Journal of Biophysical Chemistry
Vol.06 No.04(2015), Article ID:60882,27 pages
10.4236/jbpc.2015.64010
Molecular Modeling, Docking and ADMET of Dimethylthiohydantoin Derivatives for Prostate Cancer Treatment
Khaled Lotfy1,2
1Physics Department, Faculty of Science, Sohag University, Sohag, Egypt
2University College of Taiyma, Tabuk University, Tabuk, Saudi Arabia

Copyright © 2015 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 4 September 2015; accepted 1 November 2015; published 4 November 2015

ABSTRACT
In silico technique was applied to screen potential of 16 compounds of 5,5-dimethylthiohydantoin derivatives as androgen antagonist. The 3D structure of the protein was obtained from PDB database. Docking analysis of the compounds was performed using hex docking. Molecular modeling analysis exhibits relatively low LUMO-HOMO energy gap of the studied molecules, indicating that it would be kinetically stable. None of the compounds violated Lipinski’s parameters, making them potentially promising agents for biological activities. The title compounds exhibited the lowest docking energy of protein-ligand complex. Finally, the results indicate that these compounds are potentially as an androgen antagonist, and expected to be effective in prostate cancer treatment.
Keywords:
Androgen Receptor, Prostate Cancer, Molecular Modeling, Molecular Docking, ADMET

1. Introduction
Cancer can be characterized by the arrest of cell differentiation, the inhibition of apoptosis and the accelerated proliferation of cloned cells. The understanding of the mechanisms of cell-death execution and the role that they play in different diseases opens new therapeutic strategies [1] -[4] . Prostate cancer is the most frequently diagnosed malignancy in men in Western countries [5] . The prostate is an androgen-dependent organ; androgen hormones and their executor, the androgen receptor (AR), are central drivers of prostate cancer development and progression [6] - [11] . The androgen receptor (AR), located on Xq11-12, is a 110 kDa nuclear receptor that, upon activation by androgens, mediates transcription of target genes that modulate growth and differentiation of prostate epithelial cells AR signaling is crucial for the development and maintenance of male reproductive organs, including the prostate gland, as genetic males harboring loss of function AR mutations and mice engineered with AR defects do not develop prostates or prostate cancer [12] [13] . AR plays a major role in the development and maintenance of male sex characteristics by executing the biological functions of androgens. AR also has an important role in prostate cell proliferation and differentiation; and during androgen responsive prostate cancer [14] - [16] .
However, the AR is a cytoplasmic protein and is a member of the steroid/thyroid hormone receptor superfamily [17] [18] . Upon androgen binding to its receptor, the AR migrates into the nucleus, binds to specific DNA sequences called androgen response elements and modulates the transcription of target genes [17] . For this reason, the androgen receptor is a key molecular target in the etiology and progression of prostate cancer [19] - [23] . So that, manipulations of AR function of antiandrogenic drugs are the primary mode of treatment of the malignancy [14] [15] .
AR antagonists are used as a single agent (monotherapy) or in combination with castration. The latter use, referred to as “combined androgen blockade therapy”, shows significant effects by blocking adrenal androgen signals as well as suppressing the transient testosterone increase induced by GnRH analogs [24] - [29] . AR antagonists used clinically for prostate cancer include steroidal (cyproterone acetate) and nonsteroidal antagonists (flutamide, nilutamide, and bicalutamide), which all function as competitive inhibitors of AR binding to endogenous androgens testosterone and dihydrotestosterone [30] .
Although, the antiandrogens exhibit good efficacy in many cases and comprise an important part of effective therapeutics [31] - [34] . But, a considerable problem with these antiandrogens is that recurrence occurs after a short period of response [35] . Hydroxyflutamide and bicalutamide have partial agonist activities at high concentrations in vitro [36] , which may contribute to recurrence.
The thiohydantoin derivative with a carboxyl terminal side chain showed that pure antagonistic activities in vitro and oral AR antagonistic activity in vivo [37] . In this work, the molecular modeling, molecular docking and ADMET of 16 compounds of 5,5-dimethylthiohydantoin derivatives, which synthesized by [38] were conducted for the treatment of prostate cancer.
2. Experimental
2.1. Materials
The available data sets of 16 compounds were obtained from the literature [38] . The IUPAC names and structure of 5,5-dimethylthiohydantoin derivatives and are listed in Table 1 and Table 2. On the other hand, the substitute which occurs in all compounds can be seen in these tables.
2.2. Molecular Modeling
All the calculations are performed by using Gauss view 5.0 molecular visualization program and Gaussian 09 program package on the personal computer [39] . The molecular structure of the title compounds in the ground state (in a vacuum) was optimized using Semi-empirical method AM1. The frontier molecular orbital surfaces are visualized by Gauss View Molecular Visualization program [40] . All these computations were carried out for the ground states of these molecules as a single state. The dipole moment (in Debye) of the molecules and heat of formation (HOF) was directly extracted from the Gaussian09 output file. The HOMO and LUMO values were taken from the output of the Gaussian 09W calculation as molecular orbital coefficients (in a.u.).
2.3. Molecular Descriptors
Computation of molecular descriptors such as partition coefficient, topological surface area and a number of hydrogen bond acceptors & donors, was carried out using Mol inspiration online tool [41] . Using these parameters the compounds were checked for their compliance with the Lipinski rule of five [42] . Drug likeness score was computed using molsoft website [43] . The drug-likeness scores were analyzed by comparing with the earlier reports [44] . Bioactivity score predictions were also done using Mol inspiration on-line tool. Toxicity Risk Assessment, cLogP Prediction, Solubility Prediction, Molecular Weights, Drug-Likeness, Prediction and Overall Drug-Likeness Score computed using Osiris software available on-line.

Table 1. The IUPAC name of compounds from 1 to 10.
Table 2. The IUPAC name of compounds from 11 to 16.
2.4. Molecular Dynamics Simulation
In the present study, Bioinformatics tools are used, biological databases like PDB (Protein Data Bank) and the software’s like Hex [45] . The Protein Data Bank (PDB) is the single worldwide archive of structural data of biological macromolecules, established in Brookhaven National Laboratories [46] . It contains structural information of the macromolecules determined by X-ray crystallographic and NMR methods. The structure of the androgen receptor (Figure 1), which is an essential target for the hydroxyflutamide drug was retrieved from PDB (2AX6). On the other hand, most favored regions of the target structure were evaluated through Ramchandran plot analysis via PROCHECK [47] from SAVES Server (http://services.mbi.ucla.edu/SAVES/) to investigate the quality of the target structure.
