Advances in Bioscience and Biotechnology
Vol.2 No.3(2011), Article ID:5040,6 pages DOI:10.4236/abb.2011.23018
Phylogeny derived from homodimeric endonucleasecorrelates with its pre-RNA substrates
![]()
1Indian Association for the Cultivation of Science, Calcutta, India;
2Gyanxet, Calcutta, India.
E-mail: j.chakrabarti@gyanxet.com
Received 23 February 2011; revised 11 April 2011; accepted 17 April 2011.
Keywords: Phylogeny; Maximum Likelihood Method; Homodimeric Endonuclease; Noncoding RNA; Intron; Methanogen
ABSTRACT
Amongst endonuclease, the homodimeric variety is found in many prokaryotes for processing of the introns out from pre-RNAs. But as the variety and the complexity of introns rise with evolution, do the homodimeric endonuclease adapt to the changes? The correlations between evolving pre-RNAs and adapting homodimeric endonuclease in lower prokaryotes is investigated in this paper. First, we construct and observe the appearance of a long branch in the phylogeny based on homodimeric endonuclease. To appreciate the finer aspects of accelerating evolution near this long branch, we delve deeper into the pre- -RNA substrates of the endonuclease. Computational evidence of an as-yet-unreported noncoding RNA gene then emerges from this study. The capabilities of homodimeric endonuclease and the complexities of its pre-RNA substrates appear to evolve in steps together.
1. INTRODUCTION
In recent years computational approaches to annotation and investigation of noncoding RNAs have become widespread. The subject of noncoding RNAs has grown for more than half a century. It began with ribosomal RNAs and transfer RNAs, but a whole host of newer types have come up in the last couple of decades. Through the years many different aspects of the subject have been extensively studied, and the links between them analysed and established. RNA genes, especially the ribosomal ones, have been used extensively for study of phylogeny and gene-evolution [1]. The secondary structures of noncoding RNAs are complex and unique. These structural complexities and uniqueness make noncoding RNAs particularly accessible to computation. Not surprisingly, computational predictions about them have generally high accuracy [http://lowelab.ucsc.edu/ tRNAscan-SE/ and http://130.235.46.10/ARAGORN/]. The accuracy of the predictions improves many fold when the subtle links between the diverse pathways are studied and correlated [2].
In this paper we study phylogeny of lower prokaryotes based on homodimeric endonuclease. The reason for choosing homodimeric endonuclease is its close interaction with noncoding pre-RNAs; it processes the introns out of pre-RNAs. Our main interest is in methanogens because methanogens have shown promising new features amongst its tRNAs. For one, there are absolutely new tRNA genes that decode UAG stop codon [3]. For another, many of the more familiar tRNAs, found abundantly in other genomes, appear at first sight to be missing in some of the methanogens [4]. It is the search for these apparently missing tRNAs that was the subject of one of our recent investigations [5,6]. We expand our search for missing tRNAs in this paper. The homodimeric endonuclease acts on the introns of pre RNAs of methanogens [7]. It is present and active in a set of other related organisms of the euryarchaeal family. We begin with phylogeny of the members of the euryarchaeal group that have homodimeric endonuclease to look for clusters and groupings that will help us in going after some of the apparently missing tRNAs [8].
In deciphering the evolutionary history of archaea, the phylogeny was mainly based on 16s small ribosomal RNA sequence [8,9]. The 16s rRNA based tree suggests two main phyla, the euryarchaeota and crenarchaeota, their specific order of emergence, and mutual relationship among their lineages. The other phylogenetic approach, based on “whole genome”, does not recover the monophyly of euryarchaeota as halobacteriales are at the base of the archaeal tree [8]. Phylogenetics based on whole genome analysis is somewhat biased by the abundance of lateral gene transfer events that have occurred between archaea and bacteria and between the archaeal lineages [10-14]. The problem of lateral gene transfer was bypassed in archaeal tree based on only the concatenated dataset of ribosomal proteins. The concept of lateral gene transfer events between archaea and bacteria and its impact on phylogeny has recently undergone major scrutiny [15].
