Advances in Pure Mathematics
Vol.06 No.03(2016), Article ID:63912,25 pages
10.4236/apm.2016.63010
A Global Reduction Based Algorithm for Computing Homology of Chain Complexes
Madjid Allili, David Corriveau
Department of Mathematics, Bishop’s University, Sherbrooke, Canada

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 22 September 2015; accepted 21 February 2016; published 26 February 2016
ABSTRACT
In this paper, we propose a new algorithm to compute the homology of a finitely generated chain complex. Our method is based on grouping several reductions into structures that can be encoded as directed acyclic graphs. The organized reduction pairs lead to sequences of projection maps that reduce the number of generators while preserving the homology groups of the original chain complex. This sequencing of reduction pairs allows updating the boundary information in a single step for a whole set of reductions, which shows impressive gains in computational performance compared to existing methods. In addition, our method gives the homology generators for a small additional cost.
Keywords:
Homology Algorithm, Chain Complex, Homology Generators

1. Introduction
Homology has been used recently in a wide variety of applications in domains such as dynamical systems, and image processing and recognition. In dynamics, typical problems are translated into problems in topology where invariants such as the Conley index are computed using homology algorithms. In digital image analysis, topological invariants are useful in shape description, indexation, and classification. Among shape descriptors based on homology theory, there are the Morse shape descriptor [1] [2] , the Morse Connection Graph [3] , and the persistence barcodes for shape [4] . The necessity of improved algorithms appears evident as new applications of the homology computation arise in research for very large data sets. Although several algorithms and software packages have been developed for this purpose, there is still a lot of room for improvement as processing very large data sets is often very time- and memory-consuming. The classical approach to compute homology of a chain complex with integer coefficients reduces to the calculation of the Smith Normal Form (SNF) of the boundary matrices which are in general sparse [5] [6] .
Unfortunately, this approach has a very poor running-time and its direct implementation yields exponential bounds. Polynomial time algorithms for computing the SNF were given by Kannan & Bachem [7] and later improved by Chou & Collins [8] and Illiopoulos [9] . The best currently known Smith Normal Form algorithms have super cubical complexity [10] . Another generic approach is the method of reduction proposed in [5] [11] . In this method the original complex is simplified through a sequence of reductions of cells that preserve the homology groups at each step. The idea is to replace the chain complex (or even the object under study) by a smaller one with the same homology. A reduction consists of cancelling a cell B through an element of its boundary a (reduction pair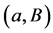 ). The two cells are suppressed from the structure of the complex and the boundary homomorphisms are updated. Direct implementations of the reduction method use unordered lists to encode the boundary homomorphisms and have cubical complexity.
). The two cells are suppressed from the structure of the complex and the boundary homomorphisms are updated. Direct implementations of the reduction method use unordered lists to encode the boundary homomorphisms and have cubical complexity.
Several algorithms based on the idea of reduction and that improve the time complexity for particular types of data sets have been designed. For cubical sets embedded in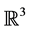 , Mrozek et al. proposed a method [12] based on the computation of acyclic subspaces by using lookup tables. Experimentally, this method performed in linear time. Another method running in quasi linear time for finite simplicial complexes embedded in
, Mrozek et al. proposed a method [12] based on the computation of acyclic subspaces by using lookup tables. Experimentally, this method performed in linear time. Another method running in quasi linear time for finite simplicial complexes embedded in 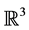 was proposed by Delfinado and Edelsbrunner in [13] . For problems dealing with higher dimensions, Mrozek and Batko developed an algorithm [14] based on the concept of coreduction which showed interesting performance results on cubical sets.
was proposed by Delfinado and Edelsbrunner in [13] . For problems dealing with higher dimensions, Mrozek and Batko developed an algorithm [14] based on the concept of coreduction which showed interesting performance results on cubical sets.
Despite the existence of efficient homology algorithms for cubical sets and lower dimension spaces, there remain many problems where the data is higher dimensional and not cubical. Generally, for these problems it is often difficult to adapt the data because the cost is too high or too complex. In this case, one has to use one of the classical methods: SNF or reduction. However, the classical methods spend most of their time in updating the boundary homomorphisms after each reduction step. This requires repetitive manipulations of the data structures that store the chain complex which incurs a high running time to accomplish the reduction.
An idea is to identify admissible reduction pairs and organize them in a structure that allows efficiently updating the boundary information in a single step for a whole set of reductions. This reduces to the minimum manipulations of the data structures that store the boundary information. Our method works as follows. Starting at an arbitrary cell A with a face a in its boundary, we identify successive adjacent admissible pairs of reduction and build the longest possible sequence originating at A. Several sequences can originate at the same cell, and each sequence can be seen as a path in a directed acyclic graph, called a reduction DAG, where nodes are reduction pairs and oriented edges denote an adjacency relation between the reduction pairs. Given a reduction DAG across a chain complex, we achieve a global simplification of the complex by performing all the reductions in the DAG at once. We will establish direct algebraic formulas that allow updating the boundary of remaining cells. The net advantage of our approach is that the boundaries are not explicitly updated after each reduction step. Instead, this information is always preserved implicitly within the data structures and processed globally for each reduction DAG allowing reducing considerably the computational time. The communication [15] contains a summary of the method developed systematically here and the preliminary experimentations that are now developed in Section 5.
2. Chain Complexes and Homology
Chain complexes are an algebraic tool for defining and computing homology. Although they are usually defined from geometrical structures such as simplicial or cellular complexes, they can also be considered in an abstract manner as pure algebraic entities. A finitely generated free chain complex 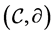 with coefficients in a ring
with coefficients in a ring 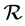 is a sequence of finitely generated free abelian groups
is a sequence of finitely generated free abelian groups 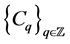 together with a sequence of homomorphisms,
together with a sequence of homomorphisms,
called boundary maps or operators, 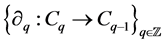 satisfying
satisfying 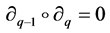 for all
for all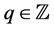 . Typically,
. Typically,
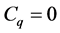 for
for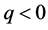 . For each p, the elements of
. For each p, the elements of 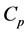 are called p-chains and the kernel of
are called p-chains and the kernel of 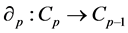
is called the group of p-cycles and denoted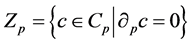 . The image of
. The image of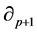 , called the group of
, called the group of
boundaries, is denoted by .
.  is a subgroup of
is a subgroup of  because of
because of
the property . The quotient groups
. The quotient groups  are the homology groups of the chain complex
are the homology groups of the chain complex .
.
Computation of Homology by Reduction of Chain Complexes
The reduction of a chain complex is a procedure that consists of removing successively pairs of generators from the bases of its chain groups while preserving the homology of the original complex. This method is originally motivated by a simple geometric idea known as the collapsing. At the algebraic level, each removal of a pair of generators that form a reduction pair is equivalent to a collection of projection maps  that send each generator of the removed pair into 0. Moreover, it projects the other cells into the remaining generators taking into account the modifications of their boundaries caused by the removal of the pair. Let
that send each generator of the removed pair into 0. Moreover, it projects the other cells into the remaining generators taking into account the modifications of their boundaries caused by the removal of the pair. Let  for each d. It is shown in [5] that
for each d. It is shown in [5] that  is a chain complex and
is a chain complex and , that is, the homologies of
, that is, the homologies of  and C coincide. To calculate the homology of the original chain complex C, the idea is to define a sequence of projections associated to the removal of pairs in all dimensions and then compute the homology of the resulting chain complex that is the image of the successive projections. A sequence of projections is complete if no other projection can be added to the sequence. Such a sequence always exits when the chain complex is finitely generated. Let
and C coincide. To calculate the homology of the original chain complex C, the idea is to define a sequence of projections associated to the removal of pairs in all dimensions and then compute the homology of the resulting chain complex that is the image of the successive projections. A sequence of projections is complete if no other projection can be added to the sequence. Such a sequence always exits when the chain complex is finitely generated. Let  be the original chain complex and
be the original chain complex and  is the final chain complex obtained after a complete sequence of projections. We already know that the two complexes have the same homology, moreover it is easily observed that if
is the final chain complex obtained after a complete sequence of projections. We already know that the two complexes have the same homology, moreover it is easily observed that if  then
then , that is, the Betti number
, that is, the Betti number  is given by the number of d-cells remaining in the complex
is given by the number of d-cells remaining in the complex .
.
Formally, a reduction pair and its associated collection of projection maps are defined as follows.
Definition 1. Let  be an abstract chain complex. A pair of generators
be an abstract chain complex. A pair of generators  such that
such that  and
and  is called a reduction pair. It induces a collection of group homomorphisms
is called a reduction pair. It induces a collection of group homomorphisms