2.4.1. Activity Prediction
The activity prediction of all molecules has been achieved by PASS server and compared with hydroxyflutamide. PASS Online predicts over 3500 kinds of biological activity, including pharmacological effects, mechanisms of action, toxic and adverse effects, interaction with metabolic enzymes and transporters, influence on gene expression, etc. Prediction is based on the analysis of structure activity-relationships for more than 250,000 biologically active substances including drugs, drug-candidates, leads and toxic compounds. The structural formula only is necessary to obtain the predicted biological activity profile for any compound.
2.4.2. HEX Docking
Hex is an interactive molecular graphics program for calculating and displaying feasible docking modes of pairs of protein and DNA molecules. Hex can also calculate protein-ligand docking; assuming the ligand is rigid, and it can superpose pairs of molecules using only knowledge of their 3D shapes [48] . It uses spherical polar fourier (SPF) correlations to accelerate the calculations and its one of the few docking programs, which have been built in graphics to view the result [49] .
The docking analysis of hydroxyflutamide with androgen receptor was carried by HEX docking software. Docking allows the scientist to virtually screen a database of compounds and predict the strongest binders based on various scoring functions. It explores ways in which two molecules, such as drugs and an enzyme; Human estrogen receptor fit together and docks to each other well, like pieces of a three-dimensional jigsaw puzzle. The molecules binding to a receptor, inhibit its function, and thus act as a drug.
The parameters used in the docking process were

Figure 1. The structure of androgen receptor (2AX6).
・ Correlation type―Shape only
・ FFT Mode―3D fast lite
・ Grid Dimension―0.6
・ Receptor range―180
・ Ligand Range―180
・ Twist range―360
・ Distance Range―40
All water molecules and ligands were removed from the proteins for docking studies.
The hydroxyflutamide and 5,5-dimethylthiohydantoin derivatives were docked with the receptor using the above parameters. Visualization of the docked pose had been done using a PyMol molecular graphics program [50] .
2.4.3. LigPlot+ v.1.4.5
Two-dimensional representations of the best docking pose for selected 5,5-dimethylthiohydantoin derivatives inside target enzyme were generated using LigPlot+ [51] . It is a computer program which generates the schematic 2-D image of the docked protein-ligand complexes. The 3-D structure of the docked complex is input as PDB file and the software produces their interacting residues and bonds. In the present study, LigPlot+ was used to identify interacting residues as well as the interacting bonds between 2AX6 and the docked inhibitors.
2.4.4. Computed Atlas of Surface Topography of Proteins (CASTp)
Binding sites and active sites of proteins and DNAs are often associated with structural pockets and cavities. CASTp server uses the weighted Delaunay triangulation and the alpha complex for shape measurements. It provides identification and measurements of surface accessible pockets as well as interior inaccessible cavities, for proteins and other molecules. It measures analytically the area and volume of each pocket and cavity, both in the solvent accessible surface (SA, Richards’ surface) and molecular surface (MS, Connolly’s surface). It also measures the number of the mouth openings, area of the openings, and circumference of mouth, lips, in both SA and MS surfaces for each pocket [52] .
2.4.5. Prediction of ADMET Properties
The ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) properties of the target compounds were calculated using some web-based applications. BBB (Blood-Brain Barrier) penetration, HIA (Human Intestinal Absorption), Caco-2 cell permeability and Ames test were calculated using admetSAR [53] .
3. Results and Discussions
3.1. Molecular Modeling
The frontier molecular orbitals for all compounds have been presented in Figures 2(a)-(d). The molecular orbital shows that, the location of possible sites responsible for electron transfer between molecules and its biological target. Thus, we can discover that how molecules react and where is the active sites in reaction. For the molecules 1-10, the HOMO are delocalized on thiohydantoin system and part of the benzene ring, while in the LUMO, the electron density predominantly are located on the benzene ring, C≡N group, thiohydantoin system except N atom which linked to benzene ring and C=O group. On the other hand, in the case of the molecules 11 and 12, the HOMO are delocalized on thiohydantoin system and part of the benzene ring, while in the LUMO, the electron density predominantly are located on the benzene ring, C≡N group, thiohydantoin system and CH2-SO2-NH2 system. However, in the case of the molecule 13, the HOMO are delocalized on thiohydantoin system, the benzene ring and O atom linked to phenyl ring, while in the LUMO, the electron density predominantly are located on the benzene ring, C≡N group, thiohydantoin system and CH2-SO2-NH2 system. In the case of molecule 14, the HOMO are delocalized on thiohydantoin system, Cl atom and part of the benzene ring, while in the LUMO, the electron density predominantly are located on the benzene ring, C≡N group, thiohydantoin system, Cl atom and SO2 system. But, the HOMO of molecule 15 are delocalized on thiohydantoin system, while in the LUMO, the electron density predominantly located are on the benzene ring, C≡N group and thiohydantoin system. However, the HOMO of molecule 16 are delocalized on thiohydantoin system, while in the LUMO, the electron density predominantly are located on the benzene ring, C≡N group and part of
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 2. (a) The 3D graph of HOMO and LUMO of 1- 4 molecules; (b) The 3D graph of HOMO and LUMO of 5 - 8 molecules; (c) The 3D graph of HOMO and LUMO of 9 - 12 molecules; (d) The 3D graph of HOMO and LUMO of 13 - 16 molecules
thiohydantoin system.
The energies of the frontier orbital (EHOMO and ELUMO) obtained with the AM1 semiempirical method are presented in Table 3. From these results, we can consider the compounds with larger values of EHOMO as being more electron donor and the compounds with smaller values of ELUMO as being more electron acceptor. From Table 3, it can be seen that, the EHOMO and ELUMO calculated values present significant differences: the values of EHOMO vary from −0.32712 au to −0.34521 au and the ELUMO values vary from −0.04819 au to −0.02938 au. HOMO-LUMO refers to the energetic difference of HOMO and LUMO. Such an energetic gap can be used as an indicator of chemical reactivity, where the low energy gap correlates to high chemical reactivity and vice versa. Considering the chemical hardness, large HOMO-LUMO gap means a hard molecule and small HOMO-LUMO gap means a soft molecule. One can also relate the stability of the molecule to hardness, which means that the molecule with least HOMO-LUMO gap means it, is more reactive. Relatively low LUMO-HOMO energy gap of the studied molecules indicates that, it would be kinetically stable. According to AM1 calculation, the heat of formation (HF) energy of the studied molecules has a negative sign (Table 3), and they are exothermic. However, the exothermic nature of HF makes the studied molecules thermodynamically stable [54] .
