Green and Sustainable Chemistry
Vol. 2 No. 4 (2012) , Article ID: 24408 , 10 pages DOI:10.4236/gsc.2012.24018
Environmentally Benign Synthetic Protocol for O-Alkylation of β-Naphthols and Hydroxy Pyridines in Aqueous Micellar Media
Department of Chemistry, Osmania University, Hyderabad, India
Email: *kcrajannaou@yahoo.com
Received August 2, 2012; revised September 5, 2012; accepted September 17, 2012
Keywords: Ultrasonically Assisted; Microwave Assisted; Green Chemistry; Micelles; β-Naphthols; Heteroaryl Hydroxy Compounds; Alcohols
ABSTRACT
Ultrasonic and microwave-assisted practical methods have been developed for the O-alkylation of aryl (β-naphthols) and Heteroaryl (hydroxy pyridines) in aqueous surfactant media in good to excellent yields. The developed methods are simple, efficient, economical and environmentally safe. Our novel methods describe a set of green methods to Williamson synthesis.
1. Introduction
The discovery, development and identification of biologically active compounds gained lot of importance in the recent years. Even though there are considerable number of adverse effects, chemists always tried to design a drug possessing maximum therapeutic application and minimum toxicity. Organic ethers are one of the most important classes of chemicals which have significant applications as herbicides, disinfectors, pharmaceuticals, plasticizers, solvents, drug intermediates and as solvents in organic synthesis. They are also used as important precursors for polymers and fragrances [1,2]. Literature survey shows that Williamson synthesis is probably one of the most common classical methods being used for the preparation of symmetrical and unsymmetrical ethers [3-11].
A good number of homogeneous Bronsted acids or Lewis acid based transition metals have also been reported as catalysts in the etherification of alcohols [5-9]. However, these systems exhibited some drawbacks including their deactivation through decomposition by the water formed during the course of reaction. In many cases these methods also reveal the accumulation of significant amount of acid at the end of reaction due to the hydrolysis of Lewis acid catalysts such as metal oxide, which upon neutralization gives considerable amount of salts. The acid waste, residual catalyst and salt material may cause several environmental problems when they are dispossed off, and some of them may be toxic. Difficulties such as catalyst recovery for reuse and neutralization of the reaction mixture also make the process tedious. Literature survey shows some reports on the use of phase transfer, polymer and clay supported catalysts for the synthesis of symmetrical and unsymmetrical ethers [12- 18]. Another general root for the synthesis of organic ethers is Mitsunobu reaction, which uses either PPh3 or polymer bound PPh3 as catalyst. Triphenyl phosphene is highly sensitive to water and toxic in nature, and the reaction demands inert and dry conditions [18-22]. Recent publications of Kumaraswamy et al. [20], Deababratha and coworkers [21,22] provide excellent bibliography and information on the synthesis of aryl, heteroaryl ethers derived from various roots.
Noteworthy drawbacks encountered in many conventional methods necessitated the search for an appropriate reaction system to enhance reaction conversion and less environmental pollution. Combinatorial synthesis has brought lot of revolution in the recent trends of organic synthesis. Many chemical reactions with organic substances conducted in the laboratory as well as in industry need organic solvents as reaction media, even though water is safe, benign, environmentally friendly and cheap compared with organic solvents. The necessity of organic solvents as reaction media in the industry sector and academic institutions [23,24], could be attributed to probably for two important reasons: 1) most of the organic substances are insoluble in water and soluble in apolar organic solvents and 2) many organic substrates, reagents and catalysts are decomposed or deactivated in aqueous medium. In spite of all these factors, the goal is to develop a novel catalytic system that uses water as a solvent for a wide range of reactions. A perusal of literature indicated that surfactants have been used to promote a variety of synthetic organic reactions. It is believed that surfactants form micelles in protic solvents such as water which act as micro reactors to enhance the reaction rates and afford very good to excellent yields of end products [25-27]. Our preliminary studies in this direction ended up fruitful results when we have performed the title reactions in aqueous micellar media. Thus, in this study, we have used a variety of anionic (SDS), cationic (CTAB) and non-ionic (Tx-100) micellar media. The first drawback of water as a solvent (the solubility) is overcome by the use of aqueous solutions of surfactants, which solubilised all the reactants.
Green or sustainable chemistry has now attained the status of a major scientific discipline, and the studies in this area have led to the development of cleaner and relatively benign chemical processes with many new technologies being developed each year. Encouraged by the “Green Chemistry strategies” of Paul Anastas and Warner [28], we have made an attempt to develop synthetic protocols for etherification reactions. Ultrasonic waves are the one of the most important tools to assist and trigger chemical reactions. Rate accelerations have been noticed in many sluggish reactions [29-35]. Thus the ultrasonically assisted reactions (USAR) became a part of green chemical reactions. The other part of non-conventional energy sources to trigger a chemical reaction is the use of microwaves [36-40]. Microwaves are electromagnetic radiation with a frequency range from 300 to 300,000 MHz, with free space wavelengths of 1 m to 1 mm. Microwaves don’t have any effect on molecular bonds or electron clouds like other parts of electromagnetic radiation such as infrared (IR) or the uv-visible radiations. The energy of microwaves is so low that only molecular rotation could be induced. In the present study we were successful to develop an acid free and solvent free synthetic protocols for etherification of phenols and hydroxy pyridines by replacing H2SO4 with a variety of micelle forming surfactants such as sodium dodecyl sulphate (SDS), cetyl trimethyl ammonium bromide (CTAB) and Triton-X 100 (Tx-100) with and without using water as reaction medium. We were also successful to conduct ultrasonically assisted (USAR) and microwave assisted reactions (MWAR).
2. Experimental Details
2.1. Typical Experimental Procedure for Etherification of β-Naphthols under Conventional Conditions
To a stirred solution of methanol (0.64 g, 20 mmol) and 0.1 M micellar solution (10 mL) was added substituted β-naphthol (1.44 g, 10 mmol) and heated up to reflux and the progress of the reaction was monitored by TLC. After the completion of reaction, the reaction mixture was diluted with dichloromethane (10 mL), and separated from aqueous layer. The organic layer was then washed with water (3 × 5 mL), dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford the crude product. The crude product was purified by column chromatography (silica gel, 100 - 200 mesh) using EtOAchexane (1:9) as eluent to obtain the desired product.
2.2. Typical Experimental Procedure for Ultrasonically-Assisted Etherification (USAS) Etherification of β–Naphthols
To a solution of methanol (0.64 g, 20 mmol)) and 0.1 M micellar solution (10 mL) was added substituted β- naphthol (1.44 g, 10 mmol) at room temperature, and sonicated at 40˚C in an ultrasonic bath. The ultrasonic bath had a frequency of 33 kHz and electric power rating of 100 W. The reaction was carried out in a round bottom flask of 50 mL capacity equipped with a mechanical agitator and the flask was suspended at the centre of the ultrasonic bath. The final products were isolated by absorbing the reaction mixture into silica gel and purifying it by column chromatography using ethyl acetate/hexane gradient.
2.3. Typical Experimental Procedure for Microwave Irradiated Etherification (MWAR) Etherification of β-Naphthols
To a solution of methanol (0.64 g, 20 mmol)) and 0.1 M micellar solution (10 mL) was added substituted β- naphthol (1.44 g, 10 mmol) and these mixtures were heated in a controlled microwave synthesizer (Biotage Initiator + SP Wave model, 0 - 200 W at 2.45 GHz, capped at 60 W during steady state) for few minutes (attains temperature 120˚C and 1 bar pressure). The final products were isolated by absorbing the reaction mixture into silica gel and purifying it by column chromatography using ethyl acetate/hexane gradient.
