Journal of Biophysical Chemistry
Vol.4 No.2(2013), Article ID:31748,10 pages DOI:10.4236/jbpc.2013.42005
A conformational B-Z DNA study monitored with phosphatemethylated DNA as a model for epigenetic dynamics focused on 5-(hydroxy)methylcytosine
![]()
Kasteel Twikkelerf 94, Tilburg, The Netherlands; h.m.buck@ziggo.nl
Copyright © 2013 Henk M. Buck. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 18 January 2013; revised 25 February 2013; accepted 8 March 2013
Keywords: B-Z DNA Transition; Artificial DNA’s; Epigenetics; Phosphorylation-Demethylation; Alzheimer
ABSTRACT
This study was directed on the Binto Z-DNA isomerization with alternating CG sequences monitored with artificial DNA model-systems based on methylation of the phosphate backbone. The chemical concept for this transition wherein shielding of the oxygen anions of the backbone phosphates plays an essential role, resulted in the preparation of the phosphatemethylated d(CpG). Even on this primitive level of only two base pair long, the B-Z conformational aspects of this selfcomplementary duplex could be described in solution with nuclear magnetic resonance (NMR) and circular dichroism (CD) measurements. The exclusivity of this choice became clear after synthesizing phosphatemethylated DNA with longer alternating CG fragments. It could be shown that conflicting conformational effects of the CG and GC fragments resulted in an overall B structure of the phosphatemethylated tetramer d(CPGPCPG). From our model considerations, it is clear that the internal stress introduced by the alternating CG sequences will be promoted by a complete shielding of the phosphate backbone. Elimination of this effect may be realized by a site-specific phosphate shielding. The role of the anti-syn isomerization of G in the CG fragments is clarified by methylation of the phosphate group. This anti-syn transition is absent in corresponding methylphosphonates, suggesting an exclusive role for base-backbone coordination via hydrogen bonding. In addition, we propose that the Binto Z-DNA interconversion may offer a mechanistic view for differences in dynamics between cytosine and its epigenetic derivative 5-methylcytosine. This mechanism has been extended to the demethylation of 5-methylcytosine and the exchange of information between the new epigenetic base, 5-hydroxymethylcytosine and the DNA backbone via an intramolecular phosphorylation. The role of 5-hydroxymethylcytosine in Alzheimer disease has been briefly discussed. In our opinion, this study can be considered as a new dynamic concept for epigenetics based on the dynamics of the B-Z transition in natural and phosphatemethylated DNA.
1. INTRODUCTION
Recently, artificial models have been developed for small molecule-DNA hybrids which show UV hyperchromicity with increasing melting temperatures and a cooperative melting behaviour through hydrophobic interactions as demonstrated by Eryazici et al. [1]. This specificity has been attributed to a reduced configurational entropy and ion-cloud sharing. Within these aspects, changes in enthalpy and entropy caused by basepair complexation are a substantial part of the hybridization affinity of artifical DNA model systems towards their natural conjugates. A structural difference between complementary strands will be strongly reflected in the overall change in entropy caused by strand adaptation induced by differences in sugar puckering and backbone manipulated DNA systems. Generally, C(2’)-endo or C(3’)-endo puckering in a modified DNA results in a distinct preference for duplexation with natural DNA or natural DNA and RNA, respectively. The C(2’)-endo ring puckering corresponds with B-DNA and the C(3’)- endo puckering with A-DNA. Sugar puckering in RNA is related to an A-conformation that enables genetic transfer from DNA via a transcriptional intermediate into translation.
As artificial models for DNA, we modified the DNA backbone by methylation of the phosphate groups, indicated as phosphatemethylated DNA. This triester demonstrates a strong hybridization affinity with its complementary natural DNA [2,3]. Our experimental data are solely based on the base interactions and sugar puckering, controlling in a distinct way the orientation of the backbone. Although phosphatemethylated DNA is an excellent candidate for duplex formation with its natural conjugate, it is unsuitable for duplex formation with RNA [4-8]. It has been shown under biological and physicalchemical conditions that replication is blocked whereas protein synthesis remains essentially unaffected. With the available data, we come to the conclusion that in the triester DNA the sugar puckering is laid down in C(2’)- endo ring puckering i.e., a B-DNA conformation. Adaptation to an A-form with C(3’)-endo puckering, as is the case in natural DNA (Band A-form), necessary for genetic transfer from DNA to RNA with an A-conformation, is unlikely for sterical reasons [4,9]. In the case of the BZ transition with its alternating C-G sequence, additional factors play an important role as site-specific phosphate shielding and sugar-base conformational changes.
We demonstrate with the B-Z transition the vital importance for genetic transfer using different (hydrogen) networks, resulting in an exchange of information between epigenetic modifications. In other words, the methylation of local lysines can prescribe CpG methylation which requires that methylation of histones and DNA via C(5) of cytosine are cooperative in carrying out an epigenetic instruction for integrating gene activity. A model is suggested for the demethylation of 5-methylcytosine and the role of 5-hydroxymethylcytosine in the exchange of genetic information.
2. METHODS AND MATERIALS
2.1. Synthesis and Characterization of Phosphatemethylated DNA
We were able to synthesize phosphatemethylated fragments with 2 - 5 bases consisting of A, T, G, and C via a stepwise (nonautomated) route following the standard phosphoramidite chemistry of Beaucage et al. [10]. As protective agents for the bases A, G, and C we used the 9-fluorenylmethoxycarbonyl (Fmoc) group [11]. After isolation of the phosphites, the base-protected triester was obtained through oxidation with t-butyl hydroperoxide. For the removal of Fmoc, triethylamine was used.
The pronounced hybridization of phosphatemethylated DNA with its complementary natural DNA (nucleotide ratio 1:1) resulted in a right-handed B-DNA duplex. From the linear relationship of the melting transitions (Tm) and the number of bases (n) for the hybridization of phosphatemethylated Cn (n = 2 - 5) and An (n = 2 - 4) with poly(dG) and poly(dT) respectively, the Tm value varies between 20˚C and 100˚C. The ratio of the Tm/n values for f (Tm,n) gives a value of 1.40, that corresponds nicely with the ratio 1.50 of the three hydrogen bridges in a CG pair versus the two hydrogen bridges in an AT pair. These hybridizations are irrespective of the chirality of phosphorus. With the experimental data, it could be shown that the Tm values of a phosphatemethylated DNA n-mer hybridized with its natural k-mer, as illustrated in Figure 1, depends on the length of the k-mer.