In the framework of density functional theory (DFT) global descriptors of chemical reactivity corresponds to global responses of systems to global perturbations (for instance, changes in the number of electrons N), whereas the external potential remains constant. Among such types of indexes, chemical potential (μ), chemical hardness (η) and softness (s) can be used as complementary tools in the description of thermodynamic aspects of chemical reactivity.
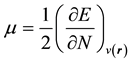 (1)
(1)
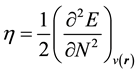 (2)
(2)
From equation (1), the first order partial derivatives of total energy (E) with respect to the number of electrons
Table 3. The frontier orbital’s energies and heat of formation.
(N) at constant external potential,
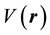 , define chemical potential (μ). On the other hand according to equation (2) the second partial derivatives of total energy (E) with respect to the number of electrons (N) at constant external potential,
, define chemical potential (μ). On the other hand according to equation (2) the second partial derivatives of total energy (E) with respect to the number of electrons (N) at constant external potential,
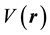 , define the global hardness (η) of the system [55] .
, define the global hardness (η) of the system [55] .
Operational schemes for the calculation of chemical hardness are based on a finite difference method and thus,
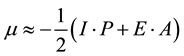 (3)
(3)
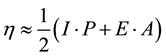 (4)
(4)
where, I・P = Ionization Potential and E・A = Electron Affinity. Using the Koopmans’ theorem in terms of the energies of highest occupied molecular orbital (EHOMO) and lowest unoccupied molecular orbital (ELUMO), according equation (5) & (6) as follow
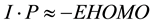 (5)
(5)
 (6)
(6)
So that, the equation 3 & 4 can be expressed as follows.
 (7)
(7)
 (8)
(8)
Electron affinity refers to the capability of the ligand to accept precisely one electron from a donor. However, in many kinds of bonding viz. covalent hydrogen bonding, partial charge transfer takes places. Softness (S) is a property of the compound that measures the extent of chemical reactivity. It is the reciprocal of hardness.
 (9)
(9)
Recently Parr et al. [56] have defined a new descriptor quantity of the global electrophilic power as an electrophilicity index (w) of the compound, which defines a quantitative classification of the global electrophilic nature of a compound. Parr et al. have proposed electrophilicity index (w) as a measure of energy lowering due to maximal electron flow between donor and acceptor. They defined electrophilicity index (w) as follows.
 (10)
(10)
This index measures the stabilization in energy when the system acquired an additional electronic charge from the environment. Electrophilicity encompasses both the abilities of an electrophile to acquire an additional electronic charge and the resistance of the system to exchange electronic charge with the environment. It contains information about both electron transfer (chemical potential) and stability (hardness) and is a better descriptor of global chemical reactivity. On the other hand, electron affinity (EA) ionization potential (IP), molecular softness, electrophilic index and electronegativity (X) were derived from these results of HOMO and LUMO as per Table 4. It is seen that the chemical potential of the title compound is negative, and it means that the compound is stable. They do not decompose spontaneously into the elements they are made up of. The hardness signifies the resistance towards the deformation of the electron cloud of chemical systems under small perturbation encountered during the chemical process.
3.2. Molinspiration Calculations
A computational study for prediction of ADME properties of all molecules is presented in Table 5. The number of rotatable bonds and Lipinski’s rule of five were also calculated [57] . The rule states that most molecules with
Table 4. Electron affinity, ionization potential, molecular softness, electrophilic index and electronegativity of the title compounds.
Table 5. Prediction of molecular properties descriptors of the title compounds.
LogP, logarithm of compound partition coefficient between n-octanol and water; TPSA, topological polar surface area; % ABS, percentage of absorption; MW, molecular weight HBA, number of hydrogen bond acceptors; HBD, number of hydrogen bond donors; Nrotb, number of rotatable bonds; Nvio, Number of violations.
good membrane permeability have logP ≤ 5, molecular weight ≤ 500, a number of hydrogen bond acceptors ≤ 10, and a number of hydrogen bond donors ≤ 5. This rule is widely used as a filter for drug-like properties. Furthermore, none of the compounds violated Lipinski’s parameters, making them potentially promising agents for biological activities. On the other hand, the number of rotatable bonds is important for conformational changes of molecules under study and ultimately for the binding with receptors or channels. It is revealed that for passing oral bioavailability criteria, a number of rotatable bonds should be ≤10 [58] . The compounds in this series, in general, possess a high number of rotatable bonds (5 - 7) and therefore, exhibit conformational flexibility. Topological Polar Surface Area (TPSA) is calculated based on the methodology published by Ertl et al., as a sum of fragment-based contributions [59] [60] in which O- and N- centered polar fragments are to be considered and calculated by surface areas that are occupied by oxygen and nitrogen atoms, and by hydrogen atoms attached to them. TPSA has been shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability and blood-brain barrier penetration. Thus, the TPSA is closely related to the hydrogen bonding potential of a compound [61] . It was found that passively absorbed molecules with a TPSA more than 140 Å are thought to have low oral availability [62] . In respect of TPSA, all the compounds were found within the limit, i.e. 140 Å, which implies that molecules are fulfilling the optimal requirement for drug absorption. TPSA was used to calculate the percentage of absorption (% ABS) following the equation:
% ABS = 109 − (0.345 × TPSA) as reported [63] . From all these parameters, it can be observed that all the title compounds exhibited a moderate % ABS ranging from 67.76% to 83.69%.
3.3. Calculation of Bioactivity Scores
The activity of all test compounds and the standard drug (Flutamide) were rigorously analyzed under four criteria of known successful drug activity in areas of GPCR ligand, ion channel modulator, kinase inhibitor and nuclear receptor ligand. For average organic molecules, the probability is that, if the bioactivity score is more than 0 then it is active if −0.5 to 0 then moderately active [64] . As per Table 6, it is readily seen that all the compounds
Table 6. Prediction of bioactivity by Molinspiration of title compounds.
GPCR = GPCR ligand, ICM = Ion channel modulator, KI = Kinase inhibitor, NRL = Nuclear receptor ligand, PI = Protease inhibitor and EI = Enzyme inhibitor. HOF = Hydroxyflutamide.
are expected to have near similar activity to standard drugs used based upon these four rigorous criteria (GPCR ligand, ion channel modulator, kinase inhibitor, and nuclear receptor ligand).
3.4. Osiris Calculations
The toxicity risks (mutagenicity, tumorigenicity, irritation, reproduction) were calculated by the methodology developed by Osiris. The toxicity risks predictor locates fragments within a molecule, which indicates a potential toxicity risk. Toxicity risk alerts are an indication that the drawn structure may be harmful concerning the risk category specified. From the data evaluated in Table 7, it is obvious that all molecules are supposed to be non-mutagenic, non-irritating with no-reproductive effects when run through the mutagenicity assessment system in comparison with the standard drug.
