90 days with deionized water, DFP (50 mg/kg/day), and CM1 (50 and 100 mg/kg/day) per oral [32]. The control group was fed with N diet throughout the study. Their body weights and food consumption details were recorded weekly.
2.3. Blood and Tissue Collections
Blood samples were collected from their tail’s veins and collected into Na-heparin tubes every 20 days. Plasma was separated immediately and kept frozen −20°C for further analyses. Packed red blood cells (RBC) were washed three times at 4000 rpm, 25ºC for 5 minutes with 5 mM phosphate-buffered saline pH 7.0 (PBS) and resuspended in the same buffer for the determination of hemoglobin (Hb) and iron concentrations. At the end of study the mice were sacrificed and anesthetized with an intraperitoneal injection of sodium phenobarbital and blood was collected from the heart left ventricles. Their hearts, livers and spleens were dissected, weighed and chopped into small pieces. The organs were kept frozen at −20°C for biochemical assay and fixed in 10% (v/v) neutralized formalin solution for pathological examinations.
2.4. Measurement of RBC Membrane Non-Heme Iron
Using ferrozine colorimetric method, assay was performed within 2 minutes to detect free iron, and for 24 hours to detect both free iron and ferritin iron [33]. The RBC suspension was mixed with cold lysis buffer (5 mM phosphate buffer, pH 8.0). The hemolysate was then centrifuged at 13,000 rpm, 4ºC for 10 minutes and this step was repeated 6 times. Ghost cell pellet was reconstituted in the PBS for the determination of non-heme iron concentration. The ghost membrane was solubilized in 0.6% (w/v) SDS in 0.2 M acetate buffer pH 4.5 and incubated with reducing reagent (0.2% ascorbic acid and 0.2% sodium dithionite in the acetate buffer). The ferrozine reagent was added to produce a colored product and optical density (OD) was measured at 563 nm. A calibration curve of Fe was made by using standard ferrous ammonium sulfate (FAS) at 0 - 200 µM. Iron content was expressed in nanomole/mg. membrane protein, of which the latter was determined based on dye-binding technique using Bio-Rad (Richmond, CA) microprotein assay reagent [34].
2.5. Quantification of Plasma NTBI Concentration
NTBI is quantified using NTA chelation/HPLC method established by Singh et al. [35] with an aluminum blocking step. In the assay procedure, plasma was incubated with a weak chelator NTA solution (80 mM at a final concentration) pH 7.0 for 30 minutes at room temperature to produce Fe3+-(NTA)2 complex. Afterward, the complex was filtered through a membrane (NanoSep®, 10-kDa cutoff, polysulfone type; Pall Life Sciences, Ann Arbor, MI, USA) and analyzed using a non-metallic HPLC system. NTBI was fractionated on a glass analytical column (ChromSep-ODS1, 100 mM × 3.0 mM, 5 μM), eluted with mobile phase solvent (3 mM CP22 in 19% acetonitrile/MOPS pH 7.0) at a flow rate of 1.0 ml/min and OD was monitored at 450 nm using a flow cell detector (SpecMonitor2300; LDC Milton-Roy Inc., Riviera Beach, FL, USA). Data analysis was conducted with BDS software (BarSpec Ltd., Rehovot, Israel). NTBI concentration represented by Fe3+-(CP22)3 peak area was determined from a calibration curve constructed from Fe3+-(NTA)2 in 80 mM NTA (0 - 16 µM).
2.6. Measurement of LPI Concentration
In principle, redox-active LPI can convert non-fluorescent dihydrorhodamine (DHR) to oxidized form rhodamine (R), resulting in an increase of fluorescence intensity (FI) [4]. Plasma sample (20 µl) was incubated with/without of 5 mM DFP at 37ºC for 30 minutes and the DHR solution containing ascorbic acid was added. Kinetics of increasing fluorescence was followed immediately for 40 minutes, with readings every 2 minutes, at 37°C using a spectrofluorometer (λexcitation 485 nm, λemission 538 nm). Slope of the FI was plotted against the reaction time between 15 - 40 minutes. A calibration curve was constructed from standard FAS solution (0 - 20 µM). Difference in rate of DHR oxidation represents a component of redox active LPI in plasma. The LPI concentration was calculated from the calibration curve relating the difference in slope with/without DFP versus standard iron concentration.
2.7. Determination of Liver Iron Content (LIC)
LIC was measured colorimetrically and expressed as μg Fe/g. tissue wet weight [36]. The liver was homogenized in the buffer containing protease inhibitor cocktail (1:10, w/v) using a glass homogenizer. The homogenate was treated with protein-precipitating agent (1 N HCl/10% trichloroacetic acid solution), mixed vigorously and heated at 95˚C for 1 hour. The liver digest was cooled down at room temperature and centrifuged at 12,000 rpm for 10 minutes. The iron in the supernatant was reacted with the chromogen solution containing 0.508 mM ferrozine, 1.5 M sodium acetate and 1.5% (v/v) thioglycolic acid for 30 minutes. OD of the colored product was measured at 562 nm. A calibration curve of Fe was made by using standard FAS (0 - 200 µM).
2.8. HPLC-Based Assay MDA Concentration
Liver was dried at 120°C in an oven overnight and weighed. The tissue (50 mg) was homogenized in the digestion solution (1.0 ml) containing 50 mM phosphate buffer pH 2.8 (0.8 ml), methanol (0.1 ml) and butyrated hydroxytoluene (BHT) (50 ppm) in an ice bath. The homogenate and plasma (0.5 ml) were mixed with 1.1 ml of 10% (w/v) trichloroacetic acid (TCA) containing BHT (50 ppm), heated at 95°C for 30 minutes and cooled down. After centrifugation at 3500 g for 10 minutes, the supernatant (0.5 ml) was mixed with the chromogenic solution containing 0.44 M H3PO4 and 0.6% (w/v) thiobarbituric acid (TBA), and heated to 95˚C for 30 minutes to produce a pink-colored product, a thiobarbituric acidreactive substance (TBARS). Using HPLC analysis [37] malondialdehyde (MDA), a lipid-peroxidation product and a fraction of TBARS, was fractioned on C18 column (Water Spherosorb ODS2 type, 250 mM × 4.3 mM, 5 μM), eluted with mobile-phase solvent (50 mM KH2PO4: methanol, 65:35 = v/v) at a flow rate of 1.0 ml/min and detected at 532 nm. Standard 1,1,3,3-tetramethoxypropane (0 - 100 μM) was used to produce a calibration curve. The curve was constructed by plotting the measured peak areas versus the concentrations and was then used to determine the liver and plasma MDA concentrations. The MDA concentrations were expressed in μM for plasma samples and in nmole⁄mg. protein for liver tissues, for which the protein concentration was determined using the dye-binding assay.