Among the methanogenic euryarchaea there are five phylogenetically divergent orders: methanobacteriales, methanococcales, methanomicrobiales, methanosarcinales and methanopyrales [16]. There appears to be two monophyly groups of methanogens, namely, methanococcales, methanomicrobiales and methanopyrales in class I; and methanomicrobiales, methanosarcinales belonging to class II. These are separated by non-methanogenic lineages, namely, thermoplasmatales, archaeoglobales and halobacteriales. An alternative hypothesis is that all common archaeal ancestors may have been methanogens, but that methanogenesis was lost in crenarchaea, and independently in all non-methanogenic euryarchaeal lineages. Clearly, the origin and evolution of methanogens is an important issue that requires new analyses. With this in view, we present here phylogenetic analyses with the sequences of homodimeric endonuclease from euryarchaeal lineages.
2. MATERIALS AND METHODS
For analysis of phylogeny the software MEGA was used. Four different phylogenetic trees were investigated based on 1) maximum likehood method, 2) neighbor joining, 3) upgma and 4) minimum evolution. We required a high level of congruence between the trees from these four different methods.
To check if tRNAser(CGA) also lies hidden in the genome, the standard and highly successful tRNA gene finding algorithms http://lowelab.ucsc.edu/tRNAscanSE/ and http://130.235.46.10/ARAGORN/ and databases were used. These algorithms also locate with high precision if the gene appears with one intron. The possibility that the gene may have more than one intron is investigated using the following algorithm. Introns in archaea have been found to occur at a few positions in tRNA. The length of an intron is bounded above by 200. Taking the consensus archaeal tRNAser(CGA) we cut them into pieces at the probable intron locations. These pieces of tRNAser(CGA) were then homology searched through the genome of Methanosaeta thermophila by varying the intervening intron length between 6 and 200. We did not assume anything about the nucleotide composition of the intron sequences.
3. RESULTS AND DISCUSSION
3.1. Phylogeny of Methanogens Based on Endonuclease
The trees resulting from endonuclease dataset are in Figure 1(a) and Figure 1(b). Figure 1(a) is based on
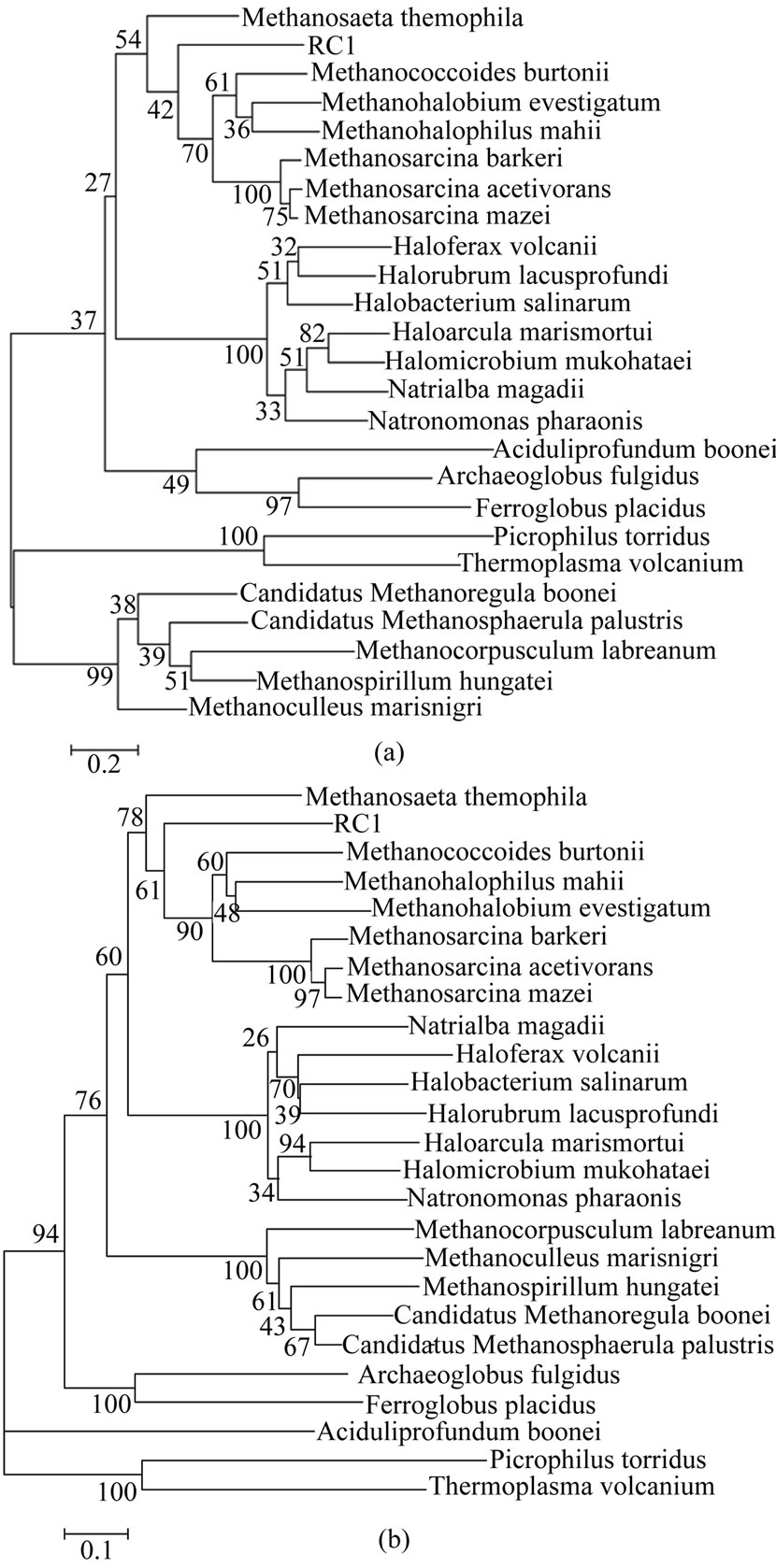
Figure 1. (a): Phylogenetic analysis (Bootstrap) of 25 Euryarchaeal species considering homodimeric endonuclease. The tree is drawn according to Maximum Likelihood model and the calculations of the best tree and the branch lengths were conducted using the program MEGA 5; (b): Phylogenetic analysis (Bootstrap) of 25 Euryarchaeal species considering homodimeric endonuclease. The tree is drawn according to neighbor-joining (NJ) and the calculations of the best tree and the branch lengths were conducted using the program MEGA 5.
