Journal of Biophysical Chemistry
Vol.5 No.2(2014), Article ID:45208,11 pages DOI:10.4236/jbpc.2014.52003
Hemin Protects U937 Cells from Oxidative Stress via Glutathione Synthesis
Yoshimi Satoh, Kyohei Oyama, Koichi Sakurai*
Division of Biochemistry, Department of Life science, Hokkaido Pharmaceutical University School of Pharmacy, Otaru, Japan
Email: *ks51@hokuyakudai.ac.jp
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 17 February 2014; revised 17 March 2014; accepted 24 March 2014
ABSTRACT
Although heme oxygenase-1 and glutathione play important roles in the antioxidant defense system, the sharing and/or cross-talking of the HO-1 and GSH system remain poorly understood. The object of this study is to determine whether the glutathione system is involved in the antioxidant function of hemin. Hydrogen peroxide decreased the viability of the human leukemic monocyte lymphoma cell line U937. When these cells were pretreated with hemin before the addition of hydrogen peroxide, cell death was prevented. An inhibitor of heme oxygenase-1 or glutathione biosynthesis significantly abolished this protective effect of hemin. These results suggest that both heme oxygenase-1 and glutathione are involved in the protective effects of hemin against U937 cell death, which was induced by hydrogen peroxide. Hemin induced an increase in glutathione levels following the upregulation of the gene expression and protein levels of glutamate-cysteine ligase catalytic subunit. The inhibitor of heme oxigenase-1 inhibited the upregulation of glutamate-cysteine ligase catalytic subunit expression. These results suggest that hemin induces glutathione synthesis through heme oxygenase-1 activation to protect cells from hydrogen peroxideinduced oxidative stress.
Keywords
Glutathione, Heme Oxygenase-1, Hemin, Oxidative Stress, Gene Expression

1. Introduction
The excessive generation of reactive oxygen species (ROS), which contain hydrogen peroxide (H2O2) can lead to several diseases, such as atherosclerosis, myocardial ischemia/reperfusion injury, diabetes mellitus and cancer [1] -[3] . All cell types have developed complex networks of antioxidant response machinery to adapt to oxidative stress conditions [4] -[6] . These responses include the induction of heat shock protein synthesis as well as the glutathione (GSH) system. Hemeoxygenase (HO)-1 is one of the primary proteins that respond to oxidative stress conditions [7] -[9] . HO-1 catalyzes the degradation of heme and leads to the production of carbon monoxide, free iron and bilirubin. Bilirubin is a potent free radical scavenger [10] [11] . Carbon monoxide has vasodilatory, anti-apoptotic activity and anti-inflammatory actions, which support the cytoprotective effects of bilirubin [9] [12] [13] . In addition, free iron induces the expression of a secondary antioxidant protein, ferritin [14] . The protective effect of HO-1 against oxidative stress has been additionally confirmed by the use of pharmacological blockers of HO-1 activity and the deletion of the HO-1 gene [15] . Hemin, which is specifically described as protoporphyrin IX containing a ferric iron ion, is a strong HO-1 inducer. The upregulation of HO-1 activity by hemin treatment has been shown to have a beneficial role in various models of cellular and tissue injury during oxidative stress [16] [17] . It is suggested that HO-1 activation may mediate these antioxidants and the protective effect of hemin.
Intracellular GSH strongly participates in detoxification reactions through its reducing potential, either directly or indirectly, as a cofactor for enzymes such as glutathione peroxidase (GPx) [6] [18] . The depletion of intracellular GSH results in oxidative stress to cardiomyocytes, hepatocytes and renal tubular epithelial cells, whereas the addition of GSH protects against cytotoxicity [19] -[21] .
Given that the HO-1 system and GSH system could potentially share an induction mechanism, which is mediated by an antioxidant response master regulator, such as the nuclear factor-erythroid-2-related factor (Nrf-2), and that the GSH system has strong antioxidant potential, the GSH system might participate in the protective effect of hemin against oxidative stress. However, the sharing and/or cross-talking of the HO-1 and GSH system remain poorly understood.
The aim of the present study is to elaborate on the connection between HO-1 induction and GSH synthesis in the protective effect of hemin. We demonstrated that hemin enhanced glutamate-cysteine ligase catalytic subunit (GCLC) expressions and GSH synthesis and that the administration of the HO-1 inhibitor abolished the expression of GCLC, which resulted in the failure of hemin to exert a cytoprotective effect. We showed here that the increase in intracellular GSH via HO-1 activation participated in the protection effect of hemin against oxidative stress.
2. Materials and Methods
2.1. Chemicals
H2O2 was obtained from Kanto Chemical Co. Inc. (Tokyo, Japan). L-Buthionine-[S,R]-Sulfoximine (BSO) was purchased from Calbiochem (San Diego, CA). The cell proliferation assay was obtained from Promega Co. (Madison, WI). Dimethyl sulfoxide (DMSO) was obtained from NakaraiTesque, Inc. (Kyoto, Japan). 5,5’-dithiobis-2-nitrobenzoic acid (DTNB) was obtained from Eastman Chemical Co. (Kingsport, TN). Fetal bovine serum (FBS) was purchased from Biosource, Inc. (Camarillo, CA). Hemin was obtained from Alfa Aesor (Ward Hill, MA). Phosphate buffer saline was obtained from Takara Bio, Inc. (Tokyo, Japan). RPMI 1640 medium and penicillin-streptomycin were obtained from Invitrogen (Carlsbad, CA). Zinc (II) protoporphyrin IX (ZnPP) was obtained from Alexis Biochemicals (San Diego, CA). Glutathione reductase from yeast (GR) and β-nicotinamide-adenine dinucleotide phosphate reduced form (NADPH) were obtained from Oriental Yeast Co., Ltd. (Tokyo, Japan). The HO-1 antibody was obtained from Assay Designs, Inc. (Ann Arbor, MI). The GCLC antibody, actin antibody, rabbit anti-goat IgG-HRP and goat anti-rabbit IgG-HRP were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The radio immune precipitation assay (RIPA) buffer and BCA assay kit were obtained from Thermo Scientific (Rockford, IL). ECL plus, Hyperfilm ECL, and an illustraTMRNAspin mini RNA isolation kit were obtained from GE healthcare (Little Chalfont, UK). A high-capacity cDNA reverse transcription kit, TaqMan Universal PCR Master Mix and TaqManGene Expression Assay were obtained from Applied Biosystems (Foster City, CA). All other chemicals that were used in this study were of the highest grade available from commercial suppliers.