3.5. The Aqueous Solubility
The aqueous solubility (S) of a compound significantly affects its absorption and distribution characteristics. Typically, a low solubility goes along with a bad absorption, and therefore the general aim is to avoid poorly soluble compounds. Our estimated logS value is a unit stripped logarithm (base 10) of a compound’s solubility measured in mol/liter. There are more than 80% of the drugs on the market have an (estimated) logS value greater than −4. In the case of title compounds, values of logS are around −4. Further, Table 8 shows the drug-likeness (DL) of title compounds which are in the comparable zone with that of standard drug used for comparison. We have calculated overall drug score (DS) for the compounds 1 - 16 and compared with that of standard Hydroxyflutamide. The drug score combines drug-likeness, mi LogP, logS, molecular weight and toxicity risks in one handy value then may be used to judge the compound’s overall potential to qualify for a drug. This value is calculated by multiplying contributions of the individual properties according to the following equation.

Table 7. Prediction of toxicity risks by Osiris of title compounds.
MUT: mutagenic; TUM: tumorigenic; IRRIT: irritant; RE: reproductive effective.
Table 8. Prediction of Bioavailability and Drug Score by Osiris of title com- pounds.
S: Solubility, DL: Drug likeness, DS: Drug Score.
where;

DS is the drug score. Si is the contributions calculated directly from mi LogP; logS, molecular weight and drug likeness (pi) via the second equation, which describes a spline curve. Parameters a and b are (1, −5), (1, 5), (0.012, −6) and (1, 0) for mi LogP, logS, molecular weight and drug-likeness, respectively. The ti is the contributions taken from the four toxicity risk types and the values are 1.0, 0.8 and 0.6 for no risk, medium risk and high risk, respectively. From this work, all compounds showed moderate to good drug score as compared with the standard drug used.
3.6. Ramachandran Plot Analysis
The Ramachandran plot analysis for androgen receptor PDB (2AX6) revealed that, the 2AX6 is an excellent choice which is in good quality to use as a target for docking studies with the title compounds. The percentage of residues in the most favored-regions was 94.1% in 2AX6 (above the cut off 75%), and the percentage of residues in additional allowed regions was 5.9% in 2AX6. This result suggests that the target enzyme’s structure was of good quality.
3.7. Prediction of Activity
PASS (Prediction of Activity Spectra) [64] is an online tool which predicts almost 900 types of activities based on the structure of a compound. The activity prediction of all title compounds has been achieved by PASS server and compared with hydroxyflutamide. As it can be seen in Table 9, the designed ligands as well hydroxyflutamide when examine in the server as a smile format shows the significant properties like “Androgen antagonist and prostate cancer treatment” which predicts that the designed ligands will probably be effective to treat prostate cancer as that of hydroxyflutamide. Based on this we decided to evaluate the in silico anti-cancer activity of the title compounds against the human androgen receptor enzyme (2AX6).
Table 9. Activity prediction of 5,5-dimethylthiohydantoin derivativesand hydroxyflutamide with PASS server.
Pa = Probability of Active, Pi = Probability of Inactive, Pa > Pi confirms significant activity
3.8. Hex Docking
Docking results between androgen receptor 2AX6 and 5,5-dimethylthiohydantoin derivatives are reported in Table 10. The molecular docking study of the title compounds with human androgen receptor shows that, all the title compounds are showing better docking score than that of hydroxyflutamide which predicts that the title compounds have the better binding affinity to the androgen receptor than hydroxyflutamide. Among all the designed compounds, the compound 8 shows the better docking score (Figure 3). This prediction leads us to believe that, the title compounds will possibly be suitable for treatment of prostate cancer.
3.9. Active Sites Identification
In order to find the active sites, the Castp Server is used. The PDB file is used as an input and the results are obtained from this tool which explains the total number of active sites present in the Query PDF file along with information on their amino acid sequence. In addition to that, it also gives their area and volume of the pockets. Pockets are empty concavities on a protein surface into which solvent (probe sphere 1.4 A) can gain access, i.e., these concavities have mouth openings connecting their interior with the outside bulk solution. Currently, shallow depressions are excluded from the calculation. In 2AX6 there are 33 pockets presented in Figure 4. On the other hand, the details of the pocket area, volume, positions and amino acids are given in Table 11. The results obtained from the Castp server revealed that, the 32nd pocket has a maximum area of 379.7 mm2 and Volumes of 473.2 mm3. Further the amino acids like ILE, MET, PHE, LEU, ALA, PHE, LEU, ALA, PHE, LEU, MET, MET, PHE, ARG, MET, VAL, MET, MET, TRP, GLN, GLY, LEU, ASN, LEU, LEU are presented in this pocket.
3.10. Lig Plot+
The interactions of each compound with the functional residues of 2AX6 demonstrated that all the ligands interact

Figure 3. Interaction and binding energy of compound 8 (a) and hydroxyflutamide (b) with androgen receptor (2AX6) respectively.
with most of the residues in the binding pocket as shown in Figure 5(a)-(d). Compound 1 had no hydrogen bond interaction with the protein, but got two external bonds with His789. Also, compound 1 got hydrophobic interaction with Leu790, Val785, Ile869, Arg786, Ser865, Lys861, Leu862 and Glu793. Also, compound 2 had no hydrogen bond interaction with the protein, but got the hydrophobic interaction with Val713, Leu712, Met894, Glu893, Glu897, Gln738, Met734 and Val716. Compound 3 was found to show two hydrogen bonds interaction with active site amino acid residues Gln902 and Met734 at a distance of 2.43 and 2.9 respectively, and the hydrophobic interaction with Lys720, Gln733, Val730, Lys822, Asp732, Ala735, Asp731 and Gln738. Compound 4 was found to show three hydrogen bonds interaction with Ala735, Gln902 and Lys822 a distance of 2.65, 3.02 and 2.84 respectively, and the hydrophobic interaction with Val911, Tyr739, Lys910, Pro817, Lys905, Lue821, Gly820, Asp732, Asp731 and Gln738. On the other hand, compound 5 had no hydrogen bond interaction with
Table 10. Docking Results of 2AX6 enzyme with 5,5-dimeth- ylthiohydantoin Derivatives.

Figure 4. The pockets in androgen receptor (2AX6).
Table 11. Pocket Information for active sites.