the maximum likelihood method; Figure (1b) uses neighbor-joining. We have cross-checked the tree using two other methods, namely, upgma and minimum evolution. The same topologies were recovered with all the four methods used for phylogenetics reconstruction, but with little variation in bootstrap values [BP]. The bootstrap values quoted below are for the neighbor-joining scheme. The endonuclease tree presented interesting similarities such as grouping of methanosarcinales and halobacteriales [60% BP] with the latter order forming a well sustained cluster [100% BP]. However endonuclease tree strongly supported the sister group of methanomicrobiales and methanosarcinales clade [76% BP], but thermophiles, halophiles and thermoacidophiles grouped, albeit with weak confidence. Moreover the endonuclease tree recovered a robust monophyly [100% BP] of three methanogens [M. barkeri, M. mazei and M. acetivorans] while the other taxa among methanosarcinales were paraphyletic with a moderate support. The apparent incongruence between methanosarcinaceae and the rest of methanosarcinales concerning the position of Methanosaeta thermophila, RC1 and M. burtonii most probably reflect a strong phylogenetic signal rather than long branch attraction [17,18]. This phylogeny indicates that M. thermophila is positioned at the base of the endonuclease tree. The tree showed that M. thermophila and RC1 as the first and second offshoot [78% BP, 61% Bp] just before methanosarcinales, whereas the mesophilic methanogen M. burtonii is grouped monophyletically with the rest of the methanococcales and methanosarcinales [90% BP]. Interestingly, M. thermophila displayed a very long branch in the endonuclease tree [paraphyletic], suggesting an acceleration of evolution of M. thermophila endonuclease protein.
3.2. Search for Missing tRNAs: Lessons from Endonuclease Phylogeny
To appreciate the acceleration of evolution of homodimeric endonuclease near M. thermophila, we delve deeper into its pre-RNA substrates. The phylogenetic trees delineate quite clearly the ‘neighbourhood’ of each element. Yet, when we look at the spectrum of RNAs, there are clear indications of “anomalies”. For instance, in NCBI the tRNAser[CGA] gene, which is present in all its closest neighbours, appears to be absent in M. thermophila. In its neighbourhood lie RC1 and M. burtonii; both have tRNAser[CGA]. Interestingly, in both RC1 and M. burtonii the corresponding tRNAser[CGA] genes have introns that are cleaved by homodimeric endonuclease. It is puzzling, therefore, that phylogeny based on homodimeric endonuclease places M. thermophila near RC1 and M. burtonii , and yet the substrate of the endonuclease is so prominently absent in M. thermophila. We are, therefore, prompted to search for the missing tRNAser [CGA] in M. thermophila.