where  and
and  is the canonical bilinear form on chains.
is the canonical bilinear form on chains.
A way to understand the global reduction of a complex is to reformulate it as an algorithm and follow an example of its execution. The following implementation is basic and simply aims at understanding the process. For instance, if the chain complex has torsion or if the dimensions are reduced in arbitrary order, then we should refer to the more general version of the reduction method described in [5] [11] . Also, we use the data structures as defined in section 4.0.
Algorithm 2. Reduction (Complex K) reduces K into  using the one-step reduction formula. Consecutive calls of this function will simplify K until it remains unchanged. Finally, if all the boundaries are null, then the homology generators are given by the remaining cells. If not, then we continue the computation of homology by using a classical method such as Smith Normal Form.
using the one-step reduction formula. Consecutive calls of this function will simplify K until it remains unchanged. Finally, if all the boundaries are null, then the homology generators are given by the remaining cells. If not, then we continue the computation of homology by using a classical method such as Smith Normal Form.
1) (Find a reduction pair)  is a reduction pair if a is a face of B with an incidence coefficient of
is a reduction pair if a is a face of B with an incidence coefficient of . If no reduction pair is found, return K unmodified. Otherwise continue. It is assumed that reduction pairs are chosen by decreasing order of dimension. First the highest dimension is reduced, then the lower dimensions.
. If no reduction pair is found, return K unmodified. Otherwise continue. It is assumed that reduction pairs are chosen by decreasing order of dimension. First the highest dimension is reduced, then the lower dimensions.
2) (Projection of K) Assign . boundary,
. boundary, . coboundary and
. coboundary and , the coeffi- cient of a in
, the coeffi- cient of a in .
.
(a) (Update cofaces of a) For all cofaces C of a, add  to C. boundary, where
to C. boundary, where  is
is , the coefficient of a in C. boundary.
, the coefficient of a in C. boundary.
(b) (Update faces of B) For all faces c of B, add  to c. coboundary, where
to c. coboundary, where  is
is , the coefficient of c in B. boundary.
, the coefficient of c in B. boundary.
(c) (Project a and B) Remove cells a and B from K.
3) (Return the result) Return the simplified K.
An example of using the reduction algorithm is illustrated in Figure 1. With the prescribed orientation of the