3. Spectroscopic Data
2-Methoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 3.91 (s, 3H), 7.15 - 7.17 (d, J = 8.8 Hz, 1 H), 7.29 - 7.36 (m, 2H), 7.43 - 7.47 (m, 1H), 7.72 - 7.77 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 55.6, 105.9, 113.7, 124.4, 126.2, 127.8, 128.2, 129.1, 129.9, 133.2, 153.8; IR (KBr) 3067, 3008, 2963, 1632, 1599, 1477, 1462, 1452, 1440, 1398, 1391, 1368, 1262, 1218, 1197, 1173, 1152, 1141, 1118, 1031, 1017, 963, 948, 874, 838, 818, 753, 743, 622, 481, 470 cm−1; MS (EI) m/z 159.2 (M)+.
2-Ethoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 1.46 (t, J = 7.2 Hz, 3H), 4.127 (d, J = 6.8 Hz, 2H), 7.14 - 7.15 (d, J = 8.8 Hz, 1H), 7.28 - 7.39 (m, 2H), 7.39 - 7.43 (m, 1H), 7.69 - 7.75 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 14.8, 64.1, 106.5, 118.9, 123.4, 126.2, 126.7, 128.2, 129.1, 129.2, 133.6, 156.8; IR (KBr) 3067, 2984, 2940, 2876, 1829, 1601, 1579, 1511, 1457, 1440, 1395, 1390, 1367, 1358, 1350, 1269, 1259, 1185, 1166, 1144, 1122, 1046, 1020, 958, 928, 873, 823, 762, 719, 623, 477 cm−1; MS (EI) m/z 173.2 (M)+.
2-Propoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 1.06 (t, J = 6.0 Hz, 3H), 1.83 - 1.86 (m, 2H), 4.01 (t, 2H), 7.10 - 7.14 (m, 2H), 7.30 - 7.41 (m, 2H), 7.69 - 7.74 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 10.5, 22.6, 69.4, 106.6, 119.1, 123.4, 126.2, 126.6, 127.6, 128.8, 129.3, 134.6, 156.7; IR (KBr) 3060, 2976, 2934, 2908, 2878, 1948, 1904, 1832, 1628, 1600, 1513, 1479, 1467, 1448, 1441, 1392, 1371, 1357, 1270, 1260, 1216, 1146, 1121, 1046, 1024, 1017, 1017, 986, 958, 916, 842, 819, 770, 746, 727, 727, 645, 623, 474; MS (EI) m/z 187 cm−1. 10 (M)+.
2-Isopropoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 1.45 (d, J = 6.0 Hz, 6H), 4.66 - 4.82 (m, 1H), 7.13 - 7.18 (m, 2H), 7.28 - 7.48 (m, 2H), 7.68 - 7.82 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 22.0, 69.8, 108.4, 119.7, 123.4, 126.2, 126.6, 127.6, 128.8, 129.3, 134.6, 155.7; IR (KBr) 3580, 2981, 2936, 2876, 1948, 1904, 1832, 1628, 1600, 1581, 1510, 1468, 1440, 1388, 1373, 1356, 1334, 1216, 1188, 1171, 1137, 1118, 1019, 974, 941, 900, 871, 842, 814, 675, 645, 623, 532 cm−1; MS (EI) m/z 187.10 (M)+.
1-Chloro-2-methoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 4.03 (s, 3H), 7.25 (d, J = 8.6 Hz, 1H), 7.37 - 7.52 (m, 1H), 7.53 - 7.61 (m, 1H), 7.79 - 7.69 (m, 2H), 8.25 (d, J = 8.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 57, 105.9, 113.7, 124.4, 126.2, 127.8, 128.2, 129.1, 129.9, 133.2, 153.8; IR (KBr) 3430, 3049, 2972, 2948, 2846, 2541, 1948, 1763, 1625, 1590, 1505, 1469, 1355, 1337, 1273, 1246, 1187, 1148, 1068, 1018, 985, 894, 865, 804, 764, 740, 657, 588, 532 cm−1; MS (EI) m/z 192 (M)+.
1-Bromo-2-methoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 4.04 (s, 3H), 7.28 (d, J = 8.6 Hz, 1H), 7.39 - 7.53 (m, 1H), 7.53 - 7.61 (m, 1H), 7.76–7.89 (m, 2H), 8.23 (d, J = 8.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 57.1, 105.9, 113.7, 124.4, 126.2, 127.8, 128.2, 129.1, 129.9, 133.2, 153.8; IR (KBr) 3430, 3045, 2970, 2941, 2841, 1620, 1594, 1500, 1466, 1454, 1351, 1334, 1270, 1245, 1185, 1153, 1134, 1061, 1021, 968, 890, 855, 803, 761, 743, 708, 644, 579, 516 cm−1; MS (EI) m/z (M-H)+ 236.10.
1-Iodo-2-methoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 4.03 (s, 3H), 7.22 (d, J = 9.0 Hz, 1H), 7.34 - 7.42 (m, 1H), 7.50 - 7.56 (m, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.84 (d, J = 9.0 Hz, 1H), 8.15 (d, J = 8.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 57.3, 87.8, 113.0, 124.4, 128.2, 128.3, 130.4, 131.3, 130.0, 135.7, 156.7; IR (KBr) 3042, 3006, 2969, 2937, 2838, 1617, 1587, 1551, 1497, 1451, 1423, 1346, 1328, 1263, 1242, 1181, 1153, 1132, 1058, 1021, 959, 887, 801, 761, 743 cm−1; MS (EI) m/z 285.06 (M)+.
1-Bromo-2-ethoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 1.38 - 1.41 (t, J = 6.8 Hz, 3H), 4.13 - 4.14 (q, J = 6.8 Hz, 2H), 7.15 - 7.17 (d, J = 8.8 Hz, 1 H), 7.29 - 7.36 (m, 2H), 7.43 - 7.47 (m, 1H), 7.78 - 7.83 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 22.8, 64.6, 105.9, 113.7, 124.4, 126.2, 127.8, 128.2, 129.1, 129.9, 133.2, 153.8.
1-Bromo-2-propoxynaphthalene: 1H NMR (400 MHz, CDCl3) δ 1.03 (t, J = 7.5 Hz, 3H), 1.80 - 1.86 (m, 2H), 4.51 (t, 7.5 Hz, 2H), 7.10 - 7.14 (m, 2H), 7.30 - 7.41 (m, 2H), 7.69 - 7.74 (m, 3H); 13C NMR (50 MHz, CDCl3) δ 10.5, 22.6, 78.4, 113.6, 123.4, 126.2, 126.6, 127.6, 128.8, 129.3, 131.3, 132.6, 154.7; MS (EI) m/z 266.04 (M)+.