We demonstrate this biophysical property within the change of f (Tm,n) for phosphatemethylated Cn hybridized with Gk. For k = 18 there is a distinct and sharp sigmoidal transition from n = 3 to n = 4. Increase of the k value results into a linear relationship between Tm and n. This is one of the fascinating properties of an allosteric-like interaction between phosphatemethylated DNA and its natural counterpart. The results are given in Figure 2.
Although these results are based on experimental data of about 25 years ago, it is for the first time that suchlike allosteric interactions of this type of hybridization are demonstrated in a straightforward manner [3,6]. This means that phosphatemethylated DNA binds cooperatively to its natural complementary DNA which involves that the natural strand adapts the modified conformation during hybridization. This allosteric effect is absent in cases where the sugar puckering and backbone conformations differ substantially from the complementary natural one. An example in the latter category is methylphosphonate Tn and poly(dA) with a maximum Tm value for n = 4. In that case the P-CH3 bond disturbs the helical structure because of stereoelectronic reasons. Both conformational aspects also play their role in the inhibition on the RNA level. In the case of phosphatemethylated Cn and poly (rG) with a maximum Tm value for n = 2, it is the difference in sugar puckering (Bversus A-conformation). For methylphosphonate Tn and poly(rA), the maximum Tm value is reached for n = 4 (deviation from the natural helical structure). Hybridization between self-complementary phosphatemethylated DNA’s may be in competition with parallel C-C duplexes for SP chirality of phosphorus, not with the RP chirality, and with parallel T-T duplexes for both SP and RP chiralities [12].
The phosphate-shielding model is used in order to afford additional information for our understanding of the B-Z dynamics in natural DNA. The results are given in the next section.
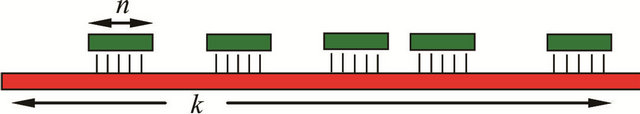
Figure 1. Representation of a phosphatemethylated DNA n-mer hybridized with its complementary natural strand of k nucleotides.
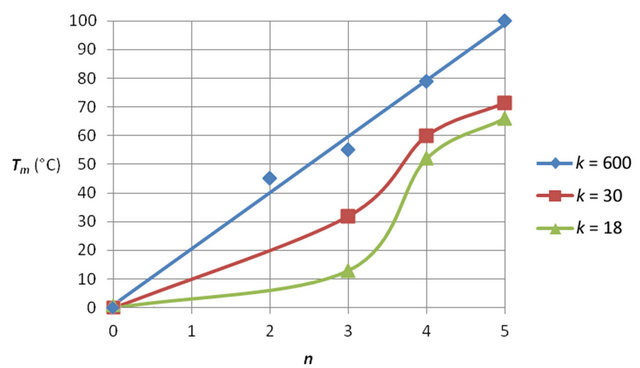
Figure 2. Melting temperature (Tm) of phosphatemethylated Cn for n = 2 - 5 hybridized with its natural complement Gk for different k values. The Tm values are obtained from [3,6].
3. THE B-Z TRANSITION IN DNA AND PHOSPHATEMETHYLATED DNA. A MODEL DESCRIPTION
3.1. B-Z Transition in DNA: Site Specific Shielding with Metal Cations. Initiation and Complexation with (Metal-) Organic Substrates
An important contribution on the salt-induced conformational change of a DNA double helix from the right-handed B-form into the zig-zag left-handed Z-form has been given by Maruyama et al. [13]. The fragments consisting of 12 base pairs with alternating CPG sequences were prepared as solute. The results show that the free energy of DNA in electrolyte solution is dominated by the electrostatic interactions between the phosphate anions and the positively charged counterions. This explains that with increasing salt concentration, the Zform becomes more stable. Other aspects with respect to the salt-induced Binto Z-DNA transition are the corresponding conformational changes. A theoretical study has been given by Kastenholtz et al. [14] using molecular dynamic simulations for a hexamer duplex with explicit solvation. Various models have been formulated for the Binto Z-dynamics. The most favoured one is the Wang model [15]. Distortion of the hydrogen-bridge interaction between the bases C and G in B-DNA is followed by rotation of the entire dC residue and concomitant anti-syn conformational change of G. Rejoining of the complementary bases results in Z-DNA. In relation to this model, we used quantum-chemical calculations starting with a rotation around the C(4’) - C(5’) bond as the result of P(V) trigonal bipyramidal (TBP) intermediate. In combination with the glycosidic C(1’)-N bond, it can be shown that the dC structure is selectively activated [16]. A conclusive model is offered by a crystallographic structure of a B-Z junction as given by Ha et al.
[17]. Then it was concluded that the Binto Z-geometry is realized by sequentially extruding of base pairs leading to free rotation by 180˚ followed by rehybridization. From our studies we suggested that the stabilization of the Z-form is the result of a site-specific interaction by cations with the phosphate anions of the DNA backbone, vide supra. Molecular mechanics calculations, directed on the distribution of the cations over the Z-DNA surface, have also demonstrated that complexation preferentially occurs with the phosphate anions of d(CPG) and not with d(GPC) motifs of the helix [14]. With 31P nuclear magnetic resonance (NMR) measurements of the natural hexamer at different concentrations of Mg2+ ions, an explicit selectivity between the different phosphates could be established. In the absence of Mg2+ ions (corresponding to B-DNA), one single resonance shift was observed. Increasing salt concentration (corresponding to Z-DNA) resulted in two well-resolved resonances with an intensity ratio of 2:3. The induced chemical shift is much larger for the three CPG phosphates than for the two GPC phosphates, indicating a favoured complexation of the Mg2+ ions in Z-DNA for the CPG parts [18]. With these NMR measurements it is clear that site-specific shielding of the oxygens of the phosphatediester linkages is of vital importance for inducing the Binto Z-transition in DNA.