Figure 1. Result of the projection of a complex after one reduction.
given complex, the boundaries of the cells in K are



The pair  is a reduction pair since the incidence number
is a reduction pair since the incidence number . It follows that
. It follows that



Clearly,  is the identity map since there is no 0-cell removed from the complex and
is the identity map since there is no 0-cell removed from the complex and  for all
for all  except for
except for . The altered boundaries in the resulting complex
. The altered boundaries in the resulting complex  are
are


To calculate the homology of the complex we continue the simplifications until  for all dimensions.
for all dimensions.
3. Computation of Homology by Grouping Reductions
The reduction algorithm chooses reduction pairs in arbitrary order and spends most of its time updating the boundaries at each reduction step. Instead of performing the reductions in arbitrary order, we organize them into reduction sequences that make up reduction directed acyclic graphs (RDAGs). We will show that this is very advantageous algorithmically.
3.1. Forming RDAGs
Starting from an arbitrary cell, we form the maximal possible directed acyclic graph (DAG) whose nodes are reduction pairs and a directed edge between two pairs  and
and  means that
means that  is adjacent to
is adjacent to  (see definition 4). A directed path in the DAG corresponds to a reduction sequence. That type of reduction sequence affects only adjacent cells to the path and leaves other cells unchanged. Information about each reduction is carried out by the reduced cells. After performing the whole set of reductions, we obtain the reduced complex by extracting the boundary information from the data structures associated to the cells visited by the reduction DAGs.
(see definition 4). A directed path in the DAG corresponds to a reduction sequence. That type of reduction sequence affects only adjacent cells to the path and leaves other cells unchanged. Information about each reduction is carried out by the reduced cells. After performing the whole set of reductions, we obtain the reduced complex by extracting the boundary information from the data structures associated to the cells visited by the reduction DAGs.
We introduce the main concepts through an example. The example details how the projections of cells are calculated given a reduction sequence. This will show what information is exactly needed to be kept about the original complex in order to be able to rebuild the reduced complex.
Example 3. Consider the complex given in Figure 2. The boundaries of the 2-cell are


Figure 2. Example of collapsing by using RDAGs.




The depicted sequence of reduction pairs originating at cell A is  and
and . The projection maps at the level of the 2-cell give the following
. The projection maps at the level of the 2-cell give the following



It follows that the projection map associated to the whole sequence is . The 2-cell A is the only cell adjacent to the sequence while the 2-cell E is not adjacent. Their respective projections with respect to the sequence are
. The 2-cell A is the only cell adjacent to the sequence while the 2-cell E is not adjacent. Their respective projections with respect to the sequence are


Each coefficient in the projection is called the coefficient of contribution of the corresponding cell to the projection of A. For instance,  is the coefficient of contribution of D in the projection of A. We can compute the coefficients
is the coefficient of contribution of D in the projection of A. We can compute the coefficients  and
and  and express concretely the projection of A:
and express concretely the projection of A:




Once a reduction DAG is built, we are interested in building the reduced complex. The cells of the new complex are the projected cells of the original complex. The projection can be used effectively to update or build the boundary operator for each projected cell. In the previous example, the boundary of  can be reconstructed from the formula of its projection
can be reconstructed from the formula of its projection

The incidence number of a given cell, say g, with respect to the boundary of the projection of A is computed by adding all the contribution coefficients of the cofaces of g present in the projection of A, each multiplied by the incidence number of g with respect to that coface. A particular coface of g can be present several times in the projection of A if it belongs to different sequences that originate at A.
For instance  is given by
is given by

since D is the only coface of g in the original complex and it is present in the projection of A. Note that the value of  is known from the original complex.
is known from the original complex.
Thus to calculate the reduced complex, it is enough to keep track of the coefficients of contribution of every reduced cell in any of the projected cells for which it contributes. In general, after a sequence of reductions is achieved, a projected cell adjacent to reduction DAGs will look as depicted in Figure 3.