2.9. Measurement of Liver Hydroxyproline Content
Hydroxyproline which is a component of collagen and an index of liver fibrosis was determined using colorimetric method [38]. Liver tissue was first hydrolyzed with 6 M HCl for 18 hours and later neutralized with 10 M NaOH. Then, the hydrolysate was mixed with chloramine T solution for 4 minutes and incubated with Ehrlich’s reagent to develop a colored product (λmax 558 nm). Liver hydroxyproline concentration was determined from a calibration curve of standard hydroxyproline and expressed as μmole/g. tissue dry weight.
2.10. Pathological Examinations
Fixed tissue sections were dehydrated with a graded series of ethyl alcohol, embedded in paraffin and finally stained with hematoxylin and eosin reagent (morphological examination) and Perls’ supravital dye (iron content) [39]. The stained slides were examined under a light microscope by an expert pathologist and photographed with a digital camera.
2.11. Statistical Analysis
Data were presented as means ± SD or mean ± SEM. Statistical significance was determined using one-way analysis of variance (ANOVA), for which p < 0.05 was considered significant.
3. RESULTS
3.1. RBC Membrane Iron Content
As shown in Figure 1 total iron (non-heme iron and non-ferritin iron) present in RBC membrane of the Fe diet-fed WT mice (29.31 ± 10.74 nmole/µg protein) on day 0 was significantly higher than that of the N diet-fed WT mice (1.45 ± 1.32 nmole/µg protein). Over 6 months, the RBC iron level was not changed in the N diet group and it was slightly increased (p > 0.05) in the Fe diet group. Interestingly, the increase of RBC iron tended to be reduced by the treatments of DFP (50 mg/kg) and CM1 (50 and 100 mg/kg), particularly on day 180 (after 180 days). However, the ferrozine method was not able to detect ferritin iron in both N diet and Fe diet groups (data not shown).
3.2. Plasma NTBI and LPI Concentrations
NTBI was not detectable in the plasma of N diet-fed mice throughout the study. At the beginning, NTBI appeared in the plasma of the mice fed with Fe diet and tended to be increased in a time-dependent manner during 6 months, indicating iron overload (Figure 2). The DFP and CM1 did not abolish the increase of plasma NTBI in the Fe diet-fed mice over 50 days; however, the chelators were effective in lowering the plasma NTBI levels after 180 days (p < 0.05). Similarly, LPI was not found in plasma of the N diet-fed WT mice and was recored at 11.23 µM in the plasma of the Fe diet-fed mice. Apparently, the DFP and CM1 chelations lowered the plasma LIP levels and the chelation was concentration dependent in the CM1 treatment (Figure 3).
3.3. Plasma MDA Concentration
Plasma MDA concentrations were not changed in the N diet group over 6 months. In comparison, plasma MDA levels after 90 days were higher in the Fe diet group
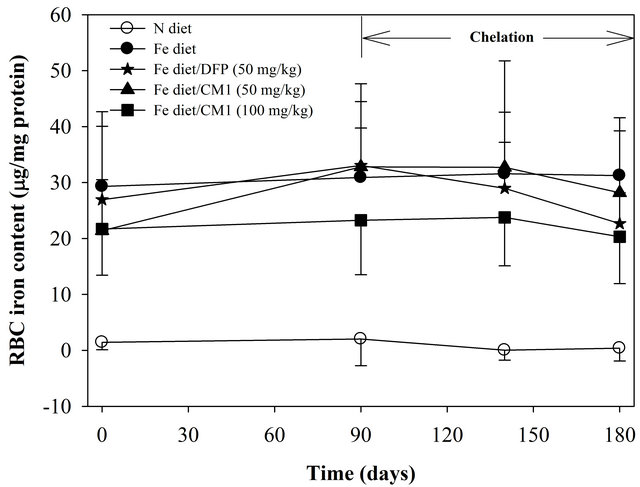
Figure 1. RBC membrane iron content in the WT mice fed with N diet (n = 15), Fe diet (n = 15), and the Fe diet together with DFP (50 mg/kg) (n = 13) and with CM1 (50 and 100 mg/kg) (n = 16 and 15, respectively) for 3 months. Data were expressed as mean ± SD. *p < 0.05 compared with the N diet group.
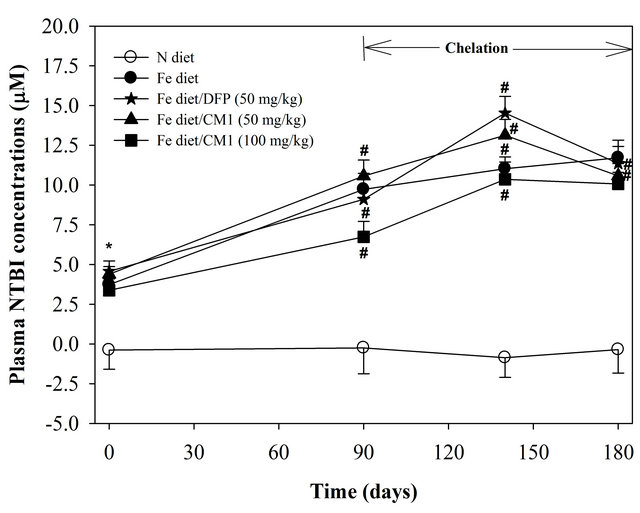
Figure 2. Plasma NTBI concentrations of the WT mice fed with N diet, Fe diet, and the Fe diet together with DFP (50 mg/kg) and with CM1 (50 and 100 mg/kg) for 3 months. Data were expressed as mean ± SD. *p < 0.05 compared with the N diet group; #p < 0.05 compared with the Fe diet group on the chelation.