1-Bromo-2-isopropoxy naphthalene: 1H NMR (400 MHz, CDCl3) δ 1.45 (d, J = 6.0 Hz, 6H), 4.61 - 4.87 (m, 1H), 7.28 (d, J = 8.6 Hz, 1H), 7.39 - 7.53 (m, 1H), 7.53 - 7.61 (m, 1H), 7.76 - 7.85 (m, 2H), 8.23 (d, J = 8.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 22.0, 69.8, 108.4, 119.7, 123.4, 126.2, 126.6, 127.6, 128.8, 129.3, 134.6, 155.7; IR (KBr) 3580, 2981, 2936, 2876, 1948, 1904, 1832, 1628, 1600, 1581, 1510, 1468, 1440, 1388, 1373, 1356, 1334, 1216, 1188, 1171, 1137, 1118, 1019, 974, 941, 900, 871, 842, 814, 675, 645, 623, 532 cm−1; MS (EI) m/z 266.10 (M)+.
4-Methoxy pyridine: 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 6.81 (d, J = 8.6 Hz, 2H), 8.43 (d, J = 8.6 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 55.1, 109.9, 151.4, 165.4, MS (EI) m/z 110.3(M)+, (B.P.168.2˚C).
3-Methoxy pyridine: 1H NMR (400 MHz, CDCl3) δ 3.84 (s, 3H), 7.34 (t, J = 8.65, J = 4.75 Hz, 1H), 7.38 (d, J = 8.65 Hz, 1H); 8.19 (d, J = 4.75 Hz, 1H); 8.32 (s, 1H); MS (EI) m/z110.3(M)+, (B.P.168.4˚C).
2-Methoxy pyridine: 1H NMR (400 MHz, CDCl3) δ 3.94 (s, 3H), 6.72 (d, J = 8.65 Hz, 1H); 6.82 (t, J = 8.65, J = 4.75 Hz, 1H), 7.59 (t, J = 8.65, J = 4.75 Hz 1H); 8.18 (d, J = 4.75 Hz, 1H); 13C NMR (50 MHz, CDCl3) δ 53.1, 110.9, 116.4, 138, 147,165.4,MS (EI) m/z 110.3 (M)+, (B.P.142.5˚C).
4-Ethoxy pyridine: 1H NMR (400 MHz, CDCl3) δ 1.34 (q, 2H), 3.94 (t, 3H), 6.81 (d, J = 8.4 Hz, 2H), 8.43 (d, J = 8.4 Hz, 2H); MS (EI) m/z 123.8(M)+, (B.P.165˚C)
6-Methoxypicolinaldehyde: 1H NMR (400 MHz, DMSO) δ 3.971 (s, 3H), 6.986 (d, J = 8.3Hz, 1H), 8.108 - 8.136 (m, 1H), 8.770 (d, J = 2.5 Hz, 1H); 9.968 (s, 1H); MS (EI) m/z 137.9(M)+.
5-Bromo-2-methoxypyrimidine: 1H NMR (400 MHz, DMSO) δ 3.915 (s, 3H), 8.765 (s, 2H); MS (EI) m/z 188.9 (M)+.
4. Results & Discussion
Aryl ethers, heteri aryl ethers in general and Naphthalene derivatives such as alkoxy naphthalenes in particular have been identified as one of the best ranges of potent antimicrobials effective against wide range of human pathogens. They occupy a central place among medicinally important compounds due to their diverse and interesting antibiotic properties with minimum toxicity. Synthesis of alkoxy naphthalenes under Williamson’s conditions (refluxing in sulfuric acid and organic solvent) is widely used method for the synthesis of aryl ethers. But this method required several hours (≥20 h) at relatively high temperature. Even though there are some reports to modify the drastic conditions of these reactions, many of them exhibited long reaction times, accumulation of unwanted by-products, which ultimately involved tedious work up procedures. Aqueous surfactants are environmentally benign, non-flammable and possesses remarkable ability to catalyze chemical transformations between some insoluble organic reactants. Encouraged by this aspect we have conducted etherification of β- naphthol and hydroxy pyridine reactions in aqueous micellar media (Scheme 1). For this purpose we have used easily available in the laboratory bench top chemicals sodium dodecyl sulfate (SDS, anionic), cetyl trimethyl ammonium bromide (CTAB, cationic) and Triton X-100 (Tx-100, non-ionic) surfactants to generate micelles in water. However, for comparison, we have also conducted etherification reactions under Williamson’s classical conditions in acidic media (Tables 1 and 2).
Results obtained under acidic and acid-free micellar conditions are compiled in Tables 1 and 2, which clearly indicate highly significant rate accelerations followed by very good yield of end products. Catalytic activity of different micelles is in the order: CTAB > SDS > Tx-100. It is believed that micelles themselves act as micro reactors. The catalytic effect of the micellar solution of CTAB may be attributed to the hydrophobic nature of organic substrates. In micellar solution, organic substrates are pushed away from water molecules towards the hydrophobic core of micelle droplets thus inducing efficient collisions between organic substrates which eventually enhance the reaction rate and result in rapid reactions in water. The hydrophobic interior of the mi-
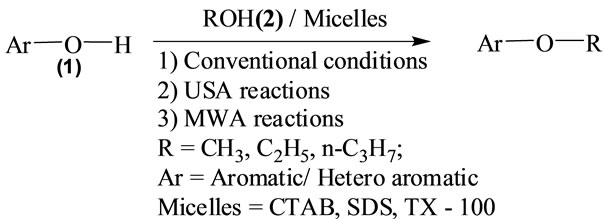
Scheme 1. Etherification of 2-naphthol and hydroxy pyridine in aqueous micellar media.
celles swiftly excludes the water molecules generated during the reaction, thus shifting the equilibrium towards the desired product that ultimately leads to an increase in the reaction yield [41-43]. This explanation is schematically represented in Figures 1 and 2.
Ultrasonic-assisted organic synthesis (USAOS) is a powerful and green approach which is being used to accelerate synthesis of organic compounds. Recent literature reports in USAOS (sonochemical synthesis) indicated that it is an environmentally benign synthesis, which minimized the use of the precious metal catalysts and led to the development of new eco-friendly protocols [29-35]. It offered the synthetic chemist a method of chemical activation which uses equipment which is relatively inexpensive. The driving force for sonochemistry is cavitation and so a general requirement is that at least one of the phases of the reaction mixture should be a liquid. The chemical effects of ultrasound do not come from a direct interaction with molecular species but arises from acoustic avitation the formation, growth, and implosive collapse of bubbles in a liquid. The observed rate and yield enhancements observed in the present study (Tables 3 and 4) could be attributed to ultrasonic cavitation effect.
Results obtained under microwave assisted synthesis (MWAS) micellar conditions are compiled in Tables 5 and 6, which clearly indicate highly remarkable rate accelerations (reaction times reduced from several (≥20) to few minutes), followed by high yields. This dramatic rate enhancement could be attributed to bulk activation of molecules, which is believed to be due to rapid superheating of the polar solvents and pressure effects [36-40].
5. Conclusion
In conclusion, this paper reports Ultrasonicand microwave-assisted O-alkylation of β-naphthols in aqueous micellar media. It describes catalytic activity of different micelles is in the order: CTAB > SDS > Tx-100. The reaction times were drastically reduced to one to two hours under sonication and few minutes under microwave conditions from several hours of classical reactions. Thus, the present protocols show rate accelerations associated with high products yields, when compared with
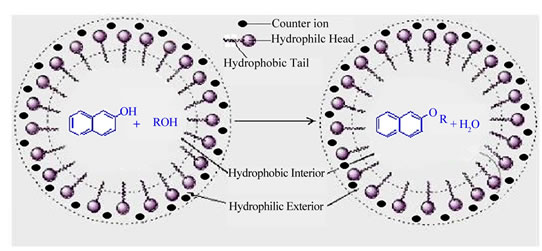
Figure 1. Schematic diagram representing the role of micelles in aqueous media.