Figure 3. A cell extended by reduction DAGs.
3.2. Projection Formulas for Grouped Reductions
We define how to organize the reductions and the complex using reduction DAGs.
Definition 4. A cell  is adjacent to the pair
is adjacent to the pair  if the cells
if the cells  and
and  are of the same dimension m and
are of the same dimension m and  is a cell with dimension
is a cell with dimension  in the boundaries of both
in the boundaries of both  and
and . A pair
. A pair  is adjacent to a pair
is adjacent to a pair  if
if  is adjacent to
is adjacent to .
.
Definition 5. A reduction DAG is a directed acyclic graph whose nodes are reduction pairs and a directed edge between two pairs  and
and  means that
means that  is adjacent to
is adjacent to .
.
Definition 6. A path P from  to
to  in a reduction DAG G is a reduction sequence
in a reduction DAG G is a reduction sequence  whose elements are nodes in G and
whose elements are nodes in G and  is adjacent to
is adjacent to , for
, for .
.
A cell A is said to be adjacent to a reduction sequence if it is adjacent to some pair of the sequence.
Definition 7. A path from  to
to  in a reduction DAG G is said to originate at
in a reduction DAG G is said to originate at  if
if  is adjacent to
is adjacent to .
.
Definition 8. A cell not appearing in any reduction DAG is called a projected cell. The cells that appear in a reduction DAG are called reduced cells.
All individual paths originating at a given cell contribute to the projection of the cell. In the following theorem, we give a formula to calculate the projection of a cell by considering the contribution of a single path and ignoring the input from other paths and sub-paths. This formula is called “path projection”.
Theorem 9. Let  be a path originating at A0. The path projection of A0 by the path P is given by
be a path originating at A0. The path projection of A0 by the path P is given by

where , and
, and  for
for .
.  is called the coefficient of contribution of
is called the coefficient of contribution of  in the path projection of A0.
in the path projection of A0.
Proof: We proceed by induction on the length of the path. For  or
or , the path projection formula is easily checked as shown in Example 3. Suppose that the path projection of
, the path projection formula is easily checked as shown in Example 3. Suppose that the path projection of  by the path of length n,
by the path of length n,  is
is

Adding a  pair of reduction
pair of reduction  to the path
to the path  so that
so that  is a face of
is a face of  leads to the
leads to the
path of length ,
, . The projection of
. The projection of  by
by  is equivalent to the pro-
is equivalent to the pro-
jection of  by the path
by the path  consisting of a single reduction pair, that is:
consisting of a single reduction pair, that is:

The last equality is due to the fact that none of the cells  contains
contains  as a face. Thus,
as a face. Thus,

Finally:

where  and
and , which proves the induction hypothesis.
, which proves the induction hypothesis.
Now, we can write

We denote by  the projection chain of the cell
the projection chain of the cell  by the path
by the path .
. 
Note that when the path  is extended by one reduction pair
is extended by one reduction pair , then the formula for the projection of
, then the formula for the projection of  by
by  is given by:
is given by:

More generally, if  is extended by a path
is extended by a path , then the formula generalizes to
, then the formula generalizes to
 (1)
(1)
where  and
and . Here
. Here .
.
Corollary 1. Let  be disjoint non overlapping paths originating at
be disjoint non overlapping paths originating at . The total projection of
. The total projection of  by
by  denoted by
denoted by

where  is the projection chain of the cell
is the projection chain of the cell  by the path
by the path .
.
Proof: It is important to mention that k cannot exceed the number of boundary faces of  which are of dimension
which are of dimension . Since the k paths are disjoint and non overlapping, each path has to start from a different face of
. Since the k paths are disjoint and non overlapping, each path has to start from a different face of . Without loss of generality, it is sufficient to prove the corollary for the case where
. Without loss of generality, it is sufficient to prove the corollary for the case where  and with paths consisting of single reduction pairs as shown in Figure 4. If we apply first the reduction by
and with paths consisting of single reduction pairs as shown in Figure 4. If we apply first the reduction by , then
, then

Applying the second reduction results in:

Since  and
and  are disjoint non overlapping, it follows that
are disjoint non overlapping, it follows that  is not a face of
is not a face of , thus
, thus

Figure 4. Reduction by two disjoint non overlapping paths.

Corollary 2. Let T be a reduction tree originating at , then the projection of
, then the projection of  by T is equal to the sum of
by T is equal to the sum of  and the projection chain of
and the projection chain of  by T.
by T.
Proof:
Typically, the trees originating at  can occur as pure paths, in which case the associated projections are given previously. Otherwise, we can find paths that share a common ancestral branch that originates at
can occur as pure paths, in which case the associated projections are given previously. Otherwise, we can find paths that share a common ancestral branch that originates at . This is seen as a bifurcation as shown in Figure 5. In this case, the ancestral branch is a path
. This is seen as a bifurcation as shown in Figure 5. In this case, the ancestral branch is a path  which is extended by two paths
which is extended by two paths  and
and . The combination of the results in Theorem 9 and Corollary 1 can be used to show that the total projection of A0 by
. The combination of the results in Theorem 9 and Corollary 1 can be used to show that the total projection of A0 by  is given by
is given by

The term  is called the projection chain of the tree.
is called the projection chain of the tree.
In general, the tree can have many bifurcations at a given node and the bifurcations can occur at different nodes for which cases, the formula given above can be easily generalized. 
One may wonder what happens when two paths are merging into a single path as illustrated in Figure 6. For this purpose, let’s consider the simple situation where two distinct reduction paths originating at A0 overlap at a certain node, which is they are extended both by the same path.
In that situation, there are two independent pure paths CB and DB and the total projection of A0 is given as follows


Figure 5. A path B bifurcates into two paths C and D.