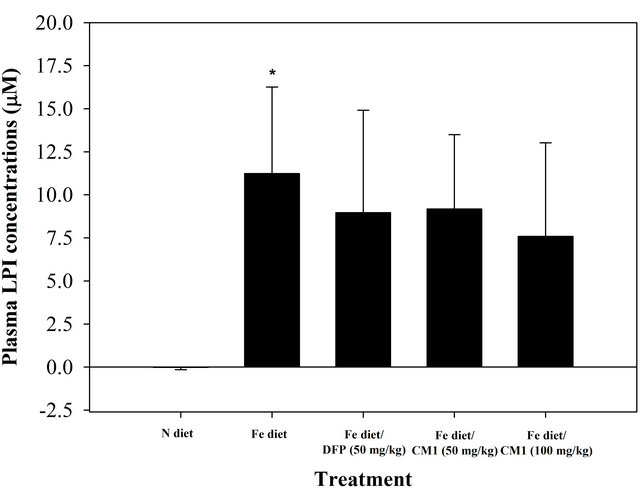
Figure 3. Plasma LPI concentrations of the WT mice fed with N diet, Fe diet, and the Fe diet together with DFP (50 mg/kg) and with CM1 (50 and 100 mg/kg) for 3 months. Data were expressed as mean ± SD. *p < 0.05 compared with the N diet group.
(1.37 ± 0.82 µM) than the N diet group (1.19 ± 0.26 µM) and tended to increase slightly after 180 days (1.53 ±0.44 µM). DFP treatment over 90 days prevented the increase of plasma MDA. Importantly, CM1 (50 and 100 mg/kg) for 90 days effectively reduced levels of plasma MDA (D = −0.32 and −0.53 μM, respectively) in a concentration-dependent manner (Table 1).
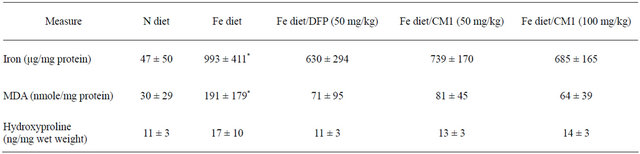
3.4. Liver Iron, MDA and Hydroxyproline Contents
For 6 months, LIC was induced to increase in the Fe diet
Table 1. Plasma MDA concentrations of WT mice fed with N diet, Fe diet, and Fe diet together with 50 mg/kg of DFP (50 mg/kg) and CM1 (50 and 100 mg/kg) for 3 months. Data are expressed as mean ± SEM.
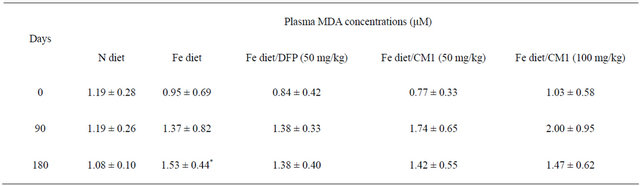
*p < 0.05 compared with the N diet group.
fed mice (993 ± 411 µmole/mg protein), which was significantly higher than that in the N diet-fed mice (47 ± 50 µmole/mg protein). DFP treatment decreased the LIC (630 ± 294 µmole/mg protein) and CM1 (50 and 100 mg/kg) and also lowered the LIC in dose-dependent manner (739 ± 170 and 685 ± 165 µmole/mg protein, respectively). Consistently, liver MDA which is a lipidperoxidation product was increased in the Fe diet-fed mice (191 ± 179 nmole/mg protein) and was significantly higher than that recorded in the N diet-fed mice (30 ± 29 nmole/mg protein). DFP and CM1 treatments effectively lowered the liver MDA and the efficiency was dependent on the CM1 dose. Hepatic hydroxyproline level was increased in parallel to the increases of hepatic iron and MDA levels. Similarly, DFP and CM1 treatments lowered the increase of the hepatic hydroxyproline content slightly (Table 2).
3.5. Pathological Examination
3.5.1. H&E Staining
In the N diet group, heart tissue showed mild hypertrophy of some myocardial fibers. Liver tissue showed a moderate degree of portal inflammation composed of lymphocytes with interface hepatitis, a mild degree of spotty necrosis (non-zonal), and was composed of lymphocytes and neutrophils. Fatty changes were not observed. The spleen tissue did not show a pathologic change. In the Fe diet group, heart tissue showed mild hypertrophy of some myocardial fibers. Liver tissue showed a moderate degree of portal inflammation composed of lymphocytes with interface hepatitis and a mild degree of spotty necrosis (non-zonal), and was composed of lymphocytes and neutrophils, as well as a mild dilatation of central veins. Neither fatty changes nor portal tract expansion were observed. In the Fe diet/DFP chelation group, heart tissue showed mild hypertrophy of some myocardial fibers. Liver tissue showed a moderate degree of spotty necrosis (centrilobular pattern with centro-central bridging necrosis) and a mild dilatation of central veins. In the Fe diet/CM1 (50 mg/kg) treatment group, the heart did not show any pathological changes. Liver tissue showed a moderate degree of spotty necrosis (centrilobular pattern with centro-central bridging necrosis), mild dilatation of central veins, microvesicular fatty change of rare hepatocytes, and no portal tract expansion. In the Fe diet/CM1 (100 mg/kg) treatment group heart tissue did not show any pathologic changes. Liver tissue showed a mild degree of spotty necrosis (non-zonal) and contained particular lymphocytes and neutrophils. Neither fatty changes nor portal tract expansion were observed (Figure 4).