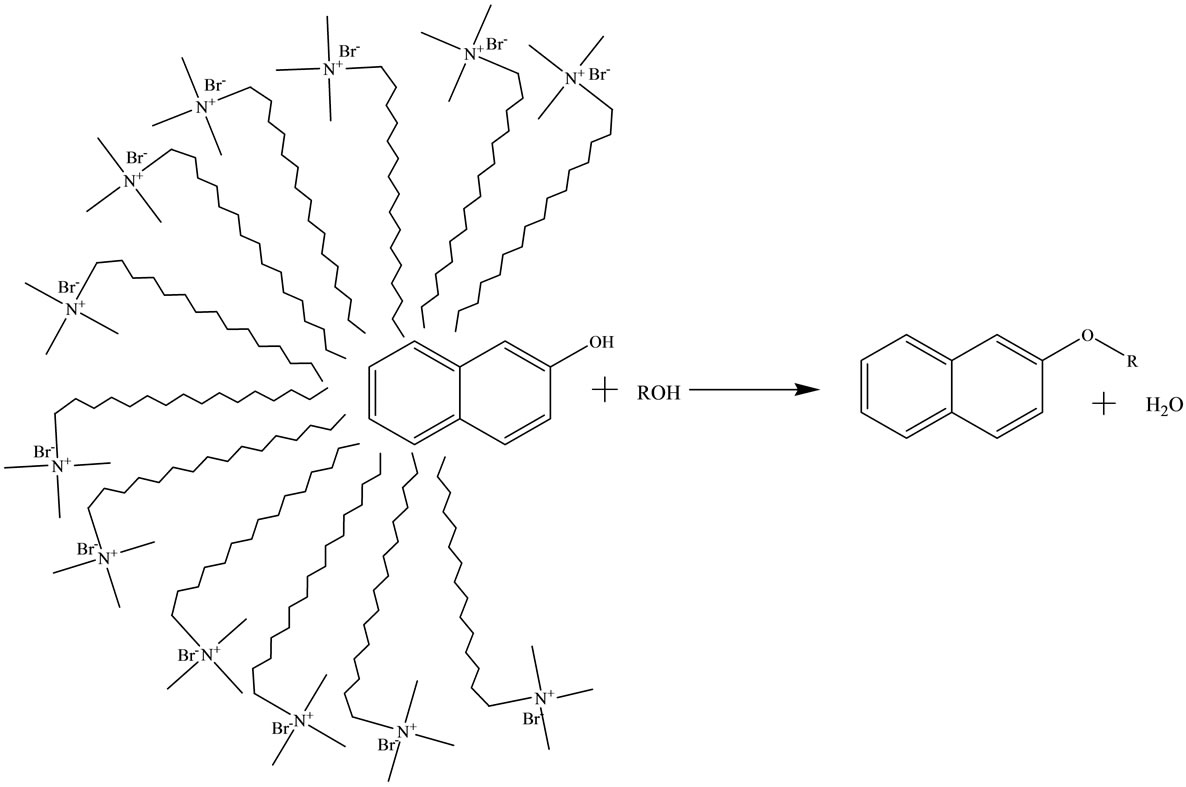
Figure 2. Schematic diagram representing the role of CTAB in aqueous media.
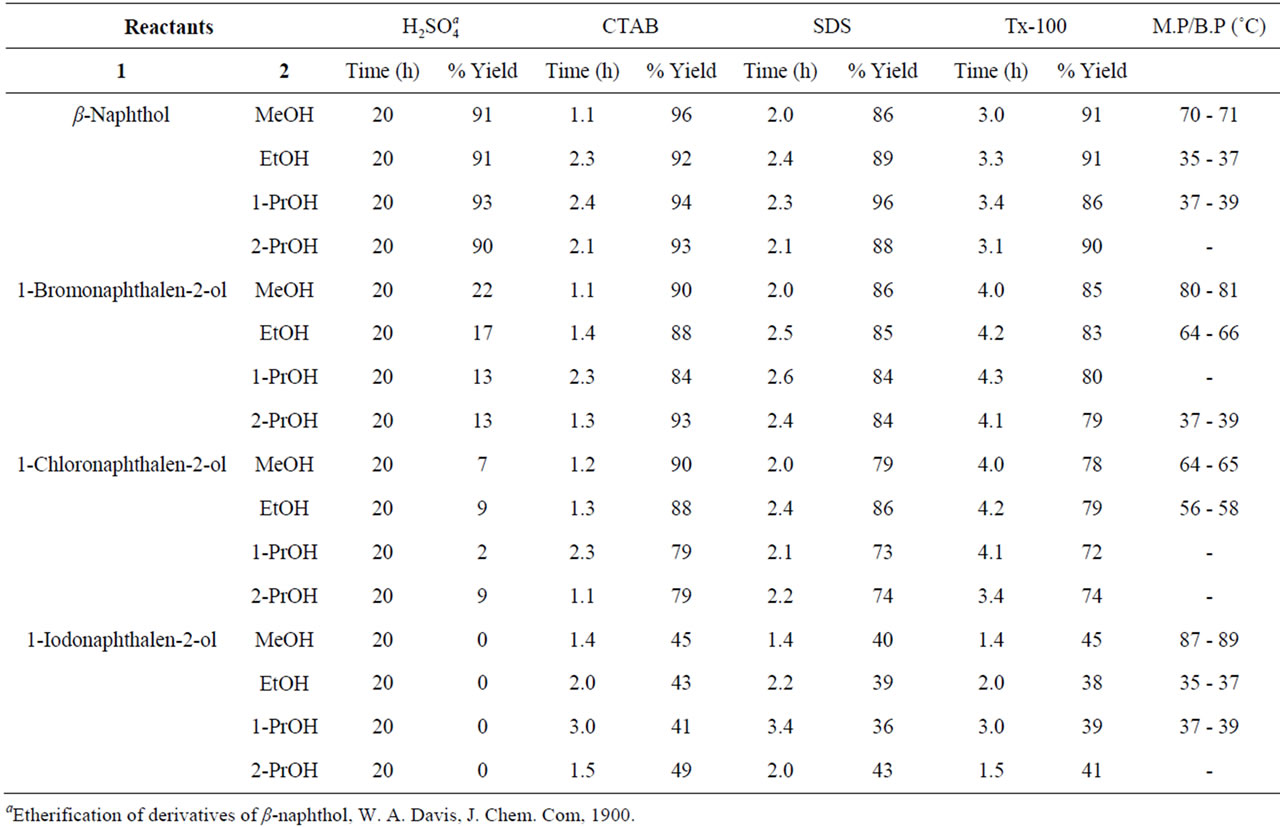
Table 1. Micellar mediated O-alkylation of β-naphthols under acidic and acid-free conditions.