Figure 6. Two paths C and D merge into a single path B.
We see from the formula that it comes down to considering each path (which is also a tree) including the shared sibling branch as pure paths contributing to the projection of A0. This comes down to considering each tree independently (see Figure 7).
Theorem 10. Let A0 be a cell adjacent to a RDAG in a chain complex C. The projection of A0 by the RDAG is equal to A0 to which we add the projection chains of A0 by all the trees in the RDAG that originate at A0, that is

where  is the collection of all the reduction trees in the RDAG originating at A0.
is the collection of all the reduction trees in the RDAG originating at A0.
Proof: Since we deal with finite complexes the number of all possible trees starting at a given cell (here A0) has to be finite. Thus, the results proved for paths in corollaries 1 and 2 and the subsequent remark regarding merging paths can be extended for the case of trees (splitting and merging) to find the formula for the projection of A0. 
We review the projection formulas of paths and trees with the example of Figure 8. There are two trees  and
and  originating at A0. The projection of A0 with respect to the tree
originating at A0. The projection of A0 with respect to the tree  can be written as
can be written as

where . The projection chain of A0 with respect to the tree
. The projection chain of A0 with respect to the tree  is given by
is given by

with . It follows that the total projection of A0 is given as the sum of both plus A0.
. It follows that the total projection of A0 is given as the sum of both plus A0.
3.3. Why Acyclic Graphs
Adjacency alone does not guarantee that cycles are not created in a reduction DAG. To ensure that every reduction sequence is associated with a well defined projection, we enforce acyclicity in the formed directed graphs. More generally, assuming that a reduction sequence  is such that
is such that  is adjacent to
is adjacent to  and
and  is adjacent to
is adjacent to  (see Figure 9), we consider extending the sequence to
(see Figure 9), we consider extending the sequence to  by setting
by setting . However, the pair
. However, the pair  is not necessarily admissible since after performing the sequence of reductions
is not necessarily admissible since after performing the sequence of reductions , the cell A0 has been modified into
, the cell A0 has been modified into 

and  occurs in the boundary of
occurs in the boundary of  as a face of both
as a face of both  and
and . Its incidence number is therefore calculated as follows
. Its incidence number is therefore calculated as follows

which is not necessarily invertible. In Figure 9,  is not invertible which means that
is not invertible which means that  is not an admissible reduction pair. In our implementation, we avoid testing if
is not an admissible reduction pair. In our implementation, we avoid testing if

Figure 7. Decomposition of a reduction DAG into two independent trees  and
and .
.


Figure 8. Example of reduction paths and the associated reduction DAG.

Figure 9. Cycles need to be avoided as reduction pairs are added in the tree.
reduction pairs are admissible or not by avoiding cycles as we build the reduction DAGs. This results in a more efficient algorithm.
3.4. Using Projection Formulas
We recapitulate the formulas with a second example, see Figure 10. The first reduction pair is . Because the cell E is adjacent to the pair
. Because the cell E is adjacent to the pair , it is marked as a projected cell. In a list associated with the reduced cell A,
, it is marked as a projected cell. In a list associated with the reduced cell A,
we save the pair  where
where . The reduction is continued from the faces of A. The
. The reduction is continued from the faces of A. The
second reduction pair is . We carry the information previously saved on the visited cofaces of b, so we
. We carry the information previously saved on the visited cofaces of b, so we
add 
 to the list associated with the cell B. Continuing from the faces of B, the
to the list associated with the cell B. Continuing from the faces of B, the