3.5.2. Perl’s Staining
In the Fe diet group, liver tissue showed numerous hemosiderin pigments deposited in sinusoidal macrophages (cluster and individual cell fashions) and hepatocytes (centrilobular pattern). Spleen tissue indicated the presence of numerous hemosiderin-laden macrophages in both red pulp and white pulp. In the Fe diet/DFP treatment group, heart tissue showed the presence of few hemosiderin-laden macrophages in the stromal tissue. Liver tissue was composed of lymphocytes and neutrophils associated with numerous hemosiderin pigments deposited in sinusoidal macrophages (cluster and individual cell fashions) and hepatocytes (centrilobular pattern). The spleen tissue showed the presence of numerous hemosiderin-laden macrophages in both red pulp and white pulp. In the Fe diet/CM1 (50 mg/kg) treatment group, liver tissue showed the existence of lymphocytes and neutrophils associated with numerous hemosiderin pigments deposited in sinusoidal macrophages (cluster and individual cell fashions) and hepatocytes (centrilobular pattern). The spleen tissue showed the presence of numerous hemosiderin-laden macrophages in both red pulp and white pulp. In the Fe diet/CM1 (100 mg/kg) treatment group, liver tissue was present in scattered hemosiderin pigments deposited in sinusoidal macro
Table 2. Liver iron, MDA and hydroxyproline contents of WT mice fed with N diet, Fe diet, and Fe diet together with 50 mg/kg of DFP (50 mg/kg) and CM1 (50 and 100 mg/kg) for 3 months. Data are expressed as mean ± SEM.
*p < 0.05 compared with the N diet group.
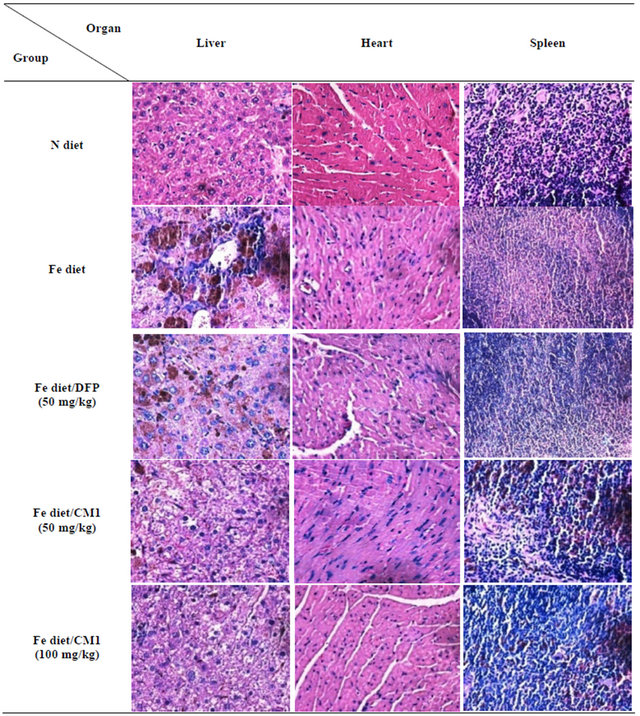
Figure 4. Hematoxylin and eosin staining of the liver, heart and spleen obtained from WT mice fed with N diet, Fe diet, and Fe diet together with 50 mg/kg of DFP (50 mg/kg) and CM1 (50 and 100 mg/kg) for 3 months.
phages (cluster and individual cell fashions) and hepatocytes. The spleen tissue showed the presence of numerous hemosiderin-laden macrophages in both red pulp and white pulp (Figure 5).
4. DISCUSSIONS
Based on strictly regulated iron homeostasis, duodenal absorption of dietary iron (inorganic and heme irons) occurs at 1 - 3 mg/day, and almost the same amount is excreted, as absorbed [40-42]. Iron overload in hereditary hemochromatosis and β-thalassemia intermedia patients is mostly caused by the increased absorption of dietary iron [43,44]. Raja and colleagues demonstrated that duodenal iron absorption of BAL C and C53BL/6 mice was modestly increased due to enhanced mucosal uptake, and their iron loading in the spleen and liver was progressive with age [45]. Administration of 3,5,5- trimethylhexanoyl ferrocene (or carbonyl iron) diet into Wistar rats resulted in a substantial increase of liver ferritin iron [46], of the macrophages and sinusoidal endothelial cells [47]. Due to the fast and progressive iron loading that occurs with ferrocene, it is the most encouraging animal model for examining the efficacy and chelation site of tested iron chelators (e.g. CP20, CP94 and DFO) [48]. aand b-Thalassemic mice also showed defective iron absorption, leading to a sustained iron overload [49,50]. b-Knockout thalassemia mice were also fed with the ferrocene diet to successfully induce severe iron overload [23,24]. Our study presents the induction of iron overload in C53BL/6 wild type mice using ferroence-supplemented diet and a therapeutic approach of
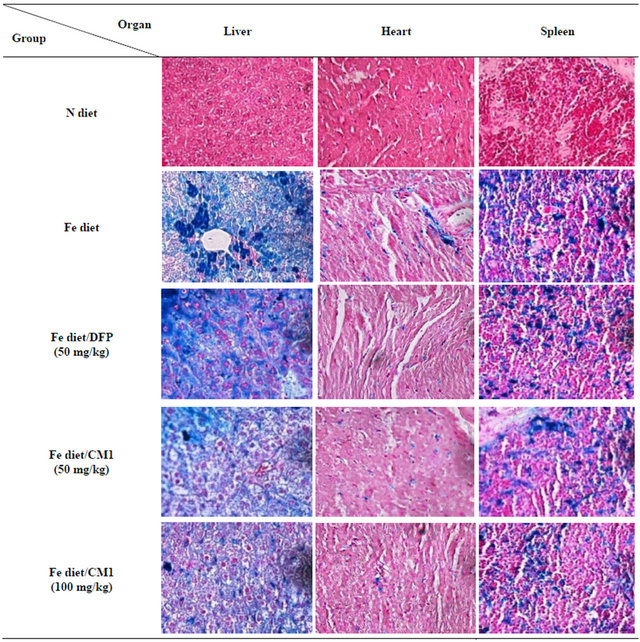
Figure 5. Perl’s Prussian blue staining of the liver, heart and spleen of WT mice fed with N diet, Fe diet, and Fe diet together with 50 mg/kg of DFP (50 mg/kg) and CM1 (50 and 100 mg/kg) for 3 months.
DFP and CM1 in the iron overload condition. After the compounds are readily absorbed into blood circulation and taken up into target organs, they would in turn chelate the toxic irons, such as plasma NTBI and LPI, red cell membrane non-heme iron and intracellular LIP and ferritin iron. Chelation of circulating NTBI can inhibit their rapid uptake into hepatocytes and cardiac myocytes [51-53], probably via Zip14 (Slc39a14) protein [54]. The CM1 chelator would be extended to evaluate its ironchelating activity in thalassemia mice and patients.