2.2. Cell Culture and Treatment
The human leukemic monocyte lymphoma cell line U937 was a kind gift from Dr. I. Kashiwakura and was cultured in RPMI 1640 medium, which contained heat-inactivated 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, and incubated at 37˚C in an atmosphere of 5% CO2/95% air as described previously [22] . U937 cells were seeded in 24-well plates containing 640 μl of culture medium at a density of 4 ´ 105 cells/well in 12-well plates containing 1.2 ml of culture medium at a density of 7.5 ´ 105 cells/well and in 6-well plates containing 3 ml of culture medium at a density of 3 ´ 106 cells/well; in addition, H2O2 was added to the medium after pretreatment with or without hemin overnight. Hemin, BSO or ZnPP was added to the medium at the same time as cell seeding unless otherwise indicated. DMSO was not observed to have an influence as a vehicle at solution concentrations of less than 0.1%.
To determine the viability of the cells, we measured the capability of cells to reduce 3-(4,5-dimethylthiazol- 2-yl)-2,5-dimethyl tetrazolium bromide (MTT) with a minor modification to a previously described protocol [20] . Briefly, after removing the medium by centrifugation at 300 ´ g for 5 min, the cells were washed twice with PBS. To assay the MTT reduction ability, fresh culture medium (50 mL) and a dye solution of MTT (7.5 mL) were added to each of the cell-containing wells and were incubated for 2 h at 37˚C. A solubilization/stop solution (50 mL) was then added for an additional overnight incubation. The absorbance readings at 570 nm (formation of formazan) and 690 nm (reference) were recorded using a microplate reader (Bio-Rad, Tokyo, Japan).
2.3. Real-Time RT-PCR
Total cellular RNA was extracted from H9c2 cells using an illustraTMRNAspin Mini RNA Isolation Kit (GE Healthcare, Buckinghamshire, UK), according to the manufacturer’s instructions, and the total RNA was quantified by absorbance at 260 nm. RNA (0.2 mg/mL) was reverse transcribed into cDNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. TaqMan PCR primers and probes for HO-1 and GCLC target genes as well as18S rRNA, which is the internal standard gene, was obtained from Applied Biosystems (Foster City, CA). TaqMan PCR was performed with 1 mL of sample cDNA in a 50-mL reaction mixture containing TaqMan Universal PCR Master Mix and the TaqMan gene expression assay. Amplification was performed using the 7500 Real Time PCR System (Applied Biosystems). Cycling conditions were 50˚C for 2 min, 95˚C for 10 min, followed by 50-cycleamplification at 95˚C for 15 s and 60˚C for 1 min. The threshold cycle number (Ct) at which each PCR amplification reaches a significant threshold level was calculated using the system software. Ct is proportional to the number of target and 18S rRNA copies existing in the reaction mixture. 18S rRNA was used to normalize mRNA expression. Data were calculated using the DDCt method.
2.4. Western Blotting
U937 cells were seeded in6-well plates and were incubated under various conditions. After the removal of the medium by centrifugation at 300 ´ g for 5 min, the cells were washed twice with PBS. The pellet was resuspended in RIPA buffer and was incubated for 30 min on ice. We passed the cells through a 5 mL syringe with a 27-gage needle 6 times to lyse the plasma membrane. The supernatant was collected by centrifugation at 22,900 ´ g for 10 min at 4˚C. Protein content was measured using the BCA protein assay kit with bovine serum albumin as the standard. A solution of the sample protein was diluted with an equal volume of 0.1 M Tris-HCl buffer at pH 6.8 containing 4% SDS, 12% 2-mercaptoethanol, 20% glycerol, and 0.01% bromophenol blue, and the mixture was boiled for 10 min. Proteins (50 mg/well) from each sample were separated on a 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane (GE Healthcare, Buckinghamshire, UK). The membrane was blocked with 5% skim milk and incubated overnight with primary antibodies at 4˚C. Probing of the membranes was performed with a polyclonal HO-1 antibody (1:5000 dilution), a polyclonal GCLC antibody (1:4000 dilution) or a polyclonal actin antibody (1:5000 dilution). After blotting, the membranes were washed with wash buffer and then incubated for 60 min at room temperature with secondary antibodies, including a donkey antirabbit IgG-conjugated HRP (1:20,000 dilution) and a rabbit anti-goat IgG-conjugated HRP (1:5000 dilution). The proteins that combined with the antibodies were detected by ECL plus Western blotting system detection reagents, and immunocomplexes were visualized using an Epi-Light UV FA1100. For the quantification of band density, films were analyzed using the Luminous Imager software (Aisin Cosmos, Toyama, Japan).
2.5. GSH Determination
Intracellular GSH was measured by the DTNB-GSSH reductase recycling assay [23] . Briefly, the cells were seeded onto 6-well plate and incubated under various conditions for 24 h. The cells were collected by centrifugation at 300 ´ g for 5 min and were washed twice with PBS. The cell pellets were resuspended in 300 mL PBS and were then sonicated with an ultrasonic disrupter (Model UR-200P, Tomy Seiko Co., Tokyo, Japan) three times for 10 s at 35 W in icewater. The lysate was centrifuged at 22,900 ´ g for 10 min at 4˚C. After incubation with 4 mM NADPH and 6 U/mL GR for 5 min at 37˚C, the supernatant was mixed with 2 mM DTNB and measured with a spectrophotometer (Shimadzu Co., Kyoto, Japan) at 412 nm. The amount of GSH in the mixture was calculated using the coefficient of the resultant product, 5-mercapto-2-nitrobenzoate: e = 15,500 M−1∙cm−1.