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 5. (a) 1a-4a) binding modes of compound 1 - 4 visualized using pymol software respectively, and 1b-4b) Ligplot + results showing the interactions of compounds 1 - 4 respectively with 2AX6; (b) 5a-8a) binding modes of compound 5 - 8 visualized using pymol software respectively, and 5b-8b) Ligplot + results showing the interactions of compounds 5 - 8 respectively with 2AX6; (c) 9a-12a) binding modes of compound 9 - 12 visualized using pymol software respectively, and 9b-12b) Ligplot + results showing the interactions of compounds 9 - 12 respectively with 2AX6; (d) 13a-16a) binding modes of compound 13 - 16 visualized using pymol software respectively, and 13b-16b) Ligplot + results showing the interactions of compounds13 - 16 respectively with 2AX6; (e) HOF a) binding modes of hydroxyflutamide visualized using pymol software and HOF b) Ligplot + results showing the interactions of hydroxyflutamide with 2AX6.
the protein, but got the hydrophobic interaction with Glu793, Lys861, Leu862, Arg786, Ill869, His789, Ser865 and Lue797. Also, compound 6 had no hydrogen bond interaction with the protein, but got the hydrophobic interaction with Glu793, Lys861, His789, Ile869, Ar786, Ser865, Lue862 and Leu797. However, compound 7 was found to show three hydrogen bonds interaction with Ala735, Gln902 and Lys822 a distance of 2.61, 3.19 and 2.85 respectively, and the hydrophobic interaction with Tyr739, Lys910, Pro817, Asp732, Asp731, Asp819 and Gln738. Moreover, compound 8 had no hydrogen bond interaction with the protein, but got the hydrophobic interaction with Lys717, Val713, Met734, Glu893, Glu897, Gln738, Met894, Leu712 and Val716. However, compound 9 had no hydrogen bond interaction with the protein but got two external bonds with Val 684 and Thr755. Furthermore, this compound got hydrophobic interaction with Trp751, Asn756, Arg752, Gln711, Val685, Gly683, Glu681, Pro682, Phe804 and Ala748. On the other hand, compound 10, was found to show one hydrogen bond interaction with Gln902 a distance of 2.72 and the hydrophobic interaction with Gln738, Ala735, Asp731, Asp732, Lys822, Lys905, Val911, Asp819 and Pro817. Compound 11 had no hydrogen bond interaction with the protein, but got the hydrophobic interaction with Pro817, Lys822, Asp731, Met734, Gln738 and Ala735. On the other hand, compound 12, was found to show one hydrogen bond interaction with Ser865 a distance of 2.77 and the hydrophobic interaction with Tyr915, Asp864, Pro868, Glu793, Glu793, Gln858, Lys861, Tyr857 and Leu797. Compound 13, was found to show one hydrogen bond interaction with Gln802 a distance of 2.23, but got two external bonds with Phe804 and Thr755, and the hydrophobic interaction with Asn756, Arg752, Trp751, Glu681, Leu805, Pro801 and Glu678. However, compound 14 had no hydrogen bond interaction with the protein, but got four external bonds with Ile869, Met787, Phe794 and Leu862, also this compound got hydrophobic interaction with Arg786, Ser865, Glu793, Leu797, Ser791, Phe747, Gly 750 and Val746. However, compound 15 had no hydrogen bonds interaction with the protein, but got hydrophobic interaction with Arg786, Ser865, Glu793, Leu797, His789, Ile869, Lys861, and Leu862. On the other hand, compound 16 was found to show one hydrogen bond interaction with Trp751 a distance of 3.13, but got the hydrophobic interaction with Pro682, Gly683, Val684, Glu681, Phe804, Pro801, Leu805, Gln802 and Glu678. The interactions of hydroxyflotamide with the functional residues of 2AX6 presented in figure in (Figure 5(e)), it shows one hydrogen bond interaction With Val 685 a distance of 2.19, but got the hydrophobic interaction with Arg752, Gly683, Val684, Pro682, Val715, Ala748, Leu744 and Gln711.
3.11. ADMET
As discussed earlier, physicochemical properties such as molecular weight MiLogP and TPSA of all a title compounds follow Lipinski’s Rule of Five. Furthermore, online software admetSAR (http://www.admetexp.org/predict/), were used to generate in silico pharmacokinetics parameters for all compounds in order to estimate their drug-like- ness properties. Various ADMET parameters are characterized for compounds 4 and hydroxyflotamide using in silico module admetSAR. The results of the analysis can be seen in Table 12. The analysis includes human intestinal absorption, blood−brain barrier penetration, Caco-2 permeability, P-glycoprotein substrate and inhibitor, CYP450 substrate and inhibitor (CYP1A2, 2C9, 2D6, 2C19, and 3A4), hERG inhibitors, AMES mutagenicity, carcinogens, fathead minnow toxicity, honey bee toxicity, aqueous solubility and Tetrahymena pyriformis toxicity. As per Table 12, hydroxyflotamide is a carcinogen as compared to the non-carcinogenic nature of the compound 4. The major limitation of hydroxyflotamide is its CYP450 3A4 substrate nature, lead to high drug−drug interaction and interruption in the metabolism of the drug combination. The compound 4 overcomes that limitation. Furthermore, the compound 4 is AMES non-toxic and non-carcinogens with compared to standard drug hydroxyflotamide.
Table 12. Prediction of ADMET profiles of the most active compound (4) and droxyflutamide.
Acute Oral Toxicity: Category III includes compounds with LD50 values greater than 500 mg/kg but less than 5000 mg/kg. Carcinogenicity (three-class): Carcinogenic compounds with TD50 (tumorigenic dose rate 50) B10 mg/kg body wt/day were assigned as “Danger,” those with TD50 [10 mg/kg body wt/day were assigned as “Warning,” and non-carcinogenic chemicals were assigned as “Non-required.” Probability indicates scale between 0 and 1. Parameters indicating difference between the most active and the least compound have been highlighted in bold letters.
4. Conclusion
The in silico study of 5,5-dimethylthiohydantoin derivatives gives promise results for using these compounds as an androgen antagonist. The title compounds bind with more competence to the binding site of similar to hydroxyflutamide. Our study has chosen 16 molecules, which demonstrate the better result in silico analysis with better binding efficiency (in terms of docking score) towards androgen receptor than that of hydroxyflutamide. Hydroxyflotamide is a carcinogen as compared to the non-carcinogenic nature of the compound 4. On the other hand, the compound 4 is AMES non-toxic and non-carcinogens with compared to standard drug hydroxyflotamide. Hence, it has been predicted that all the title compounds can possibly act as new leads for the treatment of prostate cancer as they possess androgen antagonist activity. These results may be used in future experiments to investigate the interactions of 5,5-dimethylthiohydantoin derivatives with the androgen receptor, or may be used in vivo experiments to test their effects on the abilities of treatment of prostate cancer.