RBC nonheme iron was increased in iron overload mice according to an accumulation of unpaired globin chains in thalassemic RBC. Precipitation and denaturation of the globin chain can result in the liberation of both heme and nonheme irons [55,56]. Redistribution of cellular iron is harmful to the membrane, based on an iron-catalyzed generation of ROS [57]. We found that long-term feeding of the ferrocene diet to the mice increased the amount of normal RBC membrane nonheme iron, but not cytoplasmic ferritin iron. Interestingly, the two lipophilic chelators removed the pathologic deposits of chelatable iron from the RBC membrane effectively.
Persisting plasma NTBI is rapidly transported across the hepatocyte membrane via a specific pathway [58]. Excess iron potentially damages the vital organs, such as the liver, heart and endocrine glands due to an ironcatalyzed generation of free radicals via Harber-Weiss and Fenton reactions, leading to lipid peroxidation occurring in the organs, especially in the liver. Liver inflammation is perhaps attributed to excessive iron accumulation, increased lysosomal membrane fragility, ironinduced lipid peroxidation and the direct effect of iron on collagen biosynthesis [59]. Excess reactive oxygen species (ROS) can cause progressive tissue injuries and eventually cirrhosis or hepatocellular carcinoma in iron overload patients whose iron is sequestrated predominantly in ferritin or hemosiderin [60]. In this study, hepatic iron and hydroxyproline contents were increased dramatically in the Fe diet-fed mice. Most importantly, our new chelator CM1 acted as a promising iron chelator, while DFP reduced the increases of both the iron and hydroxyproline contents. Plasma MDA level was slightly increased and hepatic MDA was markedly increased in the Fe diet-fed mice when compared to the N diet-fed mice, while liver fibrosis is likely to occur. Our recent study has demonstrated that the CM1 reduced levels of LIP in mouse hepatocyte and HepG2 cell cultures, effectively [28]. A previous study reported that reactive iron and redox ROS in the iron overloaded liver can induce fibrosis [61]. Hepatic fibrosis is an irreversible process due to an accumulation in the collagen-rich tissue [62]. Collagen concentration in liver tissue is a good index for quantifying fibrosis, hydroxyproline is a standard marker amino acid found exclusively in collagen. Excessive iron as accumulation in the liver is one of the causes of liver diseases, such as hepatitis, hepatic fibrosis and hepatocellular carcinoma. In addition, it can cause cardiomyopathies (e.g. cardiac arrhythmia and heart failure) and endocrine gland dysfunctions (e.g. diabetes, growth retardation, hypogonardism and hypoparathyroidism) [63, 64].
Excess iron may result in oxidative stress and the alteration of myocardial function because of DNA damage by hydrogen peroxide through the Fenton reaction [58, 65]. Iron deposition in the heart cells is of outstanding interest [66-68]. Heart failure is the leading cause of death among hemosiderotic b-thalassemia patients [69- 71]. In pathological examinations, iron loading for 6 months did not show any morphological changes of the liver, spleen and heart in all groups. Increased amounts of hemosiderin iron were deposited in the spleen and liver tissues of the Fe diet-fed mice when compared to the N diet-fed mice. DFP and CM1chelations were able to remove the iron deposition in the spleens and livers when compared to the results with no chelation. Our two previous studies showed green tea catechins and curcuminoids reduced iron accumulation, based on Perl’s Prussian blue staining, in the liver of the iron-loaded thalassemic mice [23,24]. Chelation with DFO removed excessive liver iron and preserved hepatocellular structure [72].
5. CONCLUSION
In conclusion, CM1 could be an effective iron chelator as it reduced excess iron deposited on erythrocyte membrane, NTBI and LPI in the plasma compartment, and excess iron accumulation in the spleen, liver and heart. CM1 can prevent or inhibit the iron-induced lipid peroxidation extracellularly and intracellulary. The CM1 is of interest for further investigation in b-thalassemia animals and patients with iron overload.
6. ACKNOWLEDGEMENTS
This work was kindly supported by the Thailand Research Fund through a Senior Research Scholar, Professor Suthat Fucharoen, MD., and the Institute of Research and Development, Government Pharmaceutical Organization, Ministry of Public Health. We deeply thank the Thalassemia Research Center, Institute of Molecular Bioscience, Mahidol University Salaya Campus for supplying thalassemic mice.
REFERENCES
- Emerit, J., Beaumont, C. and Trivin, F. (2001) Iron metabolism, free radicals, and oxidative injury. Biomedicine & Pharmacotherapy, 55, 333-339. doi:10.1016/S0753-3322(01)00068-3
- Pootrakul, P., Breuer, W., Sametband, M., Sirankapracha, P., Hershko, C. and Cabantchik, Z.I. (2004) Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron-overloaded beta-thalassemia/HbE patients treated with an oral chelator. Blood, 104, 1504- 1510. doi:10.1182/blood-2004-02-0630
- Hershko, C. (2007) Iron loading and its clinical implications. American Journal of Hematology, 82, 1147-1148. doi:10.1002/ajh.21070
- Esposito, B. P., Breuer, W., Sirankapracha, P., Pootrakul, P., Hershko, C. and Cabantchik, Z.I. (2003) Labile plasma iron in iron overload: Redox activity and susceptibility to chelation. Blood, 102, 2670-2677. doi:10.1182/blood-2003-03-0807
- Hershko, C.M., Link, G.M., Konijn, A.M. and Cabantchik, Z.I. (2005) Iron chelation therapy. Current Hematology Reports, 4, 110-116.