2.6. Statistical Analysis
Data are represented as the mean ± S.D. and were statistically analyzed by Student’s t-test or Welch’s t-test following the F-test for paired data. We also used Dunnett’s multiple comparison test after one-way analysis of variance. p < 0.05 was considered statistically significant
3. Results
3.1. Protective Effect of Hemin against H2O2 Cytotoxicity
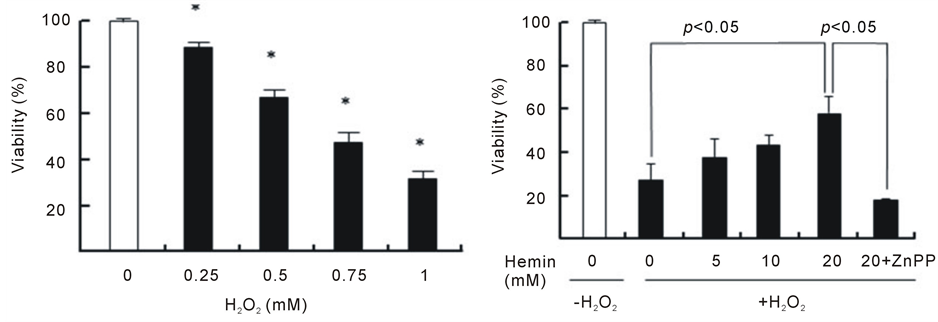
As shown in Figure 1(a), H2O2 induced a decrease in the viability of U937 cells in a dose-dependent manner during the 24 h incubation period; the viabilities were 88%, 68%, 48% and 33% at 0.25, 0.50, 0.75 and 1.0 mMH2O2, respectively. Because there was an obvious decrease in cell viabilityat an H2O2 concentration of 1.0 mM after the 24 h incubation period, this condition was chosen to induce cytotoxicity in U937 cells in the subsequent experiments. The pretreatment of cells with hemin, an inducer of HO-1 for 24 h significantly prevented the H2O2-induced cell death, and the viability concentration at 20 μM was 60% (viability without hemin was 20%) (Figure 1(b)). The pretreatment of cells with hemin in the presence of ZnPP, an inhibitor of HO-1, failed to exhibit the protective effect of hemin. We next investigated whether intracellular GSH participates in the protective effect of hemin against cell death induced byH2O2. The pretreatment of cells with hemin in the presence of BSO, an inhibitor of GSH biosynthesis, completely failed to prevent the cell death that was induced by H2O2 and markedly enhanced cytotoxicity (Figure 1(c)). BSO did not exert any cytotoxic effects at the concentrations that were used in our study. These results suggest that both HO-1 and GSH participated in the protective effect of hemin against the cell death that was induced by H2O2.
3.2. Hemin Induces the Activation of GSH Synthesis and HO-1 Expression
Figure 2 shows the changes in the GSH levels in U937 cells that were pretreated with hemin. Although no change was observed until 6 h after pretreatment with hemin, the GSH levels increased after 12 h, and the GSH levels at 24 h were approximately 2.2 times higher than that at 0 h. In the presence of BSO, intracellular GSH levels in cells that were pretreated with hemin markedly decreased (Figure 2(a)). These results suggest that hemin pretreatment induces an increase in intracellular GSH levels. We investigated the effect of hemin on the gene expression and protein levels of GCLC, which is the catalytic subunit of γ-GCS, in U937 cells. When U937 cells were pretreated with hemin for 24 h, an increase in the concentration of hemin resulted in an increase in the expression of the GCLC gene in a dose-dependent manner. At a concentration of 20 mM hemin, which showed a significant cytoprotective effect, the gene expression and protein levels of GCLC were 3.0 ± 0.1 and 2.2 ± 0.6 times higher than those of the control, respectively (Figure 2(b) and Figure 2(c)). These results suggest that hemin pretreatment of U937 cells induces the expression of the GCLC gene.
We also recorded the HO-1 gene and protein expression in cells that were pretreated with hemin. Upon pretreatment with hemin for 24 h, significant increases in HO-1gene expression were observed, and the expression levels at 5, 10 and 20 mM hemin were 19, 28 and 64 times higher than those of control cells without hemin, respectively (Figure 2(d)). Increasing the concentration of hemin resulted in increased in HO-1 protein levels in U937 cells. At a concentration of 20mM hemin, the HO-1 levels were 6.4 ± 0.1 times higher than those of control cells (Figure 2(e)). In control cells without hemin, no increase in the gene expression and protein levels of HO-1 was observed during the 24 h incubation period. These results suggest that hemin caused the increase in HO-1 expression during the 24 h treatment.
3.3. HO-1 Activity Mediates GSH Synthesis
We next investigated whether HO-1 induction by hemin pretreatment mediates the activation of GSH synthesis.
 (a)
(a)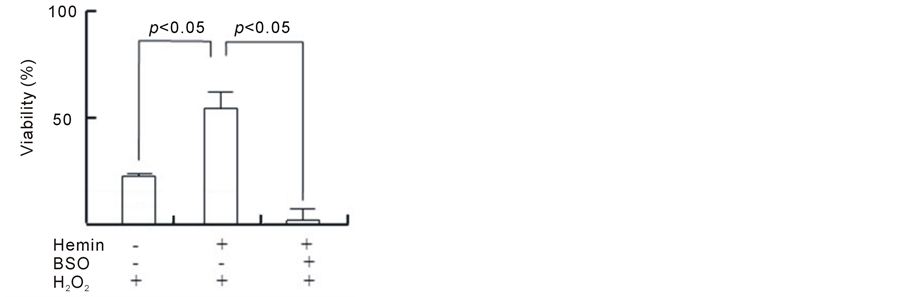 (b)
(b)
Figure 1. Effect of hemin, ZnPP and BSO on cell death induced by H2O2. U937 cells were incubated with various concentrations of H2O2 for 24 h (a). *p < 0.05 compared with control cells without H2O2. After pretreatment with 20 µM hemin and 10 µM ZnPP (b) or 0.1 mM BSO (c), U937 cells were incubated with 1 mM H2O2 for 24 h (b). Cell viability was assayed by MTT methods. Data are represented as the mean ± S.D.