The role of organic cations in the B-Z dynamics has been described in a number of papers. We mention the work of Shimada et al., using poly (L-lysine)-graft-dextran [19]. It was found that not only the cationic main chain was involved in the transition but also the dextran grafts. They propose that the side chains of the copolymer favour the electrostatic interaction between the cationic ammonium group and the phosphate anion of the backbone. Recently an interesting paper was published by Gong et al., on stabilization of Z-DNA with metallicorganic substrates as Zn(II) meso-tetrakis (N-methylpyridinium-4-yl)porphirin, ZnTMPyP [20]. In the complex structure, ZnTMPyP binds at the major groove of the Z-DNA. The stabilization occurs via the electrostatic interaction between the phosphate anions and the positive charges of the N-methylpyridinium fragments. With linear dichroism (LD) in the Soret region the preference for complexation with the Z-form was established. This specific way of complexation with Z-DNA is based on one of the crucial differences with B-DNA that Z-DNA contains only one deep helical groove.
3.2. The Band Z-Geometry in Phosphatemethylated DNA. A Specific Role for the B-Z Dynamics in Epigenetics
The effect of covalent shielding has been shown in Figure 3 with the circular dichroism (CD) spectra of the two left-handed self-complementary phosphatemethylated d(CPG)’s in combination with the two right-handed self-complementary phosphatemethylated d(GPC)’s [8, 18].
The characteristic differences in the sugar puckering for the Zand B-form of this miniduplex, namely C(2’)- endo, C(3’)-endo and C(2’)-endo, C(2’)-endo respectively, in combination with the C(4’) - C(5’) rotamers having dominant gauche-trans (dG) and gauche-gauche (dC) conformations for the Z-form and corresponding gauchegauche conformations for both bases in the B-form, were confirmed with 1H NMR analysis [9,18]. The conformation of a Z-DNA fragment is shown in Figure 4.
The difference in chirality of phosphorus in the triester has no influence on the weight of the population densities of the rotamers. The characteristic syn conformation of guanine in the Z-form was confirmed with nuclear double resonance. An indication of the stability of the diastereoisomers was obtained with the Tm values from 1H NMR (3 × 10−3 M) via the chemical shift profiles of H(6, dC) and H(1’, dG) for the duplex of d(CPG) and the chemical shift profiles of H(5, dC) and H(6, dC) for the duplex of d(GPC), in combination with UV hyperchromicity (5 × 10−6 M) measurements. The Tm values for the Z-form are significantly lower than for the B-form. For the duplex phosphatemethylated d(CPG) we found 9˚C and 13˚C for the RP and SP chirality, respectively. The corresponding d(GPC) duplex shows a transition of 27˚C for both configurations [8,18].
In that respect it is noteworthy that the molecular structure of the ammonium salt of the natural d(CPG) in the crystalline state shows a Z-DNA helix as was demonstrated by Ramakrishnan and Viswamitra [21]. The four hydrogens of the NH4+ cation play a stabilizing role in the organization of different hydrogen networks, suggesting a complex interaction for the formation of a lefthanded double helix. The major groove NH4+ cation interacts with N(7) of guanines via water bridges besides making hydrogen bond interactions directly with the anions of the phosphate groups. The NH4+ cation located in the minor groove interacts directly with the phosphate anions and water molecules. A more elaborate study was published before with the hexamer by Wang et al. [22].
This situates the molecular structure of the duplex phosphatemethylated d(CPG) measured in solution in a unique position. Apparently, besides the shielding of the phosphates and the characteristics of the sugar puckering for a left-handed natural d(CPG) duplex, the influence of coordination in solution and in the crystal state is of importance. In a search for significant hydrogen networks for the methylphosphotriester d(CPG) duplex in solution, it is of importance for understanding the dynamics of the B-Z transition to underline that reduction of phosphate-phosphate charge repulsion results in a decrease of the helix diameter, thus facilitating the anti-syn isomerization of guanine in combination with the conformational change in sugar puckering. We suggest that one of the two hydrogens of the guanine N(2) amino group (H+ donor) which is not involved in the duplex bonding with O(2) of cytosine, will be linked with one of the oxygens of the phosphotriester (H+ acceptor). An intermolecular interaction focused on the role of the guanine hydrogens can be demonstrated with the X-ray structure of 2’-deoxy-3’,5’- di-O-acetyl guanosine [23] as shown in Figure 5.
An intramolecular base-backbone hydrogen bonding has been established for the syn-nucleotide 8-bromo-2’,3’- O-isopropylidene-adenosine via O(5’)-H…N(3). The rigid complexation could be detected via a long range H(5”)- N(3) spin-spin coupling in apolar solvents [24]. This geometrical conformation is also present in the crystalline state [25]. This type of hydrogen-bond coordination may play an essential role in the anti-syn conformational change of guanine. In our opinion this interaction model may offer an explanation for the absence of a left-handed structure for a corresponding methylphosphonate DNA [26].
However, our artificial system is more complicated in its physical-chemical response than it appears at first glance. Preparing the tetrameric duplex of phosphatemethylated d(CPGPCPG) via the stepwise procedure, UV hyperchromicity measurements show relatively low temperature transitions at 18˚, 32˚, and 41˚C [18,27]. The eight diastereoisomers apparently form duplexes with different stabilities. The CD spectrum shows the overall characteristics of the B-form. The presence to some extent of a Z-form cannot be excluded. These results implicate that complete methylation of the phosphate backbones is not the appropriate way to realize a Z-geometry. It is to be expected that a subsequent increase of the
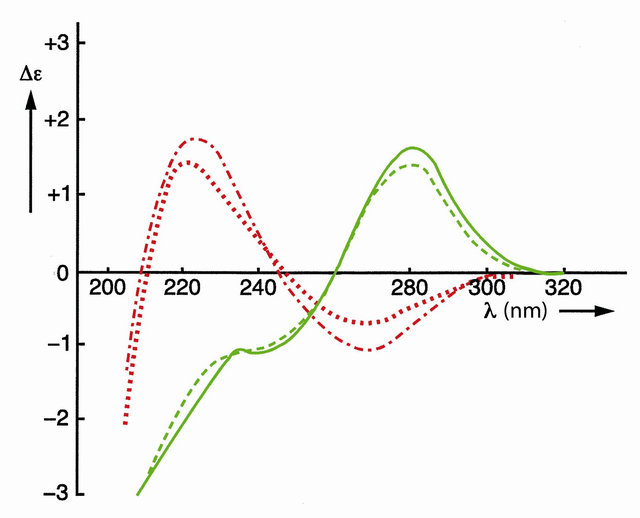
Figure 3. CD spectra of self-complementary phosphatemethylated Z-d(CPG) for RP (top) and SP, red curves, and self-complementary phosphatemethylated B-d(GPC) for RP (top) and SP, green curves. Measured in water at 4˚C.