Acknowledgements
We highly grateful to professor Medhat A. Ibrahim, headmaster of Spectroscopy Department, National Research Centre, 12311, Dokki, Cairo, Egypt, for his support to achieve the molecular modeling computation.
Cite this paper
KhaledLotfy,11, (2015) Molecular Modeling, Docking and ADMET of Dimethylthiohydantoin Derivatives for Prostate Cancer Treatment. Journal of Biophysical Chemistry,06,91-117. doi: 10.4236/jbpc.2015.64010
References
- 1. Green, D.R. and Kroemer, G. (2004) The Pathophysiology of Mitochondrial Cell Death. Science, 305, 626-629.
http://dx.doi.org/10.1126/science.1099320 - 2. Hanahan, D. and Folkman, J. (1996) Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell, 86, 353-364.
http://dx.doi.org/10.1016/S0092-8674(00)80108-7 - 3. Hartwell, L.H. and Kastan, M.B. (1994) Cell Cycle Control and Cancer. Science, 266, 1821-1828.
http://dx.doi.org/10.1126/science.7997877 - 4. Risau, W. (1997) Mechanisms of Angiogenesis. Nature, 386, 671-674.
http://dx.doi.org/10.1038/386671a0 - 5. Heidenreich, A., Bellmunt, J., Bolla, M., Joniau, S., Mason, M., Matveev, V., Mottet, N., Schmid, H.P., van der, K.T., Wiegel, T. and Zattoni, F. (2011) Guía de La EAU sobre el cáncer de próstata. Parte I: Cribado, diagnóstico y tratamiento del áncer clínicamente localizado. Actas Urológicas Españolas, 35, 501-514.
http://dx.doi.org/10.1016/j.acuro.2011.04.004 - 6. Culig, Z., Klocker, H., Bartsch, G. and Hobisch, A. (2002) Androgen Receptors in Prostate Cancer. Endocrine Related Cancer, 9, 155-170.
http://dx.doi.org/10.1677/erc.0.0090155 - 7. Culig, Z., Klocker, H., Bartsch, G. and Hobisch, A. (2001) Androgen Receptor Mutations in Carcinoma of the Prostate. American Journal of Pharmaco-Genomics, 1, 241-249.
http://dx.doi.org/10.1016/S0002-9440(10)63814-X - 8. Eder, I.E., Culig, Z., Putz, T., Nessler-Menardi, C., Bartsch, G. and Klocker, H. (2001) Molecular Biology of the Androgen Receptor: From Molecular Understanding to the Clinic. European Urology, 40, 241-251.
http://dx.doi.org/10.1159/000049782 - 9. Huggins, C. and Hodges, C.V. (1972) Studies on Prostatic Cancer: I. The Effect of Castration, of Estrogen and of Androgen Injection on Serum Phosphatases in Metastatic Carcinoma of the Prostate. CA: A Cancer Journal for Clinicians, 22, 232-240.
http://dx.doi.org/10.3322/canjclin.22.4.232 - 10. Wang, D. and Tindall, D.J. (2011) Androgen Action during Prostate Carcinogenesis. Methods in Molecular Biology, 776, 25-44.
http://dx.doi.org/10.1007/978-1-61779-243-4_2 - 11. Green, S.M., Mostaghel, E.A. and Nelson, P.S. (2012) Androgen Action and Metabolism in Prostate Cancer. Molecular and Cellular Endocrinology, 360, 3-13.
http://dx.doi.org/10.1016/j.mce.2011.09.046 - 12. McPhaul, M.J. (2002) Androgen Receptor Mutations and Androgen Insensitivity. Molecular and Cellular Endocrinology, 198, 61-67.
http://dx.doi.org/10.1016/S0303-7207(02)00369-6 - 13. Yeh, S., Tsai, M.Y., Xu, Q., Mu, X.M., Lardy, H., Huang, K.E., Lin, H., Yeh, S.D., Altuwaijri, S., Zhou, X., Xing, L., Boyce, B., Hung, M., Zhang, S., Gan, L. and Chang, C. (2002) Generation and Characterization of Androgen Receptor Knockout (ARKO) Mice: An in Vivo Model for the Study of Androgen Functions in Selective Tissues. Proceedings of the National Academy of Sciences of the United States of America, 99, 13498-13503.
http://dx.doi.org/10.1073/pnas.212474399 - 14. Hirawat, S., Budman, D.R. and Kreis, W. (2003) The Androgen Receptor: Structure, Mutations, and Antiandrogens. Cancer Investigation, 21, 400-417.
http://dx.doi.org/10.1081/CNV-120018232 - 15. Heinlein, C.A. and Chang, C. (2004) Androgen Receptor in Prostate Cancer. Endocrine Reviews, 25, 276-308.
http://dx.doi.org/10.1210/er.2002-0032 - 16. Bevan, C.L. (2005) Androgen Receptor in Prostate Cancer: Cause or Cure? Trends in Endocrinology & Metabolism, 16, 395-397.
http://dx.doi.org/10.1016/j.tem.2005.09.006 - 17. He, Y., Yin, D., Perera, M., Kirkovsky, L., Stourman, N., Li, W., Dalton, J. and Miller, D. (2002) Novel Nonsteroidal Ligands with High Binding Affinity and Potent Functional Activity for the Androgen Receptor. European Journal of Medicinal Chemistry, 37, 619-634.
http://dx.doi.org/10.1016/S0223-5234(02)01335-1 - 18. Dalton, J.T., Mukherjee, A., Zhu, Z., Kirkovsky, L. and Miller, D.D. (1998) Discovery of Nonsteroidal Androgens. Biochemical and Biophysical Research Communications, 244, 1-4.
http://dx.doi.org/10.1006/bbrc.1998.8209 - 19. Feldman, B.J. and Feldman, D. (2001) The Development of Androgen-Independent Prostate Cancer. Nature Reviews Cancer, 1, 34-45.
http://dx.doi.org/10.1038/35094009 - 20. Chen, Y., Sawyers, C.L. and Scher, H.I. (2008) Targeting the Androgen Receptor Pathway in Prostate Cancer. Current Opinion in Pharmacology, 8, 440-448.
http://dx.doi.org/10.1016/j.coph.2008.07.005 - 21. Taplin, M.E. (2008) Androgen Receptor: Role and Novel Therapeutic Prospects in Prostate Cancer. Expert Review of Anticancer Therapy, 8, 1495-1508.
http://dx.doi.org/10.1586/14737140.8.9.1495 - 22. Yap, T.A., Zivi, A., Omlin, A. and de Bono, J.S. (2011) The Changing Therapeutic Landscape of Castration-Resistant Prostate Cancer. Nature Reviews Clinical Oncology, 8, 597-610.