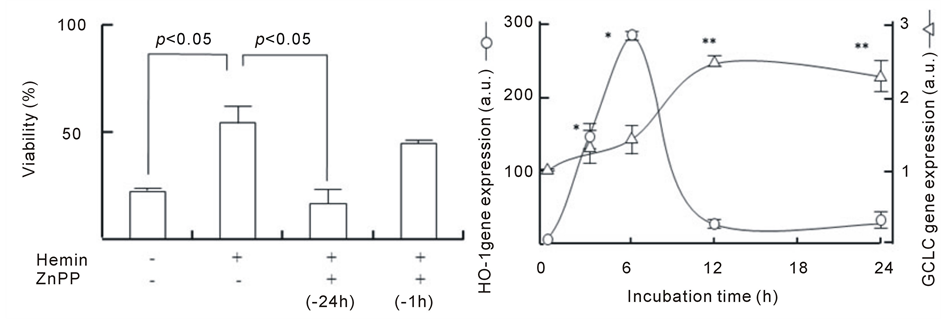
As shown in Figure 3(a), upon the addition of ZnPP at different time points during hemin pretreatment, ZnPP markedly abolished the protective action of hemin pretreatment at 24 h before the addition of H2O2. However, ZnPP treatment at 1 h before the addition of H2O2 failed to abolish the protective effect of hemin pretreatment. This result suggests that HO-1 activation an early time point during hemin pretreatment might be important for the protective effect of hemin. Figure 3(b) shows the time course of HO-1 and GCLC gene expression during hemin pretreatment. A significant increase in HO-1 expression was observed during the 3 h after the addition of hemin; the levels at 6 h were 284 ± 7 times higher than those at 0 h and decreased at 12 and 24 h (approximately 30 times). Significant increases in GCLC expression were observed at 12 h after hemin pretreatment and the levels at 12 and 24 h were 2.4 ± 0.1 and 2.3 ± 0.2 times higher than those at 0 h, respectively. GCLC upregulation follows HO-1 upregulation during hemin pretreatment. These results imply that the preceding activation of HO-1 mediates GCLC gene expression to show the protective effect of hemin.
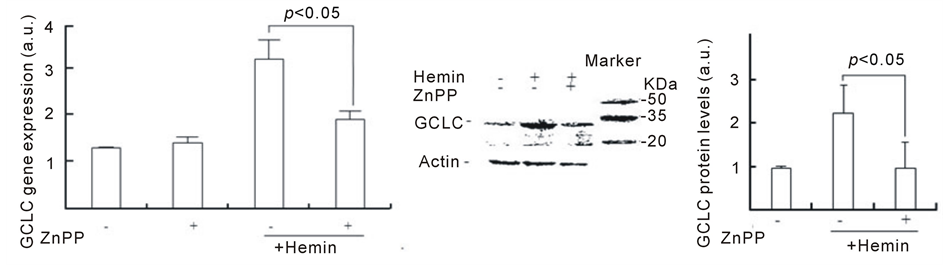
To address this hypothesis, we investigated whether the inhibition of HO-1 inhibits GSH synthesis by hemin. Figure 4 shows the effect of ZnPP on GCLC expression. Although the GCLC gene expression that was induced by hemin pretreatment for 24 h was approximately 2.5 times higher than that in control cells, ZnPP markedly inhibited the increase in GCLC gene expression (approximately 1.5 times). In control cells, ZnPP did not affect the basal GCLC expression level. Consistent with the gene expression data, the pretreatment with hemin induced increasing levels of GCLC protein, which were 2.2 ± 0.6 times higher than those in control cells at 24 h (Figure 4(b)). In the presence of ZnPP, GCLC protein levels in cells pretreated with hemin for 24 h were 1.0 ± 0.5 times higher compared with control cells, suggesting that ZnPP almost completely inhibits the upregulation of GCLC protein. Figure 4(c) shows the effect of ZnPP on intracellular GSH levels in U937 cells treated with hemin. A significant increase in intracellular GSH levels was observed at 24 h after the addition of hemin. The levels of
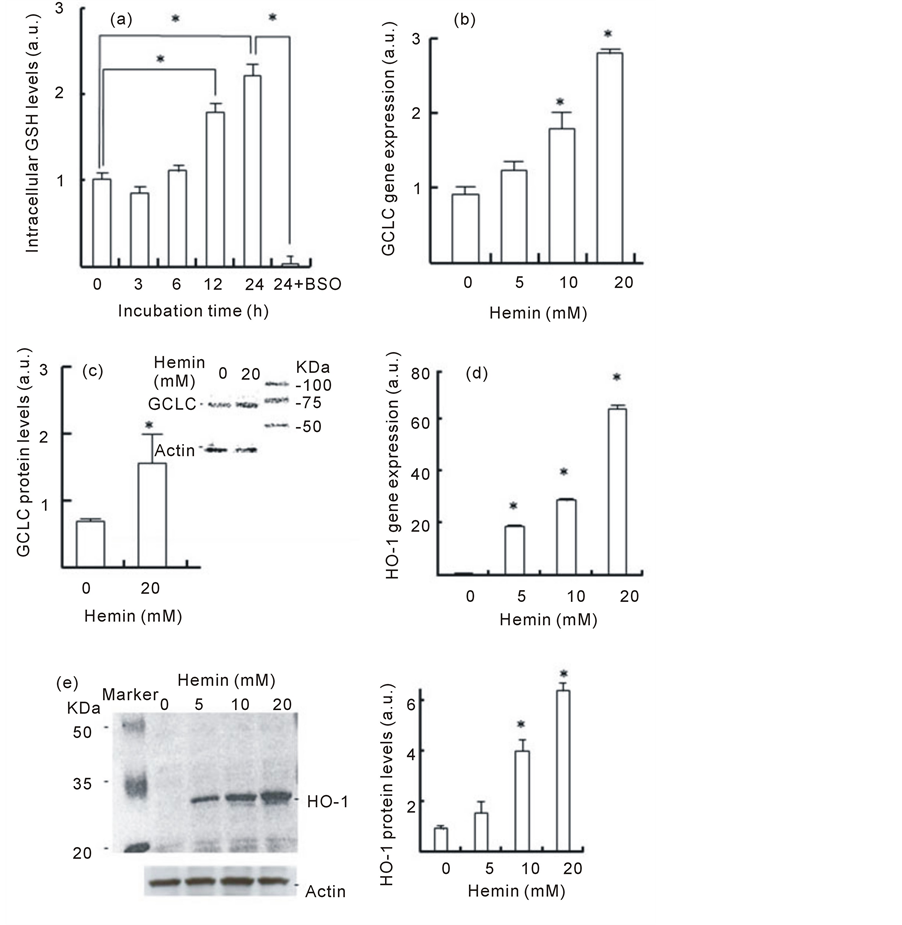
Figure 2. Effect of hemin pretreatment on GSH levels andon GCLC and HO-1 gene and protein expression. (a) U937 cells were pretreated with 20 µM hemin with or without 0.1 mM BSO for the indicated time. Data are represented as the mean ± S.D. *p < 0.05. (b, d) U937 cells were pretreated with various concentration of hemin for 24 h. The GCLC and HO-1 gene expressions were determined in real-time using RT-PCR. Data are represented as the mean ± S.D. *p < 0.05 compared with control cells without hemin. (c, e) U937 cells pretreated with 20 µM hemin for 24 h. GCLC and HO-1 protein levels were determined by Western blotting. Data are represented as the mean ± S.D. *p < 0.05 compared with control cells without hemin.