Figure 4. The conformation of a Z-DNA fragment.
number of bases may result in duplex destabilization, caused by an intrinsic conformational competition between the d(CPG) and d(GPC) parts. Likely, complete shielding of the oxygens of the phosphate backbones results to an increasing extent in inter-strand contact which will disfavour the anti-syn conformational change of guanine. If methylation follows the site-specific shielding of the Mg2+ cations, then complete methylation is the wrong strategy for the B-Z transition. These aspects will play a fundamental role in the interaction of Z-DNA binding proteins as ADAR1 which has two left-handed Z-DNA binding domains (Zα and Zβ) at its N-terminus, focused on the local hydrogen bonding of specific amino acids, Lys, Arg and Tyr (water mediated) with the phosphates. In the most simple example as is illustrated with the natural duplex d(CPGPCPGPCPG) in the B-form, the Z-form is initiated by the Zα protein that binds to BDNA and then converts it to Z-DNA stabilized by a second Zα molecule. An NMR study on these duplexes and the B-Z junction formation of DNA duplexes induced by Z-DNA binding domain of ADAR1, has been given by Lee et al. [28]. Following the profile of the Mg2+-phosphate complexation in relation to the stability of the Zform, then there may be an entrance 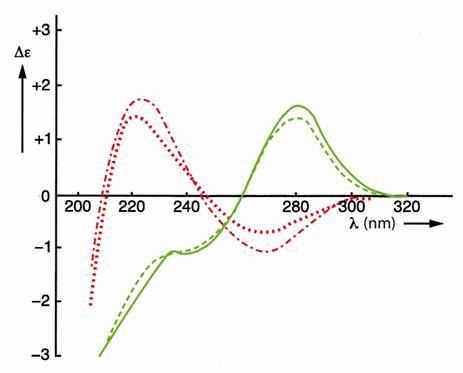 for understanding the biological role of Z-DNA in the regulation of transcription. Regions near the transcription site contain motifs favourable for forming Z-DNA. Concerning the B-Z transition in CG rich segments with conservation of the B-structure of neighbouring parts in more complex systems, it was established with X-ray and NMR that breaking of one base pair and extruding of these bases results in the separation between the B-DNA and Z-DNA segments. Therefore we suggest that the unwinding of ZDNA necessary for the transcription process will be controlled by a dynamic shift (replacement) of specific interaction sites between the phosphate and the hydrogen bonding amino acids necessary for stabilization of the Zform.
for understanding the biological role of Z-DNA in the regulation of transcription. Regions near the transcription site contain motifs favourable for forming Z-DNA. Concerning the B-Z transition in CG rich segments with conservation of the B-structure of neighbouring parts in more complex systems, it was established with X-ray and NMR that breaking of one base pair and extruding of these bases results in the separation between the B-DNA and Z-DNA segments. Therefore we suggest that the unwinding of ZDNA necessary for the transcription process will be controlled by a dynamic shift (replacement) of specific interaction sites between the phosphate and the hydrogen bonding amino acids necessary for stabilization of the Zform.
In addition we use the Binto Z-DNA dynamics as a biomolecular concept for the epigenetic coherence between histone and base methylation. This is demonstrated in the next section.
4. EPIGENETICS. A MODEL STUDY WITH 5-METHYLCYTOSINE AND 5-HYDROXYMETHYLCYTOSINE AS FIFTH AND SIXTH BASE OF THE GENOME.
4.1. 5-Methylcytosine Locations and Their Influence on Gene Expression
Epigenetic reactions accompanying the dynamics of gene expression are fundamental to cell determination and function without alteration in DNA sequence. Epigenetics is controlled by histone and base methylation. The first modification is based on methylation of the Nterminal tails of histones as mono-, diand trimethylated cation lysine residues. Demethylation of the monoand dimethylated lysine cations occurs with LSD1, a nuclear homologue of amine oxidases which are flavine dependent enzymes that utilize O2 and generate H2O2 and formaldehyde as byproducts [29]. It is suggested that trimethylated lysine turnover is accomplished by histone replacement or by an unknown demethylase specific for the trimethylated one.The methylated lysines are present as cations by protonation or fully methylation of the Nterminal site which results in a contact ion-pair with the phosphate-oxygen anion of the DNA backbone. The second molecular modification in epigenetics is DNA methylation via C(5) of cytosine. It has been shown that histone and DNA methylation provide a mechanistic link. This connection has been described with the PcG protein EZH2 which directly controls DNA methylation with DNMT as part of an epigenetic process of integrating gene-silencing networks. This involves that EZH2-mediated histone-lysine methylation (H3K27) can dictate CPG methylation [30]. A more complex example is given by the mediation of MPP8 between DNA and histone methyltransferases in which the phosphorylated protein with its negatively charged phosphate group effectuates the binding with the positively charged methylated lysine [31]. We focused on this mediation because it gives a simplified molecular description based on the mechanism of the Binto Z-DNA transition [32]. In this transition the position of the CPG pair changes from near the center to the periphery of the duplex that makes C(5) of cytosine more accessible for methylation. The impact of C(5) methylation of cytosine on the phosphate shielding has been demonstrated with the fully methylated hexamer. At a much lower critical salt concentration than in the case of the unmethylated hexamer, a Binto Z-transition takes place [33]. Then the question arises in which manner these two epigenetic modifications come to expression in a cooperative way. Most of the molecular model studies which are published are dealing with