- Taher, A.T., Musallam, K.M. and Inati, A. (2009) Iron overload: Consequences, assessment, and monitoring. Hemoglobin, 33, S46-57. doi:10.3109/03630260903346676
- Miskin, H., Yaniv, I., Berant, M., Hershko, C. and Tamary, H. (2003) Reversal of cardiac complications in thalassemia major by long-term intermittent daily intensive iron chelation. European Journal of Haematology, 70, 398-403. doi:10.1034/j.1600-0609.2003.00075.x
- Modell, B., Khan, M. and Darlison, M. (2000) Survival in beta-thalassaemia major in the UK: Data from the UK Thalassaemia Register. Lancet, 355, 2051-2052. doi:10.1016/S0140-6736(00)02357-6
- Lau, Y.L., Chan, L.C., Chan, Y.Y., Ha, S.Y., Yeung, C.Y., Waye, J.S. and Chui, D.H. (1997) Prevalence and genotypes of alphaand beta-thalassemia carriers in Hong Kong—Implications for population screening. The New England Journal of Medicine, 336, 1298-1301. doi:10.1056/NEJM199705013361805
- Economou, M., Printza, N., Teli, A., Tzimouli, V., Tsatra, I., Papachristou, F. and Athanassiou-Metaxa, M. (2010) Renal dysfunction in patients with beta-thalassemia major receiving iron chelation therapy either with deferoxamine and deferiprone or with deferasirox. Acta Haematologica, 123, 148-152. doi:10.1159/000287238
- Pippard, M.J. (1987) Iron overload and iron chelation therapy in thalassaemia and sickle cell haemoglobinopathies. Acta Haematologica, 78, 206-211. doi:10.1159/000205876
- Collins, A.F., Fassos, F.F., Stobie, S., Lewis, N., Shaw, D., Fry, M., Templeton, D.M., McClelland, R.A., Koren, G. and Olivieri, N.F. (1994) Iron-balance and dose-response studies of the oral iron chelator 1,2-dimethyl- 3-hydroxypyrid-4-one (L1) in iron-loaded patients with sickle cell disease. Blood, 83, 2329-2333.
- Freeman, A.P., Giles, R.W., Berdoukas, V.A., Talley, P.A. and Murray, I.P. (1989) Sustained normalization of cardiac function by chelation therapy in thalassaemia major. Clinical and Laboratory Haematology, 11, 299- 307. doi:10.1111/j.1365-2257.1989.tb00227.x
- Brittenham, G.M. (2003) Iron chelators and iron toxicity. Alcohol, 30, 151-158. doi:10.1016/S0741-8329(03)00101-0
- Peterson, C.M., Graziano, J.H., Grady, R.W., Jones, R.L., Vlassara, H.V., Canale, V.C., Miller, D.R. and Cerami, A. (1976) Chelation studies with 2,3-dihydroxybenzoic acid in patients with beta-thalassaemia major. British Journal of Haematology, 33, 477-485. doi:10.1111/j.1365-2141.1976.tb03566.x
- Peterson, C.M., Graziano, J.H., Grady, R.W., Jones, R.L., Markenson, A., Lavi, U., Canale, V., Gray, G.F., Cerami, A. and Miller, D.R. (1979) Chelation therapy in betathalassemia major: A one-year double blind study of 2,3- dihydroxybenzoic acid. Experimental Hematology, 7, 74- 80.
- Pippard, M.J., Jackson, M.J. and Modell, C.B. (1988) Subcutaneous diethylene triamine pentaacetic acid: Comparison with desferrioxamine in thalassemic patients with iron overload. Birth Defects Original Article Series, 23, 105-109.
- Grady, R.W., Salbe, A.D., Hilgartner, M.W. and Giardina, P.J. (1994) Results from a phase I clinical trial of HBED. Advances in Experimental Medicine and Biology, 356, 351-359. doi:10.1007/978-1-4615-2554-7_37
- Rai, B.L., Dekhordi, L.S., Khodr, H., Jin, Y., Liu, Z. and Hider, R.C. (1998) Synthesis, physicochemical properties, and evaluation of N-substituted-2-al kyl-3-hydroxy-4(1H)- pyridinones. Journal of Medicinal Chemistry, 41, 3347- 3359. doi:10.1021/jm9707784
- Srichairatanakool, S., Ounjaijean, S., Thephinlap, C., Khansuwan, U., Phisalpong, C. and Fucharoen, S. (2006) Iron-chelating and free-radical scavenging activities of microwave-processed green tea in iron overload. Hemoglobin, 30, 311-327. doi:10.1080/03630260600642666
- Thephinlap, C., Ounjaijean, S., Khansuwan, U., Fucharoen, S., Porter, J.B. and Srichairatanakool, S. (2007) Epigallocatechin-3-gallate and epicatechin-3-gallate from green tea decrease plasma non-transferrin bound iron and erythrocyte oxidative stress. Medicinal Chemistry, 3, 289- 296. doi:10.2174/157340607780620608
- Srichairatanakool, S., Thephinlap, C., Phisalaphong, C., Porter, J.B. and Fucharoen, S. (2007) Curcumin contributes to in vitro removal of non-transferrin bound iron by deferiprone and desferrioxamine in thalassemic plasma. Medicinal Chemistry, 3, 469-474. doi:10.2174/157340607781745447
- Thephinlap, C., Phisalaphong, C., Fucharoen, S., Porter, J.B. and Srichairatanakool, S. (2009) Efficacy of curcuminoids in alleviation of iron overload and lipid peroxidation in thalassemic mice. Medicinal Chemistry, 5, 474- 482. doi:10.2174/157340609789117912
- Saewong, T., Ounjaijean, S., Mundee, Y., Pattanapanyasat, K., Fucharoen, S., Porter, J.B. and Srichairatanakool, S. (2010) Effects of green tea on iron accumulation and oxidative stress in livers of iron-challenged thalassemic mice. Medicinal Chemistry, 6, 57-64. doi:10.2174/157340610791321479
- Bergeron, R.J., Wiegand, J. and Brittenham, G.M. (1999) HBED: The continuing development of a potential alternative to deferoxamine for iron-chelating therapy. Blood, 93, 370-375.
- Porter, J.B. (1996) Evaluation of new iron chelators for clinical use. Acta Haematologic, 95, 13-25. doi:10.1159/000203852
- Srichairatanakool, S., Pangjit, K. and Phisalaphong, C. (2009) Characterization and investigation of chelating activity of a novel iron chelator: 1-(N-acetyl-6-aminohexyl) -3-hydroxypyridin-4-one. Thailand Patent No. 0901000 799.