GSH were 2.8 ± 0.2 times higher than at 0 h. ZnPP dramatically inhibited the increase in GSH levels that were induced by hemin (1.3 ± 0.1 times). These results suggest that the activation of HO-1 could participate in the induction of GCLC gene expression and GSH synthesis.
4. Discussion
Cells orchestrate several antioxidative machineries in response to oxidative stress. Both HO-1 and GSH are ma-
 (a) (b)
(a) (b)
Figure 3. Involvement of HO-1 in the protective action of hemin and the time course of HO-1 and GCLC gene expression. (a) After the pretreatment with 20 µM hemin for 24 h or 10 µM ZnPP for 24 or 1 h, U937 cells were incubated with 1 mM H2O2 for 24 h. Cell viability was assayed by MTT methods; (b) U937 cells pretreated with 20 µM hemin for various times. HO-1 and GCLC gene expression were determined in real-time using RT-PCR. Data are represented as the mean ± S.D. *p < 0.05 and **p < 0.05 compared with HO-1 and GCLC gene expression at 0 h, respectively.
 (a)
(a)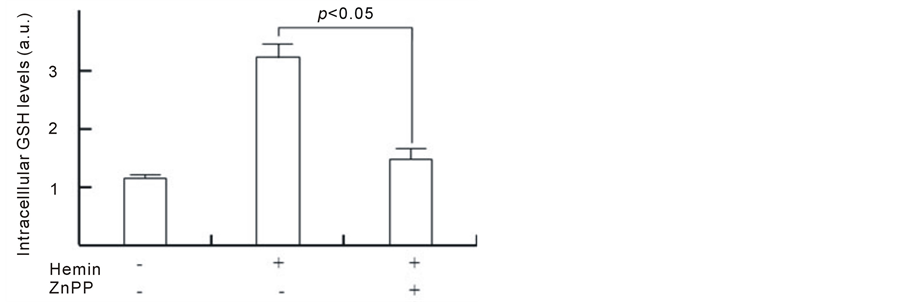 (b)
(b)
Figure 4. Effect of ZnPP on GCLC expression and increasing GSH levels induced by hemin. U937 cells were pretreated with 20 µM hemin and 10 µM ZnPP for 24 h. The GCLC gene and protein expressions were determined in real-time using RT-PCR (a) and Western blotting (b), respectively. Intracellular GSH levels were determined as described in Methods (c). Data are represented as the mean ± S.D.
jor antioxidative machinery against oxidative stress in various types of organism [15] [18] . Hemin is a ferric iron-binding porphyrin and is well-characterized as activating the expression of HO-1 in various types of cells. The present study showed that pretreatment with hemin markedly increased the expression of HO-1 and induced GSH synthesis, which in turn exhibited a cytoprotective effect against H2O2-induced cytotoxicity. This tolerance to H2O2-induced cytotoxicity was abolished by the presence of ZnPP, an inhibitor of HO-1, suggesting that HO-1 participates in the protective action of hemin. HO-1 catalyzes the degradation of heme and produces bilirubin, a potent free radical scavenger, resulting in an increase in antioxidative potential [9] -[11] . Several researchers reported that increased HO-1 activity provided cellular protection against diseases that were induced by oxidative stress, including diabetes mellitus, cardio vascular disease and atherosclerosis [24] [25] . These findings support our findings that inducing HO-1 plays an important role in the protective action against H2O2-induced oxidative damages in hemin-pretreated cells.