X-ray structures of methylated DNA as epigenetic factor. These studies were focused on the role of C(5) methylcytosine in stabilizing the duplex with a transcriptional repressor that mediates gene silencing. This epigenetic signal can be interpreted by a family of proteins that recognize symmetrically 5-methyl CPG pairs via MBD, a C(5) methyllated CPG-binding domain [34,35]. The X-ray structure of the repressor MBD-DNA complex reveals that the C(5) methyl groups make contact with a predominantly hydrophilic surface that includes tightly bound water. It is suggested that the repressor recognizes the hydration environment localized in the major groove of the DNA in which the C(5) methyl group of cytosine plays a rather unusual role as coordination center. The X-ray geometry clearly shows that the carbon-oxygen distance in C-H···O is not the result of a pure hydrogen bridge. An average value of 0.35 nm has been obtained. The orientation of the oxygens in the two coordinated water molecules (OwA and OwB) follows the tetrahedral geometry of the methyl group at C(5) in the arrangement of AwO···H-C-H···OwB. This coordination is facilitated with the assistance of the N(4) hydrogen bridge of methylcytosine via N(4)-H···OwA, and with one of the oxygens of the phosphate backbone via P-O-···H-OwB. In this intramolecular hydrogen-bridging network which is preserved by deprotonation/demethylation of the monoand dimethyl lysine cationic tails of histones or by acetylation of the appropriate histone site, MBD plays its role as coordination link between CPG pairs and the N-
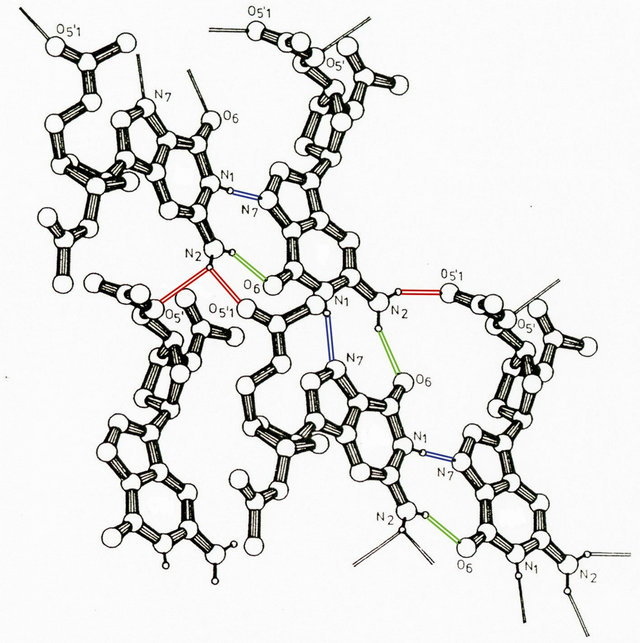
Figure 5. Projection of a part of a hydrogen bond network of acetyl guanosine. The hydrogen bonding is indicated by different colours.
terminal tails of histones. In the molecular description of this processing the (de)activation of the phosphate linkage plays an essential role. Although no direct evidence is available concerning an interaction model with DNA, a local contact ion-pair with the phosphate backbone is suggested. On the other hand from our model studies it is clear that backbone shielding via counterions and covalent shielding via methylation has a direct influence on transmission of genetic information. However, it should be mentioned that in the presented model systems a high concentration of ‘methyl marks’ is present which is in contrast with the real biological reproduction [36]. In fact the influence of a relatively small number of marks in such a system is unique. An explanation based on a molecular design suffers from a lack of information focused on the specific role of the various histones. We hypothesize that histones may play an important role in the effectiveness and optimization of the distances between the cationic methylated lysines and the negatively charged phosphate linkages by reduction of the length of the DNA backbone. Suchlike conformational changes may be the result of the molecular dynamics of a supercoiled DNA and its displacement at the interface of the histone. A recent study of van Loenhout et al. [37] on plectonemes as supercoils of DNA, based on a mechanical trick (applied streching force) and ionic strength, delivers an interesting model for DNA dynamics. Although the plectonemic coils differ from the coiling in combination with histones, they may offer additional insight in the role of the epigenetic modification originated from the N-terminal tails of histones as the mono-, diand trimethylated cation lysine residues. We focus on the influence of the ionic strength on the number of plectonemes. It has been established that increasing/decreasing salt concentration leads to a decrease/increase of these plectonemes. This observation is the result of an increase/decrease of shielding of the phosphate backbone that suppresses/promotes the electrostatic repulsion between DNA backbones. Under these conditions a certain flexibility for coiling is guaranteed, accompanied with mobility for (re)location of ion-pair bonding necessary for genetic response. Summarizing we may conclude that the overall process is a sophisticated balance of changes in enthalpy (phosphate (de)shielding) and entropy (coiling conformations and displacements) in cooperation with changes in histone geometries. It is tempting to introduce more interactive networks in combination with the specific enzymes in order to support our view on conformational transitions in DNA in relation to epigenetics. However, the complex and inconstant behaviour of the epigenetic outcome in the various biological processes makes it difficult to describe the molecular biology along the lines of bio-organic chemistry. Nevertheless we attempted to give a new mechanistic view based on the chemistry of phosphatemethylated DNA in particular directed at genetic inhibition of a specific region of natural DNA and the Binto Z-DNA conformational change. Combination of both processes results in a cooperative model which we consider as a useful mechanism for a description of molecular pathways in epigenetics. A strong indication for this cooperative model has been given by Blackledge et al. This study clearly demonstrates that CPG islands use a histone lysine demethylase for differentation [38]. A more recent example is demonstrated for the inheritance of histone methylation during DNA replication [39]. Small RNA molecules with silencing factors (siRNA) promote a regio-specific histone methylation in which the methylated lysine provides a binding site for an extending silencing network that favours DNA replication.