- Pangjit, K., Banjerdpongchai, R., Phisalaphong, C., Fucharoen, S. and Srichairatanakool, S. (2012) Efficacy of 1-(N-acetyl-6-aminohexyl)-3-hydroxypyridin-4-one (CM1) in treatment of iron-loaded hepatocyte cultures. Advances in Bioscience and Biotechnology, 3, 1060-1067. doi:10.4236/abb.2012.327129
- Jamsai, D., Zaibak, F., Khongnium, W., Vadolas, J., Voullaire, L., Fowler, K.J., Gazeas, S., Fucharoen, S., Williamson, R. and Ioannou, P.A. (2005) A humanized mouse model for a common beta0-thalassemia mutation. Genomics, 85, 453-461. doi:10.1016/j.ygeno.2004.11.016
- Jamsai, D., Zaibak, F., Vadolas, J., Voullaire, L., Fowler, K.J., Gazeas, S., Peters, H., Fucharoen, S., Williamson, R. and Ioannou, P.A. (2006) A humanized BAC transgenic/knockout mouse model for HbE/beta-thalassemia. Genomics, 88, 309-315. doi:10.1016/j.ygeno.2006.03.009
- Valerio Jr., L.G. and Petersen, D.R. (2000) Characterization of hepatic iron overload following dietary administration of dicyclopentadienyl iron (Ferrocene) to mice: Cellular, biochemical, and molecular aspects. Experimental and Molecular Pathology, 68, 1-12. doi:10.1006/exmp.1999.2278
- Ounjaijean, S., Thephinlap, C., Khansuwan, U., Phisalapong, C., Fucharoen, S., Porter, J.B. and Srichairatanakool, S. (2008) Effect of green tea on iron status and oxidative stress in iron-loaded rats. Medicinal Chemistry, 4, 365-370. doi:10.2174/157340608784872316
- Kuross, S.A., Rank, B.H. and Hebbel, R.P. (1988) Excess heme in sickle erythrocyte inside-out membranes: Possible role in thiol oxidation. Blood, 71, 876-882.
- Bradford, M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248-254. doi:10.1016/0003-2697(76)90527-3
- Singh, S., Hider, R.C. and Porter, J.B. (1990) A direct method for quantification of non-transferrin-bound iron. Analytical Biochemistry, 186, 320-323. doi:10.1016/0003-2697(90)90088-Q
- Rebouche, C.J., Wilcox, C.L. and Widness, J.A. (2004) Microanalysis of non-heme iron in animal tissues. Journal of Biochemical and Biophysical Methods, 58, 239- 251. doi:10.1016/j.jbbm.2003.11.003
- Grotto, D., Santa Maria, L.D., Boeira, S., Valentini, J., Charao, M.F., Moro, A.M., Nascimento, P.C., Pomblum, V.J. and Garcia, S.C. (2007) Rapid quantification of malondialdehyde in plasma by high performance liquid chromatography-visible detection. Journal of Pharmaceutical and Biomedical Analysis, 43, 619-624. doi:10.1016/j.jpba.2006.07.030
- Gomes, A.T., Bastos, C.G., Afonso, C.L., Medrado, B.F. and Andrade, Z.A. (2006) How variable are hydroxyproline determinations made in different samples of the same liver? Clinical Biochemistry, 39, 1160-1163. doi:10.1016/j.clinbiochem.2006.08.002
- Bancroft, J.S.A. (1982) Theory and practice of histological techniques. 2nd Edition, Churchill-Livingston, New York, 131-135.
- Aisen, P., Enns, C. and Wessling-Resnick, M. (2001) Chemistry and biology of eukaryotic iron metabolism. The International Journal of Biochemistry & Cell Biology, 33, 940-959. doi:10.1016/S1357-2725(01)00063-2
- Crichton, R.R. and Ward, R.J. (2003) An overview of iron metabolism: Molecular and cellular criteria for the selection of iron chelators. Current Medicinal Chemistry, 10, 997-1004. doi:10.2174/0929867033457566
- Conrad, M.E. and Umbreit, J.N. (2002) Pathways of iron absorption. Blood Cells, Molecules and Diseases, 29, 336- 355. doi:10.1006/bcmd.2002.0564
- Pippard, M.J., Callender, S.T., Warner, G.T. and Weatherall, D.J. (1979) Iron absorption and loading in beta-thalassaemia intermedia. Lancet, 2, 819-821. doi:10.1016/S0140-6736(79)92175-5
- McLaren, G.D., Nathanson, M.H., Jacobs, A., Trevett, D. and Thomson, W. (1991) Regulation of intestinal iron absorption and mucosal iron kinetics in hereditary hemochromatosis. Journal of Laboratory and Clinical Medicine, 117, 390-401.
- Raja, K.B., Simpson, R.J. and Peters, T.J. (1994) Intestinal iron absorption studies in mouse models of ironoverload. British Journal of Haematology, 86, 156-162. doi:10.1111/j.1365-2141.1994.tb03267.x
- Longueville, A. and Crichton, R.R. (1986) An animal model of iron overload and its application to study hepatic ferritin iron mobilization by chelators. Biochemical Pharmacology, 35, 3669-3678. doi:10.1016/0006-2952(86)90650-7
- Braumann, A., Wulfhekel, U., Dullmann, J. and Nielsen, P. (1992) Iron overload of the bone marrow by trimethylhexanoyl-ferrocene in rats. Acta Anatomica (Basel), 144, 285-295. doi:10.1159/000147318
- Florence, A., Ward, R.J., Peters, T.J. and Crichton, R.R. (1992) Studies of in vivo iron mobilization by chelators in the ferrocene-loaded rat. Biochemical Pharmacology, 44, 1023-1027. doi:10.1016/0006-2952(92)90363-N
- Van Wyck, D.B., Popp, R.A., Foxley, J., Witte, M.H., Witte, C.L. and Crosby, W.H. (1984) Spontaneous iron overload in alpha-thalassemic mice. Blood, 64, 263-266.