Figure 10. A more complicated reduction sequence.
third reduction pair to be added is . The cell c is adjacent to both the reduced cell B and the projected cell E. From this point, we continue a path
. The cell c is adjacent to both the reduced cell B and the projected cell E. From this point, we continue a path  and begin a sub-path
and begin a sub-path . The information saved with C considers the input of both paths. So we save
. The information saved with C considers the input of both paths. So we save  where
where
 and
and . But because the coefficients of contribution sum up to 0, it means
. But because the coefficients of contribution sum up to 0, it means
that C does not contribute to the projection of E. Thus, nothing is saved with C. The last reduction pair  is added to the sequence. Because, d is adjacent to the unvisited cell F, F is marked as a projected cell. The reduced cell C is also adjacent to d but no information was saved with C, so the only information to save with D
is added to the sequence. Because, d is adjacent to the unvisited cell F, F is marked as a projected cell. The reduced cell C is also adjacent to d but no information was saved with C, so the only information to save with D
is  where
where .
.
3.5. Building the Simplified Complex
Using reduction DAGs to compute the homology of a chain complex is a recursive process. At each recursion level, the algorithm simplifies the complex by constructing reduction DAGs on the complex and saving the associated projections into appropriate data structures. This is performed simultaneously for each dimension. This process eventually stops when it is impossible to add another reduction pair to any reduction DAG. In that case, the algorithm will build the associated simplified complex and continue the reduction process on the simplified complex.
The simplified complex is rebuilt from the projected cells only (reduced cells are not considered). The boundaries of the cells are updated using global projection formulas that allow to calculate the incidence numbers between cells of contiguous dimensions. Note that the reduced cells are not completely removed from the structures since they may be needed to recover homology generators expressed in terms of cells of the original complex as we explain in subsection 3.8. Contrary to the classical case where the boundary updating is done at each reduction step and may concern cells that can be reduced at a later step, a major benefit of this new approach is that the boundary updating is done only among the projected cells which often constitute a small fraction of the number of cells in the original complex.
Continuing with the example used in Figure 10, we proceed to build its simplified complex. At this point from the projection formulas we have the projection of E, which is  and the projection of F which is
and the projection of F which is . To build the simplified complex, we create two 2-dimensional cells,
. To build the simplified complex, we create two 2-dimensional cells,  and
and . We also create the projected lower dimensional cells (which is not illustrated in the example). Then, knowing that
. We also create the projected lower dimensional cells (which is not illustrated in the example). Then, knowing that , we add the projected 1-cells in the boundary of D to the boundary of
, we add the projected 1-cells in the boundary of D to the boundary of  using the correct coefficients. For example, if we suppose that
using the correct coefficients. For example, if we suppose that  is a projected cell of dimension one that was in the boundary of D in the original complex. From the structures, we know that D appears in the projection of F with an incidence coefficient of 1 (
is a projected cell of dimension one that was in the boundary of D in the original complex. From the structures, we know that D appears in the projection of F with an incidence coefficient of 1 ( ). So, we add
). So, we add  in the boundary of
in the boundary of  with a coefficient of 1. This is done for all projected cells in all dimensions.
with a coefficient of 1. This is done for all projected cells in all dimensions.
3.6. Performance Benefits of Using Reduction DAGs
We use the complex illustrated in Figure 9 as a case study. Our objective is to point out how grouping reductions differs from classical methods and leads to substantial performance benefits. Performance is measured by the number of lists updates and compared to the classical reduction algorithm (see 2). Usually, the boundary and coboundary of cells are maintained by lists data structures. Assuming ordered lists, the cost for merging two lists  and
and  is bounded by
is bounded by , where L refers to the number of elements in L. Using unordered lists is bounded by
, where L refers to the number of elements in L. Using unordered lists is bounded by .
.
1) Classical Reduction Method
(a) Reduction . According to step 2.a) of algorithm 2, we update cofaces of
. According to step 2.a) of algorithm 2, we update cofaces of . That is, for each 2-cell
. That is, for each 2-cell  in the coboundary of
in the coboundary of , we add the set of cells in the boundary of
, we add the set of cells in the boundary of  to the boundary of
to the boundary of  using the right coefficient. Therefore, this operation costs an order of
using the right coefficient. Therefore, this operation costs an order of  list updates.
list updates.
In step 2.b), we update the coboundaries of faces of . For each 1-cell
. For each 1-cell  in the boundary of
in the boundary of , we add the set of cells in the coboundary of
, we add the set of cells in the coboundary of  to the coboundary of
to the coboundary of  using the right coefficient. This time, the cost is in the order of
using the right coefficient. This time, the cost is in the order of . Taken all together, it costs an order of
. Taken all together, it costs an order of  lists updates for the first reduction.
lists updates for the first reduction.
(b) Reduction . Now
. Now , thus updating cofaces of
, thus updating cofaces of  is in the order of
is in the order of .
.
For step 2.b), updating faces of  is in the order of
is in the order of . Taken together, the cost for the second reduction is in the order of
. Taken together, the cost for the second reduction is in the order of  lists updates.
lists updates.
(c) Reduction . Now
. Now , and updating cofaces of
, and updating cofaces of  costs an order of
costs an order of  lists updates.
lists updates.
For step 2.b), updating faces of A3 amounts to a cost in the order of . The total cost for the third reduction is in the order of
. The total cost for the third reduction is in the order of .
.
Taken all together, the three reductions cost an order of  lists updates. Generally, as the re- ductions go on, the costs increase because the boundary and coboundary lists tend to grow in size.
lists updates. Generally, as the re- ductions go on, the costs increase because the boundary and coboundary lists tend to grow in size.
2) Reduction DAGs Method
(a) Reduction . The pair
. The pair , where
, where  is saved in a list maintained by
is saved in a list maintained by . The cost is one list update.
. The cost is one list update.
(b) Reduction . The pair
. The pair , where
, where  is saved in a list maintained by
is saved in a list maintained by . The cost is one list update.
. The cost is one list update.
(c) Reduction . The pair
. The pair  where
where , is saved in a list maintained by
, is saved in a list maintained by . The cost is one list update.
. The cost is one list update.
(d) Reconstruction of the simplified complex. For each non reduced 1-cell a, we scan through its cofaces of the original complex and link a with the list of projected cells previously saved by using the right coefficients. The total cost is in the order of the number of remaining 1-cells plus the size of the lists of adjacent projected cells. Note that usually, the simplification process continues on the lower dimensions. So at the end there will remain only a fraction of the lower dimensional cells (here 1-cells). The Reduction DAGs method is more efficient as long as the cost for reconstructing the simplified complex is low which our experimental results corroborate.
3.7. Minimizing Lists Updates
Different reductions lead to different computational costs (measured by the number of lists updates). We attempt to achieve a good overall performance by choosing reduction pairs that should minimize the number of updates. To select among many candidate reduction pairs, we use a heuristic that estimates their respective costs. Given a reduction pair , this cost is approximated by the number of non reduced cofaces of a (other than B) plus the size of the projection lists of each reduced coface of a.
, this cost is approximated by the number of non reduced cofaces of a (other than B) plus the size of the projection lists of each reduced coface of a.
We illustrate this on an example in Figure 11. Suppose that the 2-cell A is already reduced and has  and
and  in its list of projected cells. We consider the cost of performing the reduction
in its list of projected cells. We consider the cost of performing the reduction . The heuristic approximates this cost to three lists updates, because there is one non reduced coface of c (that is B), and the projection list of the reduced coface A has two elements
. The heuristic approximates this cost to three lists updates, because there is one non reduced coface of c (that is B), and the projection list of the reduced coface A has two elements  and
and . Indeed, performing the reduction
. Indeed, performing the reduction  amounts to inserting the pairs
amounts to inserting the pairs  and
and  into the projection list of C. We give more details about the heuristic in section 4.
into the projection list of C. We give more details about the heuristic in section 4.
3.8. Calculating Generators
An interesting aspect of using the reduction DAGs method to compute homology is that the data structures allow to extract the homology generators at a low additional cost. We explain how to proceed. Let us consider the complex of a plane quotiented by its boundary as illustrated in Figure 12. This complex is homeomorphic to a 2-sphere and the 2-cell A represents the 2-generator . The generator associated to a projected cell corresponds to the projection of the cell. As we illustrated in Figure 12, after each reduction, the projection coefficients
. The generator associated to a projected cell corresponds to the projection of the cell. As we illustrated in Figure 12, after each reduction, the projection coefficients  are saved into the projection lists maintained in each reduced cell. Generally, once the complex has been simplified to the level where all boundaries are trivial, one has to examine all projected (non-reduced) cells and find their corresponding coefficients in the projection lists of the reduced cells. Thus, to get the 2-generator associated to the 2-cell A, one has to scan through each reduced 2-cell and extract the projection coefficients associated to A.
are saved into the projection lists maintained in each reduced cell. Generally, once the complex has been simplified to the level where all boundaries are trivial, one has to examine all projected (non-reduced) cells and find their corresponding coefficients in the projection lists of the reduced cells. Thus, to get the 2-generator associated to the 2-cell A, one has to scan through each reduced 2-cell and extract the projection coefficients associated to A.
However, computing homology by reduction DAGs is a recursive process (see Figure 13) and this needs to be considered when calculating the generators. Projections correspond to generators but they can be expressed with cells of the simplified complex at any level of the recursion. However, in order for the generators to carry geometric meaning, it makes sense only to express the generators with cells from the original complex. Due to the recursive simplifications, a projected cell at a previous level of the recursion may become a reduced cell at a later level of the recursion. We illustrate this in Figure 14. In this example, there are two levels of recursion.