The search for missing/new tRNAs has to satisfy several known constraints. Archaeal tRNAs, especially the ones that are missing, are likely to have canonical [i.e., between 37 and 38] intron and/or noncanonical [at any position other than 37/38] introns. The boundary features between exons and introns require detailed attention. The exon-intron boundaries form a folded motif generically termed Bulge-Helix-Bulge [BHB]. This structure consists of two 3 nt [nucleotide] bulges on opposite strands, separated by a 4 bp central helix -- the so-called “3-4-3 motif”. The 5′ half of central helix is in exonic region; complementary 3′ half is intronic. This generic BHB, or more precisely hBHBh motif, has been observed for both canonical and noncanonical introns. For a few noncanonical introns, however, the canonical hBHBh/ motifs are not always observed. Instead a simplified hBH or HBh/ motif, including two helices [h and H or H and h/] and one bulge can be isolated [7,19-21]. Moreover in the central helix [H] of the exon-intron boundary motif for canonical introns, a few miss-pairings, such as A: C, A: G, C: U and U: U, have been observed. One thing that appears to hold with reasonable certainty is that for all types of intron, canonical or noncanonical, and for every possible BHB motif, hBHBh/ or hBH or HBh′, the cleaving sites are always located two bases away from the central helix. The cleaving of introns is catalysed by endonucleases. Recent investigations have found that in archaea all 3 types of intron cleaving endonucleases–homodimer [a2], homotetramer [a4] and heterotetramer [a2b2]-- can interact and splice the 3-4-3 structural substrate [21]. Crenarchaeal and nanoarchaeal endonucleases are heterotetrameric. Eurarchaeal endonucleases are usually homotetrameric or homodimeric, but with exceptions. M. kandleri, an euryarchaea, for instance, has heterotetrameric endonuclease. Most noncanonical introns are in crenarchaea, which have heterotetrameric endonuclease. There are a few noncanonical introns observed in euryarchaeota that have homotetrameric enzymes. However, tRNAs with noncanonical introns were not reported in euryarchaeota with homodimeric enzymes before 2007. Evidence in RC1 genome, which encodes homodimeric endonuclease, suggested the presence of noncanonical introns. Subsequently it was noticed that a similar noncanonical intron also exists in the genome of M. burtonii that also encodes homodimeric endonuclease. It was hypothesized that all three forms of endonuclease can cleave the canonical BHB, but the relaxed motifs [hBH or HBh′ or BHL] can be cleaved only by homodimer and heterotetrameric forms [21; see also http://splits.iab.keio.ac.jp/splitsdb/].
Taking a cue from the results of phylogeny based on homodimeric endonuclease, especially the close connection linking M. thermophila with RC1 and M. burtonii , we look for the missing tRNAser[CGA] gene in M. thermophila assuming it occurs in some novel way. The genome encodes homodimeric tRNA-endonuclease. Taking the consensus archaeal tRNAser[CGA] we cut the genome of M. thermophila into pieces [to take care of the introns] at all probable intron locations. These pieces of tRNAser[CGA] are then homology searched through the genome of M. thermophila. We varied the intervening intron length between 6 and 200. We did not assume anything about the nucleotide composition of the intron sequences. For M. thermophila the above procedure did identify a putative tRNAser[CGA] gene. Even though the endonuclease is homodimeric, this putative tRNAser [CGA] has two noncanonical introns, one 33 bases long in D-arm between 21 and 22; another of length 30 bases located in T-loop between 59 and 60. After the introns are removed the cloverleaf structure of tRNAser[CGA] is recovered. All the conserved bases and base-pairs of
archaeal tRNAser are precisely in place. Notable amongst them are G73, G26 and U44, the unique identity elements of tRNAser recognized by seryl tRNA synthetase [6]. Equally noteworthy are the remarkably familiar structural motifs at the exon-noncanonical intron boundaries, exonic helix [h] - bulge [B] - central helix [H], i.e. hBH, with absolutely no mismatches in the central helix. Based on this evidence we hypothesize that the sequence lying in the range 1333687-1333842 in M. thermophila genome encodes tRNASer [CGA], and that both the noncanonical introns are cleaved by the homodimeric endonuclease (Figure 2).