GSH is a tripeptide containing cysteine and is an abundant antioxidant in all organisms. Under oxidative stress conditions, GSHs are consumed by ROS and are rapidly filled primarily de novo synthesis and redox cycling [19] [21] . Gamma-glutamylcysteine synthetase (γ-GCS), which is a component in two ATP-requiring GSH synthesizing enzymes, is responsible for GSH de novo synthesis. Gamma-GCS is a heterodimer of the heavy, catalytic subunit (GCLC) and the light, regulatory subunit (GCLM). GCLC gene expression is the ratelimiting step in GSH synthesis [26] . GCLC expression is mediated by inducible machineries that respond to various stimuli, such as heat shock, oxidative stress and cytokines [15] [19] [27] . It has been demonstrated that preconditioning, such as mild oxidative stress, induces the activation of GSH de novo synthesis and exhibits cytoprotective effect against oxidative stress [28] [29] . The importance of de novo synthesis is underscored by the observations that the inhibition of GCLC can significantly deplete GSH in cultured cells and tissues within hours [30] . In the present study, we demonstrated that hemin pretreatment induced GCLC upregulation and GSH synthesis. BSO, an inhibitor of GSH biosynthesis, completely abolished the protective action of hemin pretreatment against U937 cell death that was induced by H2O2. GSH is involved in several processes, such as scavenging ROS, maintaining redox status, detoxification and regulating apoptosis [18] [19] [21] [31] . In addition, GSH and thiol compounds participate in the cell signaling response to oxidative stress [6] [32] [33] . These results suggest that the activation of GSH synthesis participates in the protective effect of hemin pretreatment against cell death that is induced by H2O2.
One of the potential master regulators that control antioxidative machineries is a transcription factor, nuclear factor-erythroid-2-related factor (Nrf-2). It has previously been demonstrated that the HO-1 gene and GCLC gene promoters have the Nrf-2 consensus sequence ARE and can be activated by Nrf-2 [34] [35] . If the HO-1 expression and GCLC expression that are induced by hemin share the same activation mechanism, then it can be expected that they would show similar expression profiles. However, we demonstrated that HO-1 and GCLC showed different expression profiles during hemin pretreatment. HO-1 gene expression reached a peak by 6 h and decreased at 12 and 24 h after hemin pretreatment. In contrast, GCLC gene expression showed no changed by 6 h, increased at 12 h and continued to be upregulated until 24 h after hemin pretreatment. In addition, we demonstrated that the inhibition of HO-1 by ZnPP inhibited the upregulation of GCLC gene expression and GSH synthesis induction. Consistent with this observation, ZnPP markedly abolished the protective action of hemin pretreatment when administered together with hemin at 24 h before the addition of H2O2. However, administering ZnPP 1 h before the addition of H2O2 failed to abolish the protective effect of hemin pretreatment. ZnPP did not affect basal GCLC gene expression in control cells that were pretreated without hemin, and BSO did not influence the gene and protein expression of HO-1. These data suggested that HO-1 could participate in the GCLC gene upregulation that was induced by hemin. We hypothesize that GCLC gene expression occurs downstream of HO-1 activated hemin. Further studies will be required to clarify the molecular mechanism of the GCLC gene upregulation that is mediated by HO-1.
In conclusion, the findings of this study are schematically represented in Figure 5. We have demonstrated for the first time that hemin exerts a cytoprotective effect via the upregulation of GSH synthesis against oxidative stress that is induced by H2O2 in U937 cells. The early event in this process appears to be an increase in HO-1activity that occurs shortly after pretreatment with hemin. HO-1 catalyzes the decomposition of hemin to CO,bilirubin and iron [9] -[12] . Carbon monoxide might activate Nrf-2, resulting in the upregulation of GCLC expression and GSH synthesis [36] . The present study proposes the mechanism of hemin-induced cytoprotective effect in which hemin activates GSH synthesis through HO-1 activation. It is interesting to speculate that the activation of Nrf-2 by several compounds, such as hemin and CO, might result in the activation of different defense pathways. To clarify these possibilities, further studies are needed.
Hemin is used for the treatment of some diseases, such as acute hepatic porphyrias, middle model thalassemia
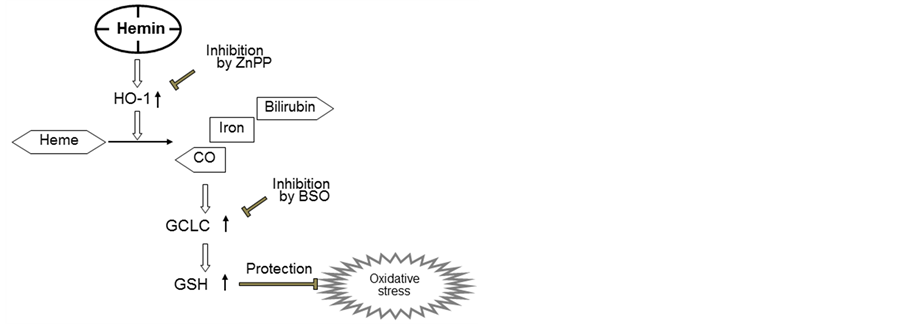
Figure 5. Schema summarizing the inhibition of H2O2-induced cytotoxicity by hemin via the up-regulation of GCLC expression through HO-1 activation..
and marrow a typicality syndrome [37] [38] . Bharucha et al. recently showed that hemin increases the plasma HO-1 protein concentration and HO-1 activity in humans [39] . The concentration of hemin used in the present study is obviously lower than the recommended therapeutic dose (3 mg/kg i.v.) for humans [40] . It is possible that the intracellular GSH in leukocytes increases after 24 h, providing hemin for the patient. Based on these findings, we propose the necessity of verifying the physiological and treated effect of GSH when HO-1 activity is increased.
Acknowledgements
The authors sincerely thank Dr. Ikuo Kashiwakura of Hirosaki University School of Health Sciences for providing cell lines and valuable regents as well as Dr. Kiyoshi Takahashi of Hokkaido Pharmaceutical University School of Pharmacy for helpful suggestions.