Figure 11. Cost of performing a reduction in terms of list updates.

Figure 12. A Complex homeomorphic to a 2-sphere.

Figure 13. Computing homology by reduction DAGs is a recursive process.

 (a) (b)
(a) (b)
 (c) (d)
(c) (d)
Figure 14. (a) Reduction 1 of the initial complex. (b) Reduction 2 of the simplified complex. (c) Initial complex after returning from the second reduction. (d) Initial complex after returning from the first reduction.
The final simplified complex is obtained after reducing the vertical edge and the 2-cell B shown in Figure 14(b). At this step, the cell A represents a 2-generator that is expressed as  (considering that after the first reduction we rename the original cells A and B as
(considering that after the first reduction we rename the original cells A and B as  and
and ). Note that in fact, the algorithm always works on the original cells and never copy nor create any new cell during the whole computation. Returning from the last recursion, we know that
). Note that in fact, the algorithm always works on the original cells and never copy nor create any new cell during the whole computation. Returning from the last recursion, we know that  and
and . The cell A represents a projected cell at both recursion levels (14(b) and 14(c)) and requires no special consideration. On the other hand, the cell B is a projected cell at the first level of recursion but becomes a reduced cell at the second recursion level. Consequently, returning from the second recursion level, we scan through each 2-cell and whenever B is encountered in one of the projection lists, it is replaced by
. The cell A represents a projected cell at both recursion levels (14(b) and 14(c)) and requires no special consideration. On the other hand, the cell B is a projected cell at the first level of recursion but becomes a reduced cell at the second recursion level. Consequently, returning from the second recursion level, we scan through each 2-cell and whenever B is encountered in one of the projection lists, it is replaced by . This is shown in Figure 14(d). Finally, the generator is expressed by
. This is shown in Figure 14(d). Finally, the generator is expressed by . In Figure 15 we show different 1- generators that we extracted from 3D models.
. In Figure 15 we show different 1- generators that we extracted from 3D models.
3.9. What If Boundaries Are Not Trivial?
In previous subsections, we stressed that all boundaries have to be trivial in order to obtain homology and its generators at the end of the reduction process. But in practice, when all reductions are done we can be in a situation