4. CONCLUSIONS
The evolving complexity of genomes involves subtle, yet unmistakable, correlations connecting the various encoded components. First, there are the protein coding parts. But, even within it are the recently discovered hidden invariant correlating patterns [22]. Then there are the effects of gene transfers, and mutations in prokaryotes
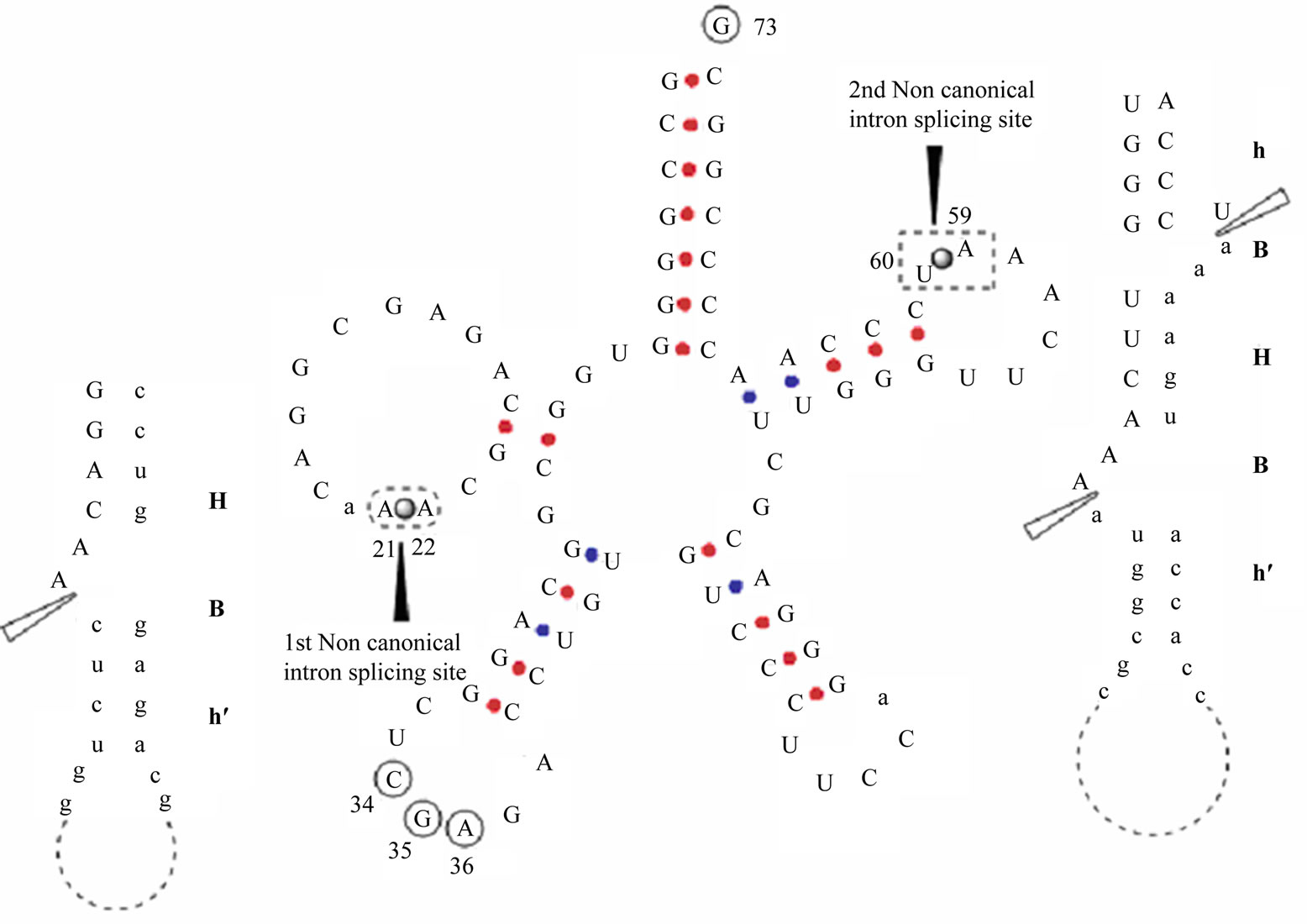
Figure 2. Shows the cloverleaf tRNASer (CGA) of Methanosaeta thermophila. It has two introns at 21 > 22 and 59 > 60. A perfect hBHBh′ pattern is present at the exon-intron boundary of 2nd non canonical intron splicing site i.e. at 59 > 60, whereas a more relaxed HBh/ motif has been recorded at the first splicing site i.e. at 21 > 22. The conserved elements (anticodon bases and the discriminator base) of this tRNA are marked in solid circles. The solid ball and the black solid arrow head marks the two non canonical splice site, one at 21 < 22 and other at 59 < 60. The hollow black bordered arrow head marks the cleavage site of endonuclease. In BHB motif, the nucleotide base shown in capital letters are part of matured tRNA i.e. exon whereas those represented in small letters falls in intronic region. The small lettered nucleotide base within the matured tRNA body are additional bases (may or may not be present) and are not part of intron.
due to their interactions with phages and other hosts [23]. To this one has to add the noncoding RNAs and their decoding and regulatory features, together forming the network of complexity. The evolution of one is delicately balanced and correlated to another in this network [21,24].
The phylogeny based on homodimeric endonuclease is new. Since the methanogens in euryarchaeal domain all have this enzyme, a finer characterization and classification emerge. While the trees derived are all in reasonable congruence with the classification based on 16s rRNA, the grouping of RC1 with M. thermophila in the neighbourhood of M. burtonii is noteworthy. Equally noteworthy is the long branch, indicative of paraphyly, for M. thermophila . We interpret it as a signal of an acceleration of evolution of endonuclease. Interestingly, while RC1 and