http://dx.doi.org/10.1038/nrclinonc.2011.117 - 23. Courtney, K.D. and Taplin, M.E. (2012) The Evolving Paradigm of Second-Line Hormonal Therapy Options for Castration-Resistant Prostate Cancer. Current Opinion in Oncology, 24, 272-277.
http://dx.doi.org/10.1097/CCO.0b013e328351059d - 24. Isbarn, H., Boccon-Gibod, L., Carroll, P.R., Montorsi, F., Schulman, C., Smith, M.R., Sternberg, C.N. and Studer, U.E. (2009) Androgen Deprivation Therapy for the Treatment of Prostate Cancer: Consider Both Benefits and Risks. European Urology, 55, 62-75.
http://dx.doi.org/10.1016/j.eururo.2008.10.008 - 25. Wirth, M.P., Hakenberg, O.W. and Froehner, M. (2007) Antiandrogens in the Treatment of Prostate Cancer. European Urology, 51, 306-313.
http://dx.doi.org/10.1016/j.eururo.2006.08.043 - 26. See, W.A., Wirth, M.P., McLeod, D.G., Iversen, P., Klimberg, I., Gleason, D., Chodak, G., Montie, J., Tyrrell, C., Wallace, D.M.A., Delaere, K.P.J., Vaage, S., Tammela, T.L.J., Lukkarinen, O., Persson, B.-E., Carrol, K. and Kolvenbag, G.J.C.M. (2002) Bicalutamide as Immediate Therapy Either Alone or as Adjuvant to Standard Care of Patients with Localized or Locally Advanced Prostate Cancer: First Analysis of the Early Prostate Cancer Program. The Journal of Urology, 168, 429-435.
http://dx.doi.org/10.1016/S0022-5347(05)64652-6 - 27. Akaza, H., Yamaguchi, A., Matsuda, T., Igawa, M., Kumon, H., Soeda, A., Arai, Y., Usami, M., Naito, S., Kanetake, H. and Ohashi, Y. (2004) Superior Anti-tumor Efficacy of Bicalutamide 80 mg in Combination with a Luteinizing Hormone-Releasing Hormone (LHRH) Agonist versus LHRH Agonist Monotherapy as First-Line Treatment for Advanced Prostate Cancer: Interim Results of a Randomized Study in Japanese Patients. Japanese Journal of Clinical Oncology, 34, 20-28.
http://dx.doi.org/10.1093/jjco/hyh001 - 28. Wirth, M.P., See, W.A., McLeod, D.G., Iversen, P., Morris, T. and Carroll, K. (2004) Bicalutamide 150 mg in Addition to Standard Care in Patients with Localized or Locally Advanced Prostate Cancer: Results from the Second Analysis of the Early Prostate Cancer Program at Median Follow-Up of 5.4 Years. The Journal of Urology, 172, 1865-1870.
http://dx.doi.org/10.1097/01.ju.0000140159.94703.80 - 29. Iversen, P., Johansson, J.E., Lodding, P., Lukkarinen, O., Lundmo, P., Klarskov, P., Tammela, T.L., Tasdemir, I., Morris, T. and Carroll, K. (2004) Bicalutamide (150 mg) versus Placebo as Immediate Therapy Alone or as Adjuvant to Therapy with Curative Intent for Early Nonmetastatic Prostate Cancer: 5.3-Year Median Follow-Up from the Scandinavian Prostate Cancer Group Study Number 6. The Journal of Urology, 172, 1871-1876.
http://dx.doi.org/10.1097/01.ju.0000139719.99825.54 - 30. Isurugi, K., Fukutani, K., Ishida, H. and Hosoi, Y. (1980) Endocrine Effects of Cyproterone Acetate in Patients with Prostatic Cancer. The Journal of Urology, 123, 180-183.
- 31. Kennealey, G.T. and Furr, B.J. (1996) Use of the Nonsteroidal Anti-Androgen Casodex in Advanced Prostatic Carcinoma. Urologic Clinics of North America, 18, 99-110.
- 32. Blackledge, G.R. (1996) Clinical Progress with a New Antiandrogen, Casodex (Bicalutamide). European Urology, 2, 96-104.
- 33. Tyrrell, C.J., Altwein, J.E., Klippel, F., Varenhorst, E., Lunglmayr, G., Boccardo, F., Holdaway, I.M., Haefliger, J.M. and Jordaan, J.P. (1991) A Multicenter Randomized Trial Comparing the Luteinizing Hormone-Releasing Hormone Analogue Goserelin Acetate Alone and with Flutamide in the Treatment of Advanced Prostate Cancer. The International Prostate Cancer Study Group. The Journal of Urology, 146, 1321-1326.
- 34. Eisenberger, M.A., Blumenstein, B.A., Crawford, E.D., Miller, G., McLeod, D.G., Loehrer, P.J., Wilding, G., Sears, K., Culkin Jr., D.J., Thompson, I.M., Bueschen, A.J. and Lowe, B.A. (1998) Bilateral Orchiectomy with or without Flutamide for Metastatic Prostate Cancer. New England Journal of Medicine, 339, 1036-1042.
http://dx.doi.org/10.1056/NEJM199810083391504 - 35. Wirth, M.P. and Froschermaier, S.E. (1997) The Antiandrogen Withdrawal Syndrome. Urological Research, 25, 67-71.
http://dx.doi.org/10.1007/BF00941991 - 36. Kemppainen,
J.A. and Wilson, E.M. (1996) Agonist and Antagonist Activities of Hydroxyflutamide and Casodex Relate to Androgen
Receptor Stabilization. Urology, 48, 157-163.
http://dx.doi.org/10.1016/S0090-4295(96)00117-3 - 37.
Tachibana, K., Imaoka, I., Shiraishi, T., Yoshino, H., Nakamura, M., Ohta, M., Kawata, H., Taniguchi, K.,
Ishikura, N., Tsunenari, T., Saito, H., Nagamuta, M., Nakagawa, T., Takanashi, K., Onuma, E. and Sato, H.
(2008) Discovery of an Orally-Active Nonsteroidal Androgen Receptor Pure Antagonist and the Structure-Activity
Relationships of Its Derivatives. Chemical & Pharmaceutical Bulletin, 56, 1555-1561.
http://doi.org/10.1248/cpb.56.1555 - 38.