References
- Finkel, T. and Holbrook, N.J. (2002) Oxidants, Oxidative Stress and the Biology of Ageing. Nature, 408, 239-247. http://dx.doi.org/10.1038/35041687
- Valko, M., Rhodes, C.J., Monocol, J., Izakovic, M. and Mazur, M. (2006) Free Radicals, Metals and Antioxidants in Oxidative Stress-Induced Cancer. Chemico-Biological Interaction, 160, 1-40. http://dx.doi.org/10.1016/j.cbi.2005.12.009
- Cooke, M.S., Evans, D.M., Dizdaroglu, M. and Lunec, J. (2003) Oxidative DNA Damage: Mechanisms, Mutation, and Disease. The FASEB Journal, 17, 1195-1214. http://dx.doi.org/10.1096/fj.02-0752rev
- Cabello, C.M., Bair 3rd, W.B. and Wondrak, G.T. (2007) Experimental Therapeutics: Targeting the Redox Achilles Heel of Cancer. Current Opinion in Investigational Drugs, 8, 1022-1037.
- Calderwood, S.K., Xie, Y., Wang, X., Khaleque, M.A., Chou, S.D., Murshid, A., Prince, T. and Zhang, Y. (2010) Signal Transduction Pathways Leading to Heat Shock Transcription. Signal Transduction Insights, 2, 13-24. http://dx.doi.org/10.4137/STI.S3994
- Tanaka, H., Sakurai, K., Takahashi, K. and Fujimoto, Y. (2003) Requirement of Intracellular Free Thiols for Hydrogen Peroxide-Induced Hypertrophy in Cardiomyocytes. Journal of Cellular Biochemistry, 89, 944-955. http://dx.doi.org/10.1002/jcb.10568
- Liu, H. and Talalay, P. (2013) Relevance of Anti-Inflammatory and Antioxidant Activities of Exemestane and Synergism with Sulforaphane for Disease Prevention. Proceedings of the National Academy of Science of the United States of America, 110, 19065-19070. http://dx.doi.org/10.1073/pnas.1318247110
- Jones, D.P. (2006) Redefining Oxidative Stress. Antioxidants and Redox Signaling, 8, 1865-1879. http://dx.doi.org/10.1089/ars.2006.8.1865
- Was, H., Dulak, J. and Jozkowicz, A. (2010) Heme Oxygenase-1 in Tumor Biology and Therapy. Current Drug Targets, 11, 1551-1570. http://dx.doi.org/10.2174/1389450111009011551
- Tenhunen, R., Marver, H.S. and Schmid, R. (1968) The Enzymatic Conversion of Heme to Bilirubin by Microsomal Heme Oxygenase. Proceedings of the National Academy of Science of the United States of America, 61, 748-755. http://dx.doi.org/10.1073/pnas.61.2.748
- Stocker, R., Yamamoto, Y., McDonagh, A.F., Glazer, A.N. and Ames, B.N. (1987) Bilirubin Is an Antioxidant of Possible Physiological Importance. Science, 235, 1043-1046. http://dx.doi.org/10.1126/science.3029864
- Ryter, S.W., Morse, D. and Choi, A.M. (2007) Carbon Monoxide and Bilirubin: Potential Therapies for Pulmonary/Vascular Injury and Disease. American Journal of Respiratory Cell and Molecular Biology, 36, 75-82. http://dx.doi.org/10.1165/rcmb.2006-0333TR
- Ryter, S.W. and Otterbein, L.E. (2004) Carbon Monoxide in Biology and Medicine. BioEssays, 26, 270-280. http://dx.doi.org/10.1002/bies.20005
- Juckett, M.B., Balla, J., Balla, G., Jessurun, J., Jacob, H.S. and Vercellotti, G.M. (1995) Ferritin Protects Endothelial Cells from Oxidized Low Density Lipoprotein in Vitro. The American Journal of Pathology, 147, 782-789.
- Yet, S.F., Melo, L.G., Layne, M.D. and Perrella, M.A. (2002) Heme Oxygenase 1 in Regulation of Inflammation and Oxidative Damage. Methods in Enzymology, 353, 163-176. http://dx.doi.org/10.1016/S0076-6879(02)53046-9
- Collino, M., Pini, A., Mugelli, N., Mastroianni, R., Bani, D., Fantozzi, R., Papucci, L., Fazi, L. and Masini, E. (2013) Beneficial Effect of Prolonged Heme Oxygenase 1 Activation in a Rat Model of Chronic Heart Failure. Disease Models & Mechanisms, 6, 1012-1020.
- Ndisang, J.F., Lane, N. and Jadhav, A. (2009) The Heme Oxygenase System Abates Hyperglycemia in Zucker Diabetic Fatty Rats by Potentiating Insulin-Sensitizing Pathways. Endocrinology, 150, 2098-2108. http://dx.doi.org/10.1210/en.2008-0239
- Meister, A.J. (1988) Glutathione Metabolism and Its Selective Modification. The Journal of Biological Chemistry, 263, 17205-17208.
- Noctor, G., Gomez, L., Vanacker, H. and Foyer, C.H. (2002) Interactions between Biosynthesis, Compartmentation and Transport in the Control of Glutathione Homeostasis and Signalling. Journal of Experimental Botany, 53, 1283- 1304. http://dx.doi.org/10.1093/jexbot/53.372.1283
- Sakurai, K. and Cederbaum, A.I. (1998) Oxidative Stress and Cytotoxicity Induced by Ferric-Nitrilotriacetate in HepG2 Cells That Express Cytochrome P450 2E1. Molecular Pharmacology, 54, 1024-1035.