M. burtonii both have tRNAser[CGA], in M. thermophila it remains unreported. Since pre tRNAser[CGA] in RC1 and M. burtonii are substrates of homodimeric endonuclease, its complete absence in M. thermophila is inexplicable. We interpret it as a signal of the accelerating capabilities of the endonuclease to search anew for tRNAser[CGA] in M. thermophila.
tRNAser[CGA] is characterized by a large number of unique features. First, its secondary cloverleaf structure is so intricate. And on this coverleaf are special identity elements at very well defined locations [6]. These make the search well tailored for precision computation. We hypothesize that the sequence lying in the range 1333687-1333842 in M. thermophila genome encodes tRNASer [CGA]. It meets all the features of this tRNA from other methanogens. This case, however, is somewhat new for homodimeric endonuclease in that the pretRNA has two noncanonical introns. The secondary structural motifs at the exon-intron boundaries are of the types found and experimentally established earlier, and the central helices are perfectly matched. The hypothesis, therefore, is predicated on the premise that the capabilities of the endonuclease grow in step with the evolving intronic complexity of its pre RNA substrate.
REFERENCES
- Chen, K., Eargle, J., Sarkar, K., Gruebele, M. and Luthey-Schulten, Z. (2010) Functional role of ribosomal signatures. Biophysical Journal, 99, 3930-3940. doi:10.1016/j.bpj.2010.09.062
- Sachidanandam, R. (2005) RNAi as a bioinformatic consumer. Briefings in Bioinformatics, 6, 146-162. doi:10.1093/bib/6.2.146
- Srinivasan, G., James, C. and Krzycki, J. (2002) Pyrrolysine encoded by UAG in archaea: Charging of a UAGdecoding specialized tRNA. Science, 296, 1459-1462. doi:10.1126/science.1069588
- Kohrer, C., Srinivasan, G., Mandal, D., Mallick, B., Ghosh, Z., Chakrabarti, J. and RajBhandary, U. (2008) Identification and characterization of a tRNA decoding the rare AUA codon in Haloarcula marismortui. RNA, 14, 1-10.
- Das, S., Mitra, S., Sahoo, S. and Chakrabarti, J. (2011) Novel hybrid encodes both continuous and split tRNA genes. Journal of Biomolecular Structure and Dynamics, 28, 827-831.