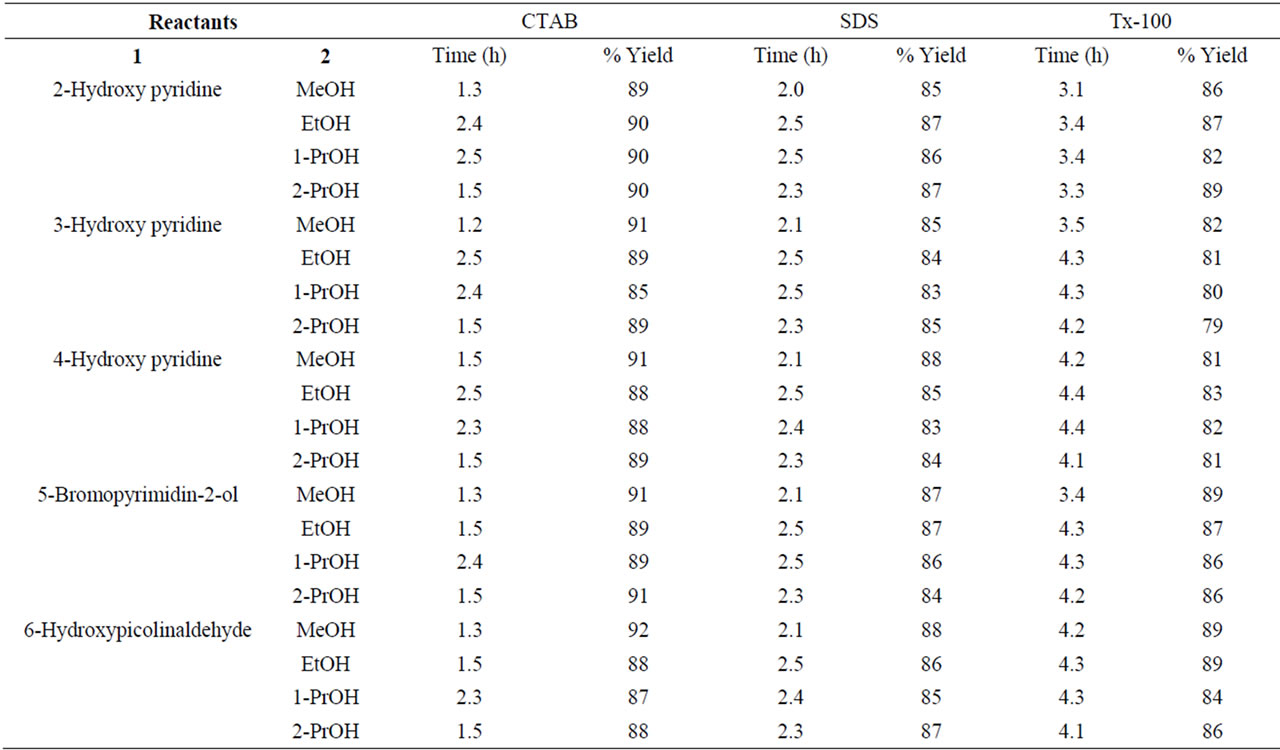
Table 2. Micellar mediated O-alkylation of hydroxy pyridines under conventional conditions.
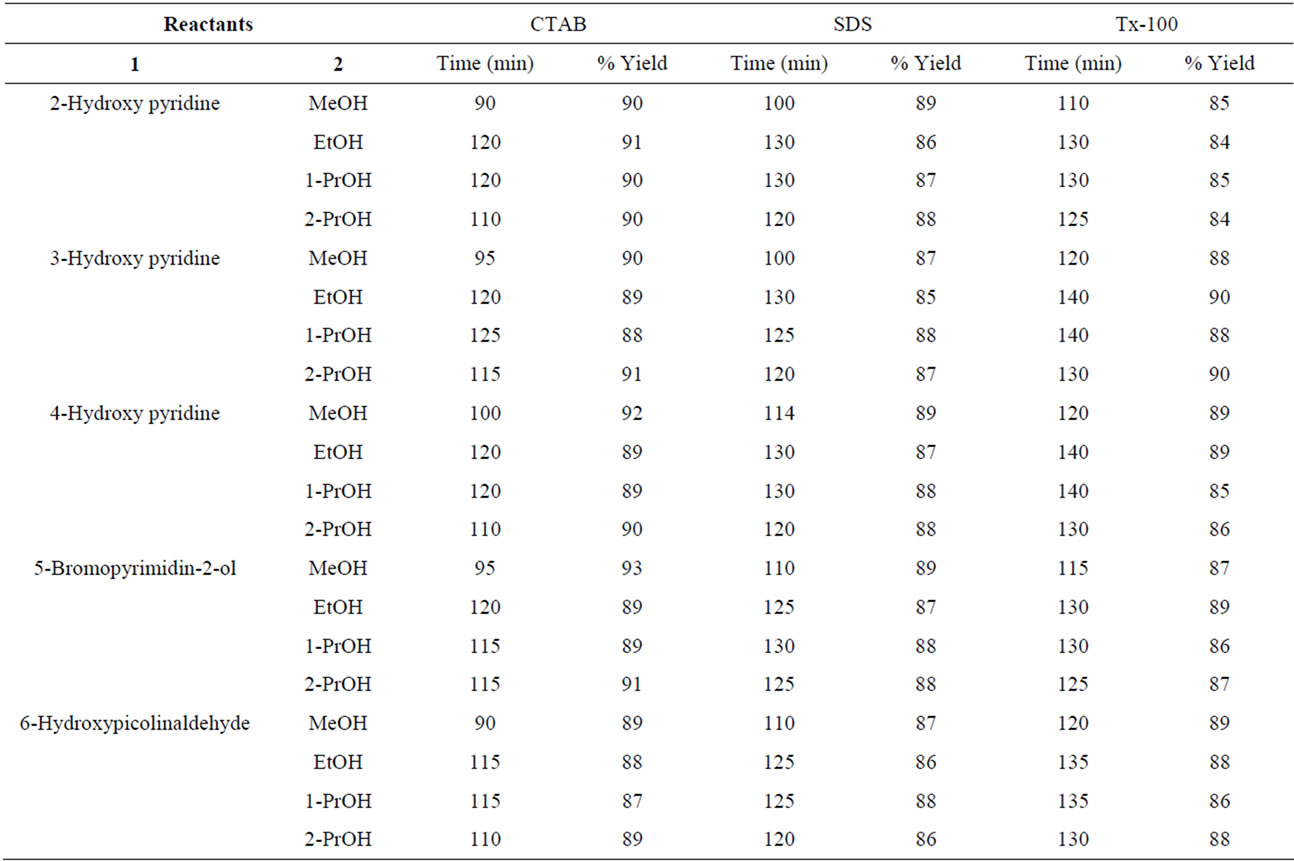
Table 3. Ultrasonically-assisted O-alkylation of hydroxy pyridines under solvent-free conditions.