Yoshino, H., Sato, H., Tachibana, K., Shiraishi, T., Nakamura, M., Ohta, M., Ishikura, N., Nagamuta, M., Onuma,
E., Nakagawa, T., Arai, S., Ahn, K.H., Jung, K.Y. and Kawata, H. (2010) Structure-Activity Relationships of
Bioisosteric Replacement of the Carboxylic Acid in Novel Androgen Receptor Pure Antagonists. Bioorganic &
Medicinal Chemistry, 18, 3159-3168.
http://dx.doi.org/10.1016/j.bmc.2010.03.036 - 39. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, J.M., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, O., Foresman, J.B., Ortiz, J.V., Cioslowski, J. and Fox, D.J. (2009) Gaussian 09, Revision A. Gaussian, Inc., Wallingford.
- 40. Dennington, R., Keith, T. and Millam, J. (2007) Gauss View, Version 4.1.2. Semichem Inc., Shawnee Mission.
- 41.
Lipinski, C.A., Lombardo, F., Dominy, B.W. and Feeney, P.J. (1997) Experimental and
Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development
Settings. Advanced Drug Delivery Reviews, 64, 4-17.
http://dx.doi.org/10.1016/j.addr.2012.09.019 - 42.
Molinspiration Cheminformatics (Homepage on the Internet). Nova Ulica, SK-900 26 Slovensky Grob, Slovak Republic.
http://www.molinspiration.com
http://www.Molinspiration.Com/cgi-bin/properties - 43. Molsoft Offers Software Tools and Services in Lead Discovery, Modeling, Cheminformatics, Bioinformatics.
http://molsoft.com/mprop/ - 44. Chen, G., Zheng, S., Luo, X., Shen, J., Zhu, W., Liu, H., Gui, C., Zhang, J., Zheng, M., Puah, C.M., Chen, K. and Jiang, H. ( 2005) Focused Combinatorial Library Design Based on Structural Diversity, Druglikeness and Binding Affinity Score. Journal of Combinatorial Chemistry, 7, 398-406.
http://dx.doi.org/10.1021/cc049866h - 45.
http://hex.loria.fr/hex.php. - 46. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. (2000) The Protein Data Bank. Nucleic Acids Research, 28, 235-242.
http://dx.doi.org/10.1093/nar/28.1.235 - 47. Laskowski, R.A., MacArthur, M.W., Moss, D.S. and Thornton, J.M. (1993) PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. Journal of Applied Crystallography, 26, 283-291.
http://dx.doi.org/10.1107/S0021889892009944 - 48. Ritchie, W.D. and Kemp, G.J.L. (2000) Protein Docking Using Spherical Polar Fourier Correlations. Proteins: Structure, Function, and Genetics, 39, 178-194.
- 49. Ritchie, W.D. (2003) Evaluation of Protein Docking Predictions Using Hex 3.1 in CAPRI Rounds 1 and 2. Proteins: Structure, Function, and Bioinformatics, 52, 98-106.
http://dx.doi.org/10.1002/prot.10379 - 50. Delano, W.L. (2002) The PyMOL Molecular Graphics System. De Lano Scientific, San Carlos.
- 51. Laskowski, R.A. and Swindells, M.B. (2011) LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. Journal of Chemical Information and Modeling, 51, 2778-2786.
http://dx.doi.org/10.1021/ci200227u - 52. Zhao, Y.H., Abraham, M.H., Le, J., Hersey, A., Luscombe, C.N., Beck, G. and Sherborne, B. (2002) Rate-Limited Steps of Human Oral Absorption and QSAR Studies. Pharmaceutical Research, 19, 1446-1457.
http://dx.doi.org/10.1023/A:1020444330011 - 53. Cheng, F., Li, W., Zhou, Y., Shen, J., Wu, Z., Liu, G., Lee, P. and Tang, Y. (2012) admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. Journal of Chemical Information and Modeling, 52, 3099-3105.
http://dx.doi.org/10.1021/ci300367a - 54. Awad, M.K., Khairau, K.S. and Diab, M.A. (1994) Theoretical Investigations of the Stability of Degradation Products of Polystyrene and Poly(4-vinylpyridine). Polymer Degradation and Stability, 46, 165-170.
http://dx.doi.org/10.1016/0141-3910(94)90048-5 - 55. Ghanty, T.K. and Ghosh, S.K. (1996) A Density Functional Approach to Hardness, Polarizability, and Valency of Molecules in Chemical Reactions. The Journal of Physical Chemistry, 100, 12295-12298.
http://dx.doi.org/10.1021/jp960276m - 56. Parr, R.G., Szentpaly, L.V. and Liu, S.J. (1999) Electrophilicity Index. Journal of the American Chemical Society, 121, 1922-1924.
http://dx.doi.org/10.1021/ja983494x - 57. Lipinski, C.A., Lombardo, F., Dominy, B.W. and Feeney, P. (2012) Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Advanced Drug Delivery Reviews, 64, 4-17.
http://dx.doi.org/10.1016/j.addr.2012.09.019 - 58. Veber, D.F., Johnson, S.R., Cheng, H.Y., Smith, B.R., Ward, K.W. and Kapple, K.D. (2002) Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. Journal of Medicinal Chemistry, 45, 2615-2623.
http://dx.doi.org/10.1021/jm020017n - 59. Ertl, P., Rohde, B. and Selzer, P. (2000) Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. Journal of Medicinal Chemistry, 43, 3714-3717.
- 60. Pervez, A., Meshram, J., Tiwari, V., Sheik, J., Dongre, R., Youssoufi, M.H., et al. (2010) Pharmacophores Modeling in Terms of Prediction of Theoretical Physico-Chemical Properties and Verification by Experimental Correlations of Novel Coumarin Derivatives Produced via Betti’s Protocol. European Journal of Medicinal Chemistry, 45, 4370-4378.
http://dx.doi.org/10.1016/j.ejmech.2010.06.004 - 61. Clark, D.E. (1999) Rapid Calculation of Polar Molecular Surface Area and Its Application to the Prediction of Transport Phenomena. 2. Prediction of Blood-Brain Barrier Penetration. Journal of Pharmaceutical Sciences, 88, 815-821.
http://dx.doi.org/10.1021/js980402t - 62. Lalitha, P. and Sivakamasundari, S. (2010) Calculation of Molecular Lipophilicity and Druglikeness for Few Heterocycles. Oriental Journal of Chemistry, 26, 135-141.
- 63. Verma, A. (2012) Lead Finding from Phyllanthus debelis with Hepatoprotective Potentials. Asian Pacific Journal of Tropical Biomedicine, 2, S1735-S1737.
http://dx.doi.org/10.1016/s2221-1691(12)60486-9 - 64. Lagunin, A., Stepanchikova, A., Filimonov, D. and Poroikov, V. (2000) PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics, 16, 747-748.
http://dx.doi.org/10.1093/bioinformatics/16.8.747