- Garrick, L.M., Strano-Paul, L.A., Hoke, J.E., KirdaniRyan, L.A., Alberico, R.A., Everett, M.M., Bannerman, R.M. and Garrick, M.D. (1989) Tissue iron deposition in untransfused beta-thalassemic mice. Experimental Hematology, 17, 423-428.
- Chua, A.C., Ingram, H.A., Raymond, K.N. and Baker, E. (2003) Multidentate pyridinones inhibit the metabolism of nontransferrin-bound iron by hepatocytes and hepatoma cells. European Journal of Biochemistry, 270, 1689- 1698. doi:10.1046/j.1432-1033.2003.03525.x
- Chua, A.C., Olynyk, J.K., Leedman, P.J. and Trinder, D. (2004) Nontransferrin-bound iron uptake by hepatocytes is increased in the Hfe knockout mouse model of hereditary hemochromatosis. Blood, 104, 1519-1525. doi:10.1182/blood-2003-11-3872
- Porter, J.B., Rafique, R., Srichairatanakool, S., Davis, B.A., Shah, F.T., Hair, T. and Evans, P. (2005) Recent insights into interactions of deferoxamine with cellular and plasma iron pools: Implications for clinical use. Annals of the New York Academy of Sciences, 1054, 155- 168. doi:10.1196/annals.1345.018
- Liuzzi, J.P., Aydemir, F., Nam, H., Knutson, M.D. and Cousins, R.J. (2006) Zip14 (Slc39a14) mediates nontransferrin-bound iron uptake into cells. Proceedings of the National Academy of Sciences of the United States of America, 103, 13612-13617. doi:10.1073/pnas.0606424103
- Shinar, E. and Rachmilewitz, E.A. (1990) Oxidative denaturation of red blood cells in thalassemia. Seminars in Hematology, 27, 70-82.
- Scott, M.D., van den Berg, J.J., Repka, T., Rouyer-Fessard, P., Hebbel, R.P., Beuzard, Y. and Lubin, B.H. (1993) Effect of excess alpha-hemoglobin chains on cellular and membrane oxidation in model beta-thalassemic erythrocytes. Journal of Clinical Investigation, 91, 1706-1712. doi:10.1172/JCI116380
- Browne, P.V., Shalev, O., Kuypers, F.A., Brugnara, C., Solovey, A., Mohandas, N., Schrier, S.L. and Hebbel, R.P. (1997) Removal of erythrocyte membrane iron in vivo ameliorates the pathobiology of murine thalassemia. Journal of Clinical Investigation, 100, 1459-1464. doi:10.1172/JCI119666
- Andrews, N.C. (1999) Disorders of iron metabolism. The New England Journal of Medicine, 341, 1986-1995. doi:10.1056/NEJM199912233412607
- Bacon, B.R. and Britton, R.S. (1989) Hepatic injury in chronic iron overload. Role of lipid peroxidation. Chemico-Biological Interactions, 70, 183-226. doi:10.1016/0009-2797(89)90045-8
- [61] Jensen, P.D. (2004) Evaluation of iron overload. British Journal of Haematology, 124, 697-711. doi:10.1111/j.1365-2141.2004.04838.x
- [62] Ramm, G.A. and Ruddell, R.G. (2005) Hepatotoxicity of iron overload: Mechanisms of iron-induced hepatic fibrogenesis. Seminars in Liver Disease, 25, 433-449. doi:10.1055/s-2005-923315
- [63] Popper, H. and Uenfriend, S. (1970) Hepatic fibrosis. Correlation of biochemical and morphologic investigations. The American Journal of Medicine, 49, 707-721. doi:10.1016/S0002-9343(70)80135-8
- [64] Kushner, J.P., Porter, J.P. and Olivieri, N.F. (2001) Secondary iron overload. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology. Education Program, 47-61.
- [65] Weatherall, D.J. (1998) Pathophysiology of thalassaemia. Baillière’s Clinical Haematology, 11, 127-146. doi:10.1016/S0950-3536(98)80072-3
- [66] Bartfay, W.J. and Bartfay, E. (2000) Iron-overload cardiomyopathy: evidence for a free radical—Mediated mechanism of injury and dysfunction in a murine model. Biological Research for Nursing, 2, 49-59. doi:10.1177/109980040000200106
- [67] Schellhammer, P.F., Engle, M.A. and Hagstrom, J.W. (1967) Histochemical studies of the myocardium and conduction system in acquired iron-storage disease. Circulation, 35, 631-637. doi:10.1161/01.CIR.35.4.631
- [68] Kremastinos, D.T., Toutouzas, P.K., Vyssoulis, G.P., Venetis, C.A. and Avgoustakis, D.G. (1984) Iron overload and left ventricular performance in beta thalassemia. Acta Cardiologica, 39, 29-40.
- [69] Sonakul, D., Pacharee, P. and Thakerngpol, K. (1988) Pathologic findings in 76 autopsy cases of thalassemia. Birth Defects Original Article Series, 23, 157-176.
- [70] Bannerman, R.M., Keusch, G., Kreimer-Birnbaum, M., Vance, V.K. and Vaughan, S. (1967) Thalassemia intermedia, with iron overload, cardiac failure, diabetes mellitus, hypopituitarism and porphyrinuria. The American Journal of Medicine, 42, 476-486. doi:10.1016/0002-9343(67)90276-8
- [71] Aldouri, M.A., Wonke, B., Hoffbrand, A.V., Flynn, D.M., Ward, S.E., Agnew, J.E. and Hilson, A.J. (1990) High incidence of cardiomyopathy in beta-thalassaemia patients receiving regular transfusion and iron chelation: reversal by intensified chelation. Acta Haematologica, 84, 113- 117. doi:10.1159/000205046
- [72] Kremastinos, D.T., Tiniakos, G., Theodorakis, G.N., Katritsis, D.G. and Toutouzas, P.K. (1995) Myocarditis in beta-thalassemia major. A cause of heart failure. Circulation, 91, 66-71. doi:10.1161/01.CIR.91.1.66
- [73] Cohen, A., Martin, M. and Schwartz, E. (1984) Depletion of excessive liver iron stores with desferrioxamine. British Journal of Haematology, 58, 369-373. doi:10.1111/j.1365-2141.1984.tb06096.x
NOTES
*Corresponding author.