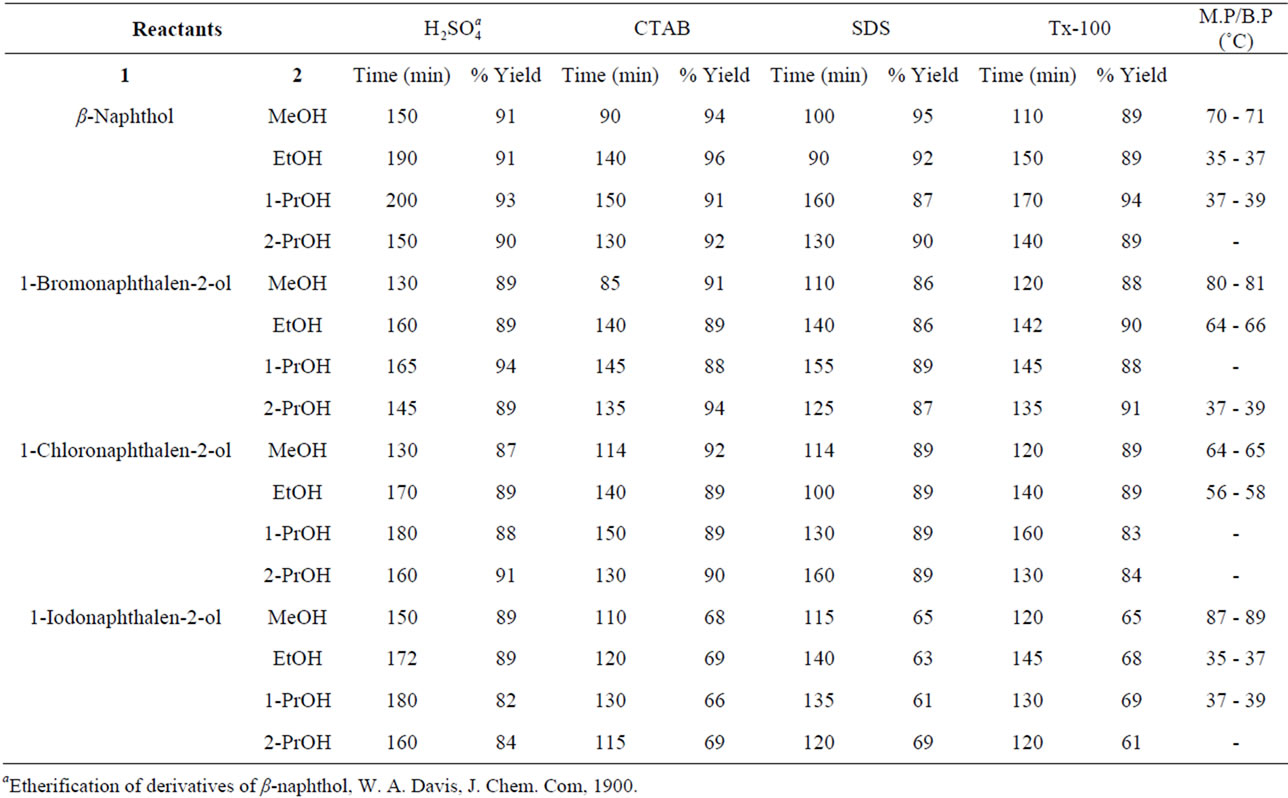
Table 4. Ultrasonically-assisted O-alkylation of β-naphthols under solvent-free conditions.
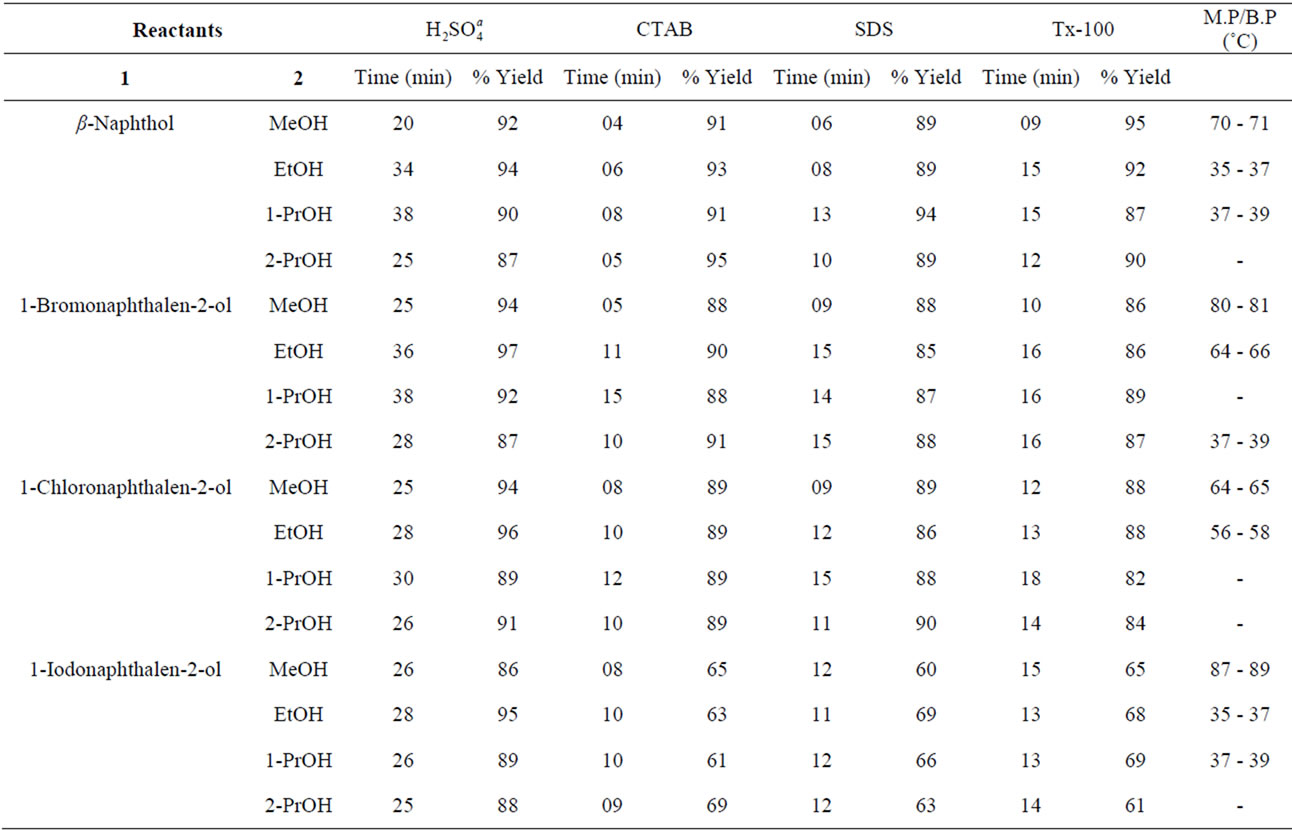
Table 5. Microwave-assisted O-alkylation of β-naphthols under solvent-free conditions.