- Mallick, B., Chakrabarti, J., Sahoo, S., Ghosh, Z. and Das, S. (2005) Identity elements of archaeal tRNA. DNA Research, 12, 235-246. doi:10.1093/dnares/dsi008
- Abelson, J., Trotta, C.R. and Li, H. (1998) tRNA splicing. Journal of Biological Chemistry, 273, 12685-12688. doi:10.1074/jbc.273.21.12685
- Wolf, Y.I., Rogozin, I.B., Grishin, N.V. and Koonin, E.V. (2002) Genome trees and the tree of life. Trends in Genetics, 18, 472-479. doi:10.1016/S0168-9525(02)02744-0
- Woese, C.R. (1987) Bacterial evolution. Microbiology and Molecular Biology Reviews, 51, 221-271.
- Aravind, L., Tatusov, R.L., Wolf, Y.I., Walker, D.R. and Koonin, E.V. (1998) Evidence for massive gene exchange between archaeal and bacterial hyperthermophiles. Trends in Genetics, 14, 442-444. doi:10.1016/S0168-9525(98)01553-4
- Nelson, K.E., Clayton, R.A., Gill, S.R., et al. (1999) Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritime. Nature, 399, 323-329. doi:10.1038/20601
- Ochman, H., Lawrence, J.G. and Groisman, E.A. (2000) Lateral gene transfer and the nature of bacterial innovation. Nature, 405, 299-304. doi:10.1038/35012500
- Gogarten, J.P., Doolittle, W.F. and Lawrence, J.G. (2002) Prokaryotic evolution in light of gene transfer. Molecular Biology and Evolution, 19, 2226-2238.
- Jain, R., Rivera, M.C., Moore, J.E. and Lake, J.A. (2002) Horizontal gene transfer in microbial genome evolution. Theoretical Population Biology, 61, 489-495. doi:10.1006/tpbi.2002.1596
- Zhaxybayeva, O., Swithers, K.S., Lapierre, P., Fournier, G.P., Bickhart, D.M., DeBoy, R.T., Nelson, K.E., Nesbo, C. L., Ford Doolittle, W.J., Gogarten, P. and Noll, K.M. (2009) On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proceedings of the National Academy of Sciences, 106, 5865-5870. doi:10.1073/pnas.0901260106
- Garrity, G. (2001) Bergey’s manual of systematic bacteriology. Springer-Verlag, Berlin.
- Smith, D.R., Doucette-Stamm, L.A., Deloughery, C., et al. (1997) Complete genome sequence of Methanobacterium thermoautotrophicum DH, functional analysis and comparative genomics. Journal of Bacteriology, 179, 7135-7155.
- Slesare, A.I., Mezhevaya, K.V., Makarova, K.S., et al. (2002) The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proceedings of the National Academy of Sciences, 99, 4644-4649. doi:10.1073/pnas.032671499
- Kjems, J., Leffers, H., Olsen,T. and Garrett, R.A. (1989) A unique tRNA intron in the variable loop of the extreme thermophile Thermophilum pendens and its possible evolutionary implications. Journal of Biological Chemistry, 264, 17834-17837.
- Li, H. and Abelson, J. (2000) Crystal structure of a dimeric archaeal splicing Endonuclease. Journal of Molecular Biology, 302, 639-648. doi:10.1006/jmbi.2000.3941
- Tocchini-Valentini, G.D., Fruscoloni, P. and TocchiniValentini, G.P. (2005) Coevolution of tRNA intron motifs and tRNA endonuclease architecture in Archaea. Proceedings of the National Academy of Sciences, 102, 15418-15422.
- Mittal, A. and Jayaram, B. (2011) Backbones of folded proteins reveal novel invariant amino acid neighborhoods. Journal of Biomolecular Structure and Dynamics, 28, 443-454.
- Ghosh, Z., Mallick, B. and Chakrabarti, J. (2009) Cellular versus microRNAs in host-virus interaction. Nucleic Acids Research, 37, 1035-1048. doi:10.1093/nar/gkn1004
- Giulio, M.D. (1999) The non-monophyletic origin of the tRNA molecule. Journal of Theoretical Biology, 197, 403-414. doi:10.1006/jtbi.1998.0882