4.2. 5-Hydroxymethylcytosine Locations and their Influence on Gene Expression. A Model for Demethylation of 5-Methylcytosine
5-Hydroxymethylcytosine is a new epigenetic modification resulting by the oxidation of 5-methylcytosine at CPG sites by the TET family of cytosine oxygenases [40]. It is possible that 5-hydroxymethylcytosine represents a route to demethylate 5-methylcytosine via 5-formyl and 5-carboxycytosine [41]. Various studies have been shown that 5-hydroxymethylcytosine is present in high levels in the brain [40,42,43]. The lower affinity for 5-methyl CPG binding proteins suggests that 5-hydroxymethylcytosine might have a different role in the regulation of gene expression and may not be restricted to 5-methyl CPG. Nearest-neighbour analyses have shown that in embryonic stem cells about one fifth of total 5-methylcytosine is present in 5-methyl CPT, 5-methyl CPA and to a minor extent in 5-methyl CPC sequences, whereas somatic tissues have neglible non-methylated 5-CPG [44]. Molecular dynamics simulations of the binding mode of SRA-DNA complexes consisting of 5-methylcytosine and 5-hydroxymethylcytosine provide a structural explanation for recognition of 5-hydroxymethylcytosine [45]. It was found that UHRF1, a key factor in methyl conservation, leads to a structural differentation between the cytosines. The geometric orientation of 5-hydroxymethylcytosine that fits in the binding pocket of the UHRF1 SRA domain is in accordance with the expected extra stabilization introduced by hydrogen bonding of the hydroxyl group. This interaction is facilitated by a conserved water molecule in the SRA binding pocket that stabilizes the overall hydrogen bonding including the specific amino acid sites and the backbone of the DNA. In the case of the methylcytosine, the conserved water molecule in the vicinity of the methylcytosine only participates with the SRA domain and the phosphate backbone, vide supra, in which the methyl groups make contact with a predominantly hydrophilic surface that includes tightly bond water. Effectuating this model description for a more direct interaction of 5-hydroxymethylcytosine with the phosphate backbone, a route for a temporary phosphorylation of 5-hydroxymethylcytosine may be opened. In fact this intermediate state affords a stringent link between histone and 5-hydroxymethylcytosine. This transition structure consists of a P(V) TBP in which the OH group of the 5-hydroxymethylcytosine and one of the anionic oxygens of the phosphate backbone coordinated with the positively charged N-terminal tail of a lysine residue occupy the apical positions of the TBP. This model must be considered as a concept for an on-off mechanism in the exchange of information. The fact that the relevant molecular sites are in apical locations of the TBP involves that the dynamic reversibility is guaranteed [46]. We consider this model as an intramolecular base-backbone interaction in which the phosphorylation is activated via a P(V) TBP intermediate. It is tempting to construct a similar transition geometry for 5-carboxycytosine, resulting in decarboxylation. An illustration is given in Figure 6.
In fact phosphorylation of 5-hydroxymethylcytosine may also result (in part) in the formation of cytosine with formaldehyde as leaving group. Such a process may be responsible for the initiation of protein Tau into globular amyloid-like aggregates in vitro and in vivo [47] resulting in Alzheimer’s disease. These model considerations may be helpful in understanding these complex biomolecular interactions.
5. DISCUSSION
The site-specific shielding of the backbone phosphates in Z-DNA is of importance for understanding its stability. The significance of the introduction of modified DNA’s as the methylphosphotriester DNA has been demonstrated. Flexibility in conformational adaptation of ZDNA within Band A-structures has given sufficient support for their interplay in cellular processes as controlling transcription activity. Modified Z-DNA as methylphosphotriester is (only) observed on a two base-pair level.
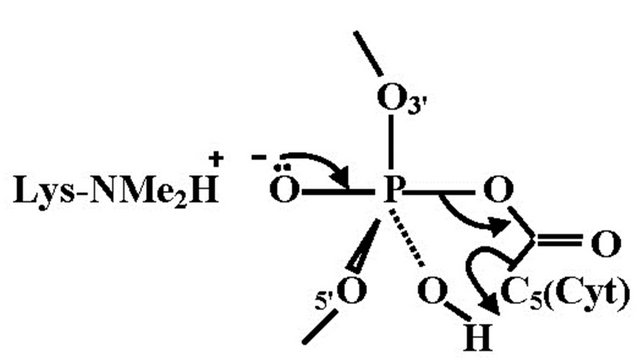
Figure 6. The TBP geometry for decarboxylation of 5-carboxycytosine. The equatorial positions represent the DNA backbone and the P-OH group, in which the hydrogen facilitates the dissociation of cytosine by C(5) protonation.
Longer CG fragments of these triesters can be considered as distorted B-conformations with decreasing duplex stability. The triesters prepared with the stepwise nonautomated procedure hybridize with their complementary natural DNA and not with RNA.
We demonstrated the vital importance for genetic transfer by different (hydrogen) networks, suggesting a complex interaction between two epigenetic modifications. In other words, the methylation of local lysines can prescribe CPG methylation which requires that methylation of histones and DNA via C(5) of cytosine are cooperative in carrying out an epigenetic instruction for integrating gene activity. As a biomolecular concept for the epigenetic coherence between histone and base methylation, we used the Binto Z-DNA dynamics in combination with the hybridization properties of phosphatemethylated DNA.
REFERENCES
- Eryazici, I., Yildirim, I., Schatz, G.C. and Nguyen, S.T. (2012) Enhancing the melting properties of small molecule-DNA hybrids through designed hydrophobic interactions: An experimental-computational study. Journal of the American Chemistry Society, 134, 7450-7458. doi:10.1021/ja300322a
- Koole, L.H., van Genderen, M.H.P., Reiniers, R.G. and Buck, H.M. (1987) Enhanced stability of Watson & Crick duplex structure by methylation of the phosphates groups in one strand. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 90, 41-46.
- Moody, M.R., van Genderen, M.H.P. and Buck, H.M. (1990) Thermodynamics of polymer duplexes between phosphate-methylated DNA and natural DNA. Biopolymers, 30, 609-618. doi:10.1002/bip.360300513
- van Genderen, M.H.P., Koole, L.H. and Buck, H.M. (1988) Duplex stability of hybrids between phosphatemethylated DNA and natural RNA. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 91, 53-57.
- van Genderen, M.H.P. (1989) Structure and stability of phosphate-methylated DNA duplexes: model systems for specific DNA-protein interaction and conformational transmission. Thesis, Eindhoven University of Technology, Eindhoven.