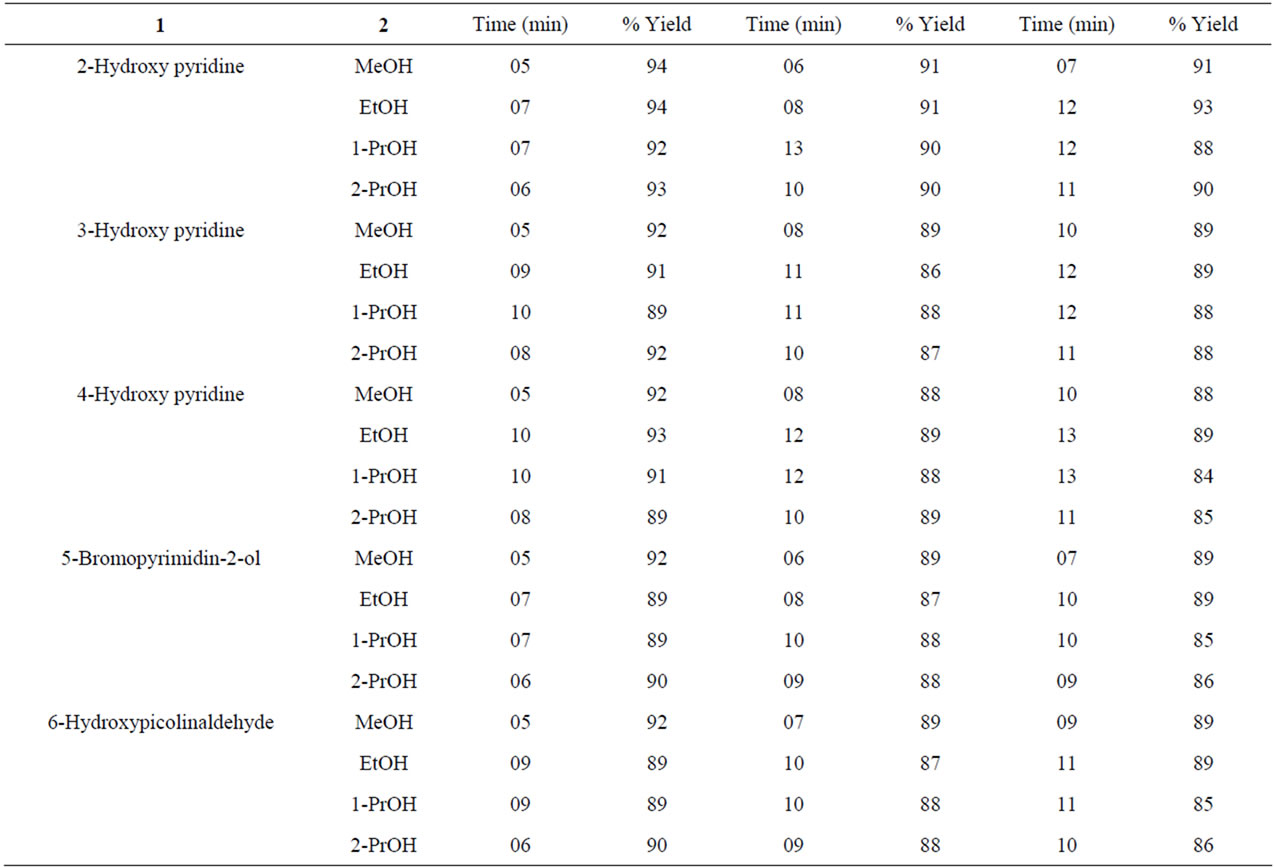
Table 6. Microwave-assisted O-alkylation of hydroxy pyridines under solvent-free conditions.
the similar reactions performed under classical conditions. Water is not only an inexpensive and environmentally benign solvent, but also plays an important role in reactivity and selectivity. Surfactants catalyze the reaction efficiently with short reaction times without using any harmful organic reagents and solvents.
6. Acknowledgements
The authors are thankful to Professor P. K. Saiprakash for constant encouragement and Heads of the Chemistry Department at Osmania University and Nizam College, Hyderabad for facilities.
REFERENCES
- W. H. Miles and K. B. Connell, “Synthesis of Methyl Diantilis, a Commercially Important Fragrance,” Journal of Chemical Education, Vol. 83, No. 2, 2006, p. 285. doi:10.1021/ed083p285
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. D. Pina, “From Glycerol to Value-Added Products,” Angewandte Chemie International Edition, Vol. 46, No. 24, 2007, pp. 4434-4440. doi:10.1002/chin.200737271
- N. Baggett, “Comprehensive Organic Chemistry,” In: D. Barton, W. D. Ollis and J. F. Stoddart, Eds., Comprehensive Organic Chemistry, 1st Edition, Pergaman, Oxford, 1979, p. 819.
- J. March, “Advanced Organic Chemistry, Reactions, Mechanism and Structure,” 4th Edition, Wiley, New York, 1992, p. 386.
- S. Kim, K. N. Chung and S. Yang, “Direct Synthesis of Ethers via Zinc Chloride-Mediated Etherification of Alcohols in Dichloroethane,” The Journal of Organic Chemistry, Vol. 52, No. 17, 1987, pp. 3917-3919. doi:10.1021/jo00226a036
- L. Karas and W. J. Piel, “Kirk-Othmer Encyclopedia of Chemical Technology,” 4th Edition, Wiley, New York, 1992, p. 860
- P. Salehi, N. Iranpoor and F. K. Behbahani, “Selective and Efficient Alcoholyses of Allylic, Secondaryand Tertiary Benzylic Alcohols in the Presence of Iron (III),” Tetrahedron, Vol. 54, No. 5-6, 1998, pp. 943-948. doi:10.1016/S0040-4020(97)10350-7
- G. V. M. Sharma and A. K. Mahalingam,” A Facile Conversion of Alcohols into p-Methoxybenzyl Ethers (PMBEthers) Using p-Methoxybenzyl Alcohol-Yb(OTf)3,” The Journal of Organic Chemistry, Vol. 64, No. 24, 1999, pp. 8943-8944. doi:10.1021/jo990563x
- K. J. Miller and M. M. Abu-Omar, “Palladium-Catalyzed SN1 Reactions of Secondary Benzylic Alcohols: Etherification, Amination, and Thioetherification,” European Journal of Organic Chemistry, Vol. 2003, No. 7, pp. 1294-1299. doi:10.1002/ejoc.200390185
- C. Siswanto, T. Battal, O. E. Schuss and J. F. Rathman, “Synthesis of Alkylphenyl Ethers in Aqueous Surfactant Solutions by Micellar Phase-Transfer Catalysis. 1. Single-Phase Systems,” Langmuir, Vol. 13, No. 23, 1997, pp. 6047-6052. doi:10.1021/la960487i
- F. Li, Q. R. Wang, Z. B. Ding and F. G. Tao, “Microwave-Assisted Synthesis of Diaryl Ethers without Catalyst,” Organic Letters, Vol. 5, No. 12, 2003, pp. 2169-2171. doi:10.1021/ol0346436
- H. Firouzabadi, N. Iranpoor and A. A. Jafari, “Facile Preparation of Symmetrical and Unsymmetrical Ethers From Their Corresponding Alcohols Catalyzed by Aluminumdodecatangstophosphate (AlPW12O40), as a Versatile and a Highly Water Tolerant Lewis Acid,” Journal of Molecular Catalysis A: Chemical, Vol. 227, No. 1-2, 2005, pp. 97-100. doi:10.1016/j.molcata.2004.09.078
- A. Corma and M. Renz, “A General Method for the Preparation of Ethers Using Water-Resistant Solid Lewis Acids,” Angewandte Chemie International Edition, Vol. 46, No. 1-2, 2007, pp. 298-300. doi:10.1002/anie.200604018
- K. T. V. Rao, P. S. N. Rao, P. S. Saiprasad and N. Lingaiah, “Cesium Exchanged Heteropoly Tungstate Supported on Zirconia as an Efficient and Selective Catalyst for the Preparation of Unsymmetrical Ethers,” Catalysis Communications, Vol. 10, No. 10, 2009, pp. 1394-1397. doi:10.1016/j.catcom.2009.03.004
- J. Pozniczek, A. Micek-llnicka, A. Lubanska and A. Bielanski, “Catalytic Synthesis of Ethyl-Tert-Butyl Ether on Dawson Type Heteropolyacid,” Applied Catalysis A: General, Vol. 286, No. 1, 2005, pp. 52-60. doi:10.1016/j.apcata.2005.02.035
- T. Ollevier and T. M. Mwene-Mbeja, “Bismuth Triflate Catalyzed Claisen Rearrangement of Allyl Naphthyl Ethers,” Tetrahedron Letters, Vol. 47, No. 24, 2006, pp. 4051-4055. doi:10.1016/j.tetlet.2006.03.193
- Y. Q. Cao and B. G. Pei, “Etherification of Phenols Catalysed by Solid-Liquid Phase Transfer Catalyst PEG 400 without Solvent,” Synthetic Communications, Vol. 30, No. 10, 2000, pp. 1759-1766. doi:10.1080/00397910008087221
- T. Mitsudome, T. Matsuno, S. Sueoka, T. Mizugaki, K. Jitsukawa and K. Kaneda, “Direct Synthesis of Unsymmetrical Ethers from Alcohols Catalyzed by Titanium Cation-Exchanged Montmorillonite,” Green Chemistry, Vol. 14, No. 3, 2012, pp. 610-613. doi:10.1039/c2gc16135d