- Dickinson, D.A. and Forman, H.J. (2002) Cellular Glutathione and Thiols Metabolism. Biochemical Pharmacology, 64, 1019-1026. http://dx.doi.org/10.1016/S0006-2952(02)01172-3
- Sundström, C. and Nilsson, K. (1976) Establishment and Characterization of a Human Histiocytic Lymphoma Cell Line (U-937). The International Journal of Cancer, 17, 565-577. http://dx.doi.org/10.1002/ijc.2910170504
- Anderson, M.E. (1985) Determination of Glutathione and Glutathione Disulfide in Biological Samples. Methods in Enzymology, 113, 548-555. http://dx.doi.org/10.1016/S0076-6879(85)13073-9
- Morse, D., Lin, L., Choi, A.M. and Ryter, S.W. (2009) Heme Oxygenase-1, A Critical Arbitrator of Cell Death Pathways in Lung Injury and Disease. Free Radical Biology and Medicine, 47, 1-12. http://dx.doi.org/10.1016/j.freeradbiomed.2009.04.007
- Abraham, N.G. and Kappas, A. (2008) Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacological Reviews, 60, 79-127.
- Franklin, C.C., Backos, D.S., Moharet, I., White, C.C., Forman, J. and Kavanagh, T.J. (2009) Structure, Function, and Post-Translational Regulation of the Catalytic and Modifier Subunits of Glutamate Cysteine Ligase. Molecular Aspects of Medicine, 30, 86-98. http://dx.doi.org/10.1016/j.mam.2008.08.009
- An, J.H. and Blackwell, T.K. (2003) SKN-1 links C. Elegans Mesendodermal Specification to a Conserved Oxidative Stress Response. Genes & Development, 17, 1882-1893.
- Uranga, R.M., Katz, S. and Salvador, G.A. (2013) Enhanced Phosphatidylinositol 3-Kinase (PI3K)/Akt Signaling Has Pleiotropic Targets in Hippocampal Neurons Exposed to Iron-Induced Oxidative Stress. The Journal of Biological Chemistry, 288, 19773-19784.
- Yamamoto, H., Yamamoto, Y., Yamagami, K., Kume, M., Kimoto, S., Toyokuni, S., Uchida, K., Fukumoto, M. and Yamaoka, Y. (2000) Heat-Shock Preconditioning Reduces Oxidative Protein Denaturation and Ameliorates Liver Injury by Carbon Tetrachloride in Rats. Research in Experimental Medicine, 199, 309-318.
- Gilbert, H.F. (1990) Molecular and Cellular Aspects of Thiol-Disulfide Exchange. Advances in Enzymology and Related Areas of Molecular Biology, 63, 69-172. http://dx.doi.org/10.1002/9780470123096.ch2
- Circu, M.L. and Aw, T.Y. (2008) Glutathione and Apoptosis. Free Radical Research, 42, 689-706.
- Yang, Y.C., Lii, C.K., Lin, A.H., Yeh, Y.W., Yao, H.T., Li, C.C., Liu, K.L. and Chen, H.W. (2011) Induction of Glutathione Synthesis and Heme Oxygenase 1 by the Flavonoids Butein and Phloretin is Mediated through the ERK/Nrf2 Pathway and Protects Against Oxidative Stress. Free Radical Biology and Medicine, 51, 2073-2081. http://dx.doi.org/10.1016/j.freeradbiomed.2011.09.007
- Ichimura, T., Ito, M., Takahashi, K., Oyama, K. and Sakurai, K. (2011) Involvement of Mitochondrial Swelling in Cytochrome C Release from Mitochondria Treated with Calcium and Alloxan. Journal of Biophysical Chemistry, 2, 10-18. http://dx.doi.org/10.4236/jbpc.2011.21002
- Balogum, E., Hoque, M., Gong, P., Killeen, E., Green, C.J., Foresti, R., Alam, J. and Motterlini, R. (2003) Curcumin Activates the Haem Oxygenase-1 Gene via Regulation of Nrf2 and the Antioxidant-Responsive Element. Biochemical Journal, 371, 887-895. http://dx.doi.org/10.1042/BJ20021619
- Kim, Y.C., Masutani, H., Yamaguchi, Y., Itoh, K., Yamamoto, M. and Yodoi, J. (2001) Hemin-Induced Activation of the Thioredoxin Gene by Nrf2. A Differential Regulation of the Antioxidant Responsive Element by a Switch of Its Binding Factors. The Journal of Biological Chemistry, 276, 18399-18406. http://dx.doi.org/10.1074/jbc.M100103200
- Li, M.H., Jang, J.H., Na, H.K., Cha, Y.N. and Surh, Y.J. (2007) Carbon Monoxide Produced by Heme Oxygenase-1 in Response to Nitrosative Stress Induces Expression of Glutamate-Cysteine Ligase in PC12 Cells via Activation of Phosphatidylinositol 3-Kinase and Nrf2 Signaling. The Journal of Biological Chemistry, 282, 28577-28586.
- Timonen, T.T. and Kauma, H. (1992) Therapeutic Effect of Hemearginate in Myelodysplastic Syndromes. European Journal of Haematology, 49, 234-238. http://dx.doi.org/10.1111/j.1600-0609.1992.tb00054.x
- Volin, L., Ruutu, T., Knuutila, S. and Tenhunen, R. (1988) Heme Arginate Treatment for Myelodysplastic Syndromes. Leukemia Research, 12, 423-431. http://dx.doi.org/10.1016/0145-2126(88)90062-8
- Bharucha, A.E., Kulkarni, A., Choi, K.M., Camilleri, M., Lempke, M., Brunn, G.J., Gibbons, S.J., Zinsmeister, A.R. and Farrugia, G. (2010) First-in-Human Study Demonstrating Pharmacological Activation of Heme Oxygenase-1 in Humans. Clinical Pharmacology & Therapeutics, 87, 187-190. http://dx.doi.org/10.1038/clpt.2009.221
- Anderson, K.E. (2003) Approaches to Treatments and Prevention of Human Porphyrias. In: Kadish, K.M., Smith, K.M. and Guilard, R., Eds., The Porphyrin Handbook, Medical Aspects of Porphyrins, Volume 14, Academic Press, San Diego, 247-275. http://dx.doi.org/10.1016/B978-0-08-092388-8.50016-9
NOTES

*Corresponding author.