- van Genderen, M.H.P., Koole, L.H. and Buck, H.M. (1989) Hybridization of phosphatemethylated DNA and natural oligonucleotides. Implications for protein-induced DNA duplex destabilization. Recueil Travaux Chimiques des Pays-Bas, 108, 28-35. doi:10.1002/recl.19891080106
- Buck, H.M. (1996) Phosphate-methylated DNA: A unique oligodeoxynucleotide as compared with other modified DNAs. Proceedings of the Koninklijke Nederlandse Akademie van Wetenschappen, 99, 145-153.
- Buck, H.M. (2004) The chemical and biochemical properties of methylphosphotriester DNA. Nucleosides, Nucleotides and Nucleic Acids, 23, 1833-1847. doi:10.1081/NCN-200040620
- Buck, H.M. (2007) The chemical and biochemical properties of methylphosphotriester DNA and RNA in comparison with their corresponding methylphosphonates. A dynamic model description. Nucleosides, Nucleotides and Nucleic Acids, 26, 205-222. doi:10.1080/15257770601112812
- Beaucage, S.L. and Caruthers, M.H. (1981) Deoxynucleoside phosphoramidites—A new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Letters, 22, 1859-1862. doi:10.1016/S0040-4039(01)90461-7
- Koole, L.H., Moody, H.M., Broeders, N.L.H.L., Quaedflieg, P.J.L.M., Kuijpers, W.H.A., van Genderen, M.H.P., Coenen, A.J.J.M., van der Wal, S. and Buck, H.M. (1989) Synthesis of phosphate-methylated DNA fragments using 9-fluorenylmethoxycarbonyl as transient base protecting group. Journal of Organic Chemistry, 54, 1657-1664. doi:10.1021/jo00268a030
- Koole, L.H., van Genderen, M.H.P. and Buck, H.M. (1987) A parallel right handed duplex of the hexamer d(TPTPTPTPTPT) with phosphate triester linkages. Journal of the American Chemistry Society, 109, 3916-3921. doi:10.1021/ja00247a015
- Maruyama, Y., Yoshida, N. and Hirata, F., (2010) Revisiting the salt-induced conformational change of DNA with 3D-RISM theory. Journal of Physical Chemistry B, 114, 6464-6471. doi:10.1021/jp912141u
- Kastenholz, M.A., Schwartz, T.U. and Hünenberger, P.H. (2006) The transition between the B and Z conformations of DNA investigated by targeted molecular dynamics simulations with explicit solvation. Biophysical Journal, 91, 2976-2990. doi:10.1529/biophysj.106.083667
- Wang, A.H.-J., Quigley, G.J., Kolpak, F.J., Crawford, J.L., van Boom, J.H., van der Marel, G. and Rich, A. (1979) Molecular structure of a left-handed double helical DNA fragment at atomic resolution. Nature, 282, 680-686. doi:10.1038/282680a0
- van Lier, J.J.C., Smits, M.T. and Buck, H.M. (1983) B-Z Transition in methylated DNA: A quantum-chemical study. European Journal of Biochememistry, 132, 55-62. doi:10.1111/j.1432-1033.1983.tb07324.x
- Ha, S.C., Lowenhaupt, K., Rich, A., Kim, Y.-G. and Kim, K.K. (2005) Crystal structure of a junction between BDNA and Z-DNA reveals two extruded bases. Nature, 437, 1183-1186. doi:10.1038/nature04088
- Quaedflieg, P.J.L.M., Koole, L.H., van Genderen, M.H.P. and Buck, H.M. (1989) A structural study of phosphatemethylated d(CPG)n and d(GPC)n DNA oligomers. Implication of phosphate shielding for the isomerization of B-DNA into Z-DNA. Recueil Travaux Chimiques des Pays-Bas, 108, 421-423. doi:10.1002/recl.19891081107
- Shimada, N., Yamamoto, M., Kano, A. and Maruyama A. (2010) Cationic graft copolymer as a DNA B-Z transition inducer: Effect of copolymer structure. Biomacromolecules, 11, 3043-3048. doi:10.1021/bm100870b
- Gong, L., Jang, Y.J., Kim, J. and Kim, S.K. (2012) Zform DNA specific binding geometry of Zn(II) mesotetrakis (N-methylpyridinium-4-yl) porphyrin probed by linear dichroism spectroscopy. Journal of Physical Chemistry B, 116, 9619-9626. doi:10.1021/jp3041346
- Ramakrishnan, B. and Viswamitra, M.A. (1988) Crystal and molecular structure of the ammonium salt of the dinucleoside monophosphate d(CPG). Journal of Biomolecular Structure & Dynamics, 6, 511-523. doi:10.1080/07391102.1988.10506504
- Wang, A.H.-J., Quigley, G.J., Kolpak, F.J., van der Marel, G., van Boom, J.H. and Rich, A. (1981) Left-handed double helical DNA: Variations in the backbone conformation. Science, 211, 171-176. doi:10.1126/science.7444458
- Koole, L.H., Buck, H.M., Kanters, J.A. and Schouten, A. (1988) Molecular conformation of 2’-deoxy-3’,5’-di-Oacetyl guanosine. Crystal structure and high resolution proton nuclear magnetic resonance investigations. Canadian Journal of Chemistry, 66, 2634-2639. doi:10.1139/v88-413
- Koole, L.H., de Boer, H., de Haan, J.W., Haasnoot, C.A.G., van Dael, P. and Buck, H.M. (1986) Intramolecular basebackbone association in 8-bromo-2’,3’-O-isopropylideneadenosine. Detection of an O(5’)-H···N(3) spin-spin coupling. Journal of the Chemical Society, Chemical Communications, 4, 362-364. doi:10.1039/c39860000362
- Fujii, S., Fujiwara, T. and Tomita, K. (1976) Structural studies on the two forms of 8-bromo-2’,3’-O-isopropylideneadenosine. Nucleic Acids Research, 3, 1985-1996. doi:10.1093/nar/3.8.1985
- Callahan, L., Han, F.-S., Watt, W., Duchamp, D., Kézdy, F.J. and Agarwal, K. (1986) B-to Z-DNA transition probed by oligonucleotides containing methylphosphonates. Proceedings of the National Academy of Sciences USA, 83, 1617-1621. doi:10.1073/pnas.83.6.1617
- Rijkers, D. (1990) Synthesis of model systems for B-Z transition of DNA. Graduate Study, Eindhoven University of Technology, Eindhoven.