- T. Ashok Rao, D. Dutta and G. I. Georg, “Polymer-Bound Triphenylphosphine as Traceless Reagent for Mitsunobu Reactions in Combinatorial Chemistry: Synthesis of Aryl Ethers from Phenols and Alcohols,” Tetrahedron Letters, Vol. 39, No. 48, 1998, pp. 8751-8754. doi:10.1016/S0040-4039(98)01988-1
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. P. Kumar, “Mitsunobu and Related Eactions: Advances and Applications,” Chemical Reviews, Vol. 109, No. 6, 2009, pp. 2551-2651. doi:10.1021/ cr800278z
- M. Debabrata and S. L. Buchwald, “Cu-Catalyzed Arylation of Phenols: Synthesis of Sterically Hindered and Heteroaryl Diaryl Ethers,” The Journal of Organic Chemistry, Vol. 75, No. 5, 2010, pp. 1791-1794. doi:10.1021/jo9026935
- M. D. Maiti, “Chemoselectivity in the Cu-Catalyzed O-Arylation of Phenols and Aliphatic Alcohols,” Chemical Communications, Vol. 47, No. 29, 2011, pp. 8340-8342. doi:10.1039/c1cc12694f
- P. A. Grieco, “Organic Synthesis in Water,” Blacky Academic and Professional, London, 1998. doi:10.1007/978-94-011-4950-1
- C. J. Li and T. H. Chan, “Organic Reactions in Aqueous Media,” John Wiley and Sons, New York, 1997.
- J. H. Fendler and E. J. Fendler, “Catalysis in Micellar and Macromolecular Systems,” Academic Press, London, 1975.
- D. A. Sabatini, R. C. Knox and J. H. Harwell, “Surfactant-Enhanced Subsurface Remediation,” American Chemical Society, Washington DC, 1994.
- F. M. Menger, J. U. Rhee and H. K. Rhee, “Applications of Surfactants to Synthetic Organic Chemistry,” The Journal of Organic Chemistry, Vol. 40, No. 25, 1975, pp. 3803-3805. doi:10.1021/jo00913a051
- P. T. Anastas and J. C. Warner, “Green Chemistry: Theory and Practice,” Oxford University Press, New York, 1998.
- T. J. Mason, “Chemistry with Ultrasound,” Elsevier Science Publishers Ltd., England, 1990.
- A. Kottror-earou and M. R. Hoffman, “Ultrasonic Irradiation of p-Nitrophenol in Aqueous Solution,” The Journal of Physical Chemistry, Vol. 95, No. 9, 1991, pp. 3630- 3638. doi:10.1021/j100162a037
- K. S. Suslick, “Ultrasound, Its Chemical, Physical and Biological Effects,” VCH Publishers, Inc., Deerfield Beach, 1988.
- M. A. Margulis, “Advances in Sonochemistry,” In: T. J. Mason, Ed., Advances in Sonochemistry 1 Edition, Greenwich Connection, London, 1990, p. 49.
- T. J. Mason, “Ultrasound in Synthetic Organic Chemistry,” Chemical Society Reviews, Vol. 26, No. 6, 1997, pp. 443-451. doi:10.1039/cs9972600443
- G. Cravotto and P Cintas, “Power Ultrasound in Organic Synthesis: Moving Cavitational Chemistry from Academia to Innovative and Large-Scale Applications,” Chemical Society Reviews, Vol. 35, No. 2, 2006, pp. 180-196. doi:10.1039/b503848k
- H. Fillion and J. L. Luche, “Ch.9: Selected Experiments,” In: J. L. Luche, Ed., Synthetic Organic Sonochemistry, Plenum, New York, 1998 and References Therein.
- P. Lidström, J. Tierney, B. Wathey and J. Westman, “Microwave Assisted Organic Synthesis—A Review,” Tetrahedron, Vol. 57, No. 45, 2001, pp. 9225-9283. doi.org/10.1016/S0040-4020(01)00906-1
- C. O. Kappe, “Controlled Microwave Heating in Modern Organic Synthesi,” Angewandte Chemie International Edition, Vol. 43, No. 46, 2004, pp. 6250-6284. doi:10.1002/anie.20040065
- C. R. Strauss and R. W. Trainor, “Developments in Microwave-Assisted Organic Chemistry,” Australian Journal of Chemistry, Vol. 48, No. 10, 1995, pp. 1665-1692. doi:10.1071/CH9951665
- A. Manvar, P. Bochiya, V. Virsodia, R. Khunt and A. Shah, “Microwave-Assisted and Zn[l-Proline]2 Catalyzed Tandem Cyclization under Solvent Free Conditions: Rapid Synthesis of Chromeno[4,3-C]Pyrazol-4-Ones,” Journal of Molecular Catalysis A: Chemical, Vol. 275, No. 1-2, 2007, pp. 148-152. doi:10.1016/j.molcata.2007.05.039
- A. Sharma, P. Appukkuttan and E. Van der Eycken, “Microwave-Assisted Synthesis of Medium-Sized Heterocycles,” Chemical Communications, Vol. 48, No. 11, 2012, pp. 1623-1637. doi:10.1039/c1cc15238f
- L.-M. Wang, N. Jiao, J. Qiu, J.-J. Yu, J.-Q. Liu, F.-L. Guo and Y. Liu, “Sodium Stearate-Catalyzed Multicomponent Reactions for Efficient Synthesis of Spirooxindoles in Aqueous Micellar Media,” Tetrahedron, Vol. 66, No. 1, 2010, pp. 339-343. doi:10.1016/j.tet.2009.10.091
- Y. Watanabe, K. Sawada and M. Hayashi, “A Green Method for the Self-Aldol Condensation of Aldehydes Using Lysin,” Green Chemistry, Vol. 12, No. 3, 2010, pp. 384-386. doi:10.1039/b918349c
- H. Firouzabadi, N. Iranpoor and A. Garzan, “Highly Efficient Halogenation of Organic Compounds with Halides Catalyzed by Cerium(III) Chloride Heptahydrate Using Hydrogen Peroxide as the Terminal Oxidant in Water,” Advanced Synthesis & Catalysis, Vol. 347, No. 11-12, 2005, pp. 1925-1928. doi:10.1002/adsc.200900124
NOTES
*Corresponding author.