- Lee, Y.-M., Kim, H.-E., Park, C.-J., Lee, A.-R., Ahn, H.- C., Cho, S.J., Choi, K.-H., Choi, B.-S. and Lee, J.-H. (2012) NMR study on the B-Z junction formation of DNA duplexes induced by Z-DNA binding domain of human ADAR1. Journal of the American Chemistry Society, 134, 5276-5283. doi:10.1021/ja211581b
- Shi, Y., Lan, F., Matson, C., Mulligan, P., Whetstine, J.R., Cole, P.A., Casero, R.A. and Shi, Y. (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell, 119, 941-953. doi:10.1016/j.cell.2004.12.012
- Viré, E., Brenner, C., Deplus, R., Blanchon, L., Fraga, M., Didelot, C., Morey, L., van Eynde, A., Bernard, D., Vanderwinden, J.-M., Bollen, M., Esteller, M., Di Croce, L., de Launoit, Y. and Fuks, F. (2006) The polycomb group protein EZH2 directly controls DNA methylation. Nature, 439, 871-874. doi:10.1038/nature04431
- Chang, Y., Sun, L., Kokura, K., Horton, J.R., Fukuda, M., Espejo, A., Izumi, V., Koomen, J.M., Bedford, M.T., Zhang, X., Shinkai, Y., Fang, J. and Cheng, X. (2011) MPP8 mediates the interactions between DNA methyltransferase Dnmt3a and H3K9 methyltransferase GLP/ G9a. Nature Communications, 2, 533. doi:10.1038/ncomms1549
- Buck, H.M. (2011) DNA systems for B-Z transition and their significance as epigenetic model: The fundamental role of the methyl group. Nucleosides, Nucleotides and Nucleic Acids, 30, 918-944. doi:10.1080/15257770.2011.620580
- Fujii, S., Wang, A.H.-J., van der Marel, G., van Boom, J.H. and Rich, A. (1982) Molecular structure of (m5dC-dG)3: The role of the methyl group on 5-methyl cytosine in stabilizing Z-DNA. Nucleic Acids Research, 10, 7879-7892. doi:10.1093/nar/10.23.7879
- Mayer-Jung, C., Moras, D. and Timsit, Y. (1998) Hydration and recognition of methylated CPG steps in DNA. European Molecular Biology Organization Journal, 17, 2709-2718.
- Ho, K.L., McNae, I.W., Schmiedeberg, L., Klose, R.J., Bird, A.P. and Walkinshaw, M.D. (2008) MeCP2 binding to DNA depends upon hydration at methyl-CPG. Molecular Cell, 29, 525-531. doi:10.1016/j.molcel.2007.12.028
- McEwen, K.R. and Ferguson-Smith, A.C. (2010) Distinguishing epigenetic marks of developmental and imprenting regulation. Epigenetics & Chromatin, 3, 2. doi:10.1186/1756-8935-3-2
- Van Loenhout, M.T.J., de Grunt, M.V. and Dekker, C. (2012) Dynamics of DNA supercoils. Science, 338, 94-97. doi:10.1126/science.1225810
- Blackledge, N.P., Zhou, J.C., Tolstorukov, M.Y., Farcas, A.M., Park, P.J. and Klose, R.J. (2010) CPG islands recruit a histone H3 lysine 36 demethylase. Molecular Cell, 38, 179-190. doi:10.1016/j.molcel.2010.04.009
- Li, F., Martienssen, R. and Can, W.Z. (2011) Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature, 475, 244-248. doi:10.1038/nature10161
- Jin, S.G., Wu, X., Li, A.X. and Pfeifer, G.P. (2011) Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Research, 39, 5015-5024. doi:10.1093/nar/gkr120
- Ito, S., Shen, L., Dai, Q., Wu, S.C., Collins, L.B., Swenberg, J.A., He, C. and Zhang, Y. (2011) Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science, 333, 1300-1303. doi:10.1126/science.1210597
- Chouliaras, L., van den Hove, D.L.A., Kennis, G., Keitel, S., Hof, P.R., van Os, J., Steinbusch, H.W.M., Schmitz, C. and Rutten, B.P.F. (2012) Age-related increase in levels of 5-hydroxymethylcytosine in mouse hippocampus is prevented by caloric restriction. Current Alzheimer Research, 9, 536-544.
- van den Hove, D.L.A., Chouliaras, L. and Rutten B.P.F. (2012) The role of 5-hydroxymethylcytosine in aging and Alzheimer’s disease: Current status and prospects for the future studies. Current Alzheimer Research, 9, 545-549.
- Tahiliani, M., Koh, K.P., Shen, Y., Pastor, W.A., Bandukwala, H., Brudno, Y., Agarwal, S., Iyer, L.M., Liu, D.R., Aravind, L. and Rao, A. (2009) Conversion of 5- methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930-935. doi:10.1126/science.1170116
- Frauer, C., Hoffmann, T., Bultmann, S., Casa, V., Cardoso, C., Antes, I. and Leonhardt, H. (2011) Recognition of 5- hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One, 6, e21306. doi:10.1371/journal.pone.0021306
- Castelijns, M.M.C.F., Schipper, P., van Aken, D. and Buck, H.M. (1981) Dynamic equilibriums between pentavalent protonated oxyphosphoranes and their isomeric tetravalent enol phosphonium ions via interand intramolecular proton transfer. Journal of Organic Chemistry, 46, 47-53. doi:10.1021/jo00314a010
- Nie, C.L., Wei, Y., Chen, X., Liu, Y.Y., Dui, W., Liu, Y. Davies, M.C., Tendler, S.J. and He, R.G. (2007) Formaldehyde at low concentration induces protein tau into globular amyloid-like aggregates in vitro and in vivo. PloS One, 2, e629. doi:10.1371/journal.pone.0000629