 (a) (b) (c)
(a) (b) (c)

 (d) (e) (f)
(d) (e) (f)
Figure 15. (a)-(c) The 3D models of a chain, two kissing children and a Buddha. (d)-(f) The calculated holes (one dimensional generators).
where some of the boundaries remain non trivial. For example, this may be due to the presence of torsion.
In Definition 1, the pair  is defined as a reduction pair if
is defined as a reduction pair if . But this condition is in fact too
. But this condition is in fact too
restrictive. In order for  to be a group homomorphism,
to be a group homomorphism,  has to be an integer. This is satisfied
has to be an integer. This is satisfied
when  divides
divides  for every
for every . The easiest and most efficient way to guarantee this is to choose B and a such that
. The easiest and most efficient way to guarantee this is to choose B and a such that . Thus, for performance considerations, we designed the reduction DAGs algorithm to consider only reduction pairs with
. Thus, for performance considerations, we designed the reduction DAGs algorithm to consider only reduction pairs with  coefficients. But the drawback is that once all reductions are done, it is not guaranteed that all boundaries will be trivial. There may remain reduction pairs such that
coefficients. But the drawback is that once all reductions are done, it is not guaranteed that all boundaries will be trivial. There may remain reduction pairs such that  but
but  divides
divides . In that case, one can modify the present algorithm to test for divisibility and continue the computations with the modified version. Another possibility is to have recourse to a classical algorithm such as the Smith Normal Form. Note that at this level most of the cells generating the complex have already been reduced to a small number.
. In that case, one can modify the present algorithm to test for divisibility and continue the computations with the modified version. Another possibility is to have recourse to a classical algorithm such as the Smith Normal Form. Note that at this level most of the cells generating the complex have already been reduced to a small number.
4. Data Structures and Algorithms
The first data structure represents a chain complex.
ChainComplex:
E: two dimensional array of cells organized by their respective dimension, that is E[d][i] denotes the i-th d-cell.
In our implementation, the pointer to a cell is used to identify the cell. These identifiers are saved in E. The main benefit to using pointers is that when we build the simplified complex, no new cell is created. Only the pointers of the projected cells are copied into the simplified complex.
Cell:
boundary/coboundary: list of faces/cofaces.
state: a flag taking one of the values in {NORMAL, REDUCED, PROJECTED, VISITED}.
projCells: list to save the PROJECTED cells and their associated coefficients that project onto the given cell when it is REDUCED.
nbUpdates: approximates the number of updates to a projCells list when the given cell is reduced by one of its cofaces.
Initially, all cells are set to the NORMAL state. REDUCED is assigned to the cells in a reduction sequence and PROJECTED is assigned to the cells adjacent to a reduction sequence. The VISITED flag is assigned to the d-cells that don’t have any  coface that can form an admissible reduction pair. A reduction pair
coface that can form an admissible reduction pair. A reduction pair  is admissible if
is admissible if  and adding the pair to the reduction DAG does not create a cycle. The flag helps to avoid testing more than once if the given d-cells have an admissible reduction pair.
and adding the pair to the reduction DAG does not create a cycle. The flag helps to avoid testing more than once if the given d-cells have an admissible reduction pair.
The projCells list is used by the REDUCED cells to keep track of the coefficients of contribution of every PRO- JECTED cell for which it contributes. For example, if we use the sequence of reduction pairs illustrated in Figure
2, then B.projCells and C.projCells will respectively contain  and
and  where
where  and
and .
.
To build a sequence of reduction pairs, nbUpdates is used to select reduction pairs that should minimize the amount of updates to the projCells list. The number of updates is approximated by the number of non reduced cofaces plus the number of entries in the projCells lists of REDUCED cofaces.
We now give the principal steps of the algorithm.
HOM_RDAG(ChainComplex K) returns the Betti numbers of a chain complex K by using reduction DAGs.
1) (Initialization) For all cells in K, set its state to NORMAL, empty projCells, set nbUpdates to the number of cofaces.
2) (Reduce cells) Proceed by decreasing order of dimension. Order the d-cells by increasing value of nb- Updates. Following this order, start a reduction DAG (call BuildRDAG()) from each cell whose state is NOR- MAL.
For dimension 0, change remaining NORMAL 0-cells to PROJECTED.
3) (Build the simplified complex) Call BuildSimplifiedComplex(K). This method extracts the boundary information from the data structures to build a simplified complex .
.
Continue to recursively simplify  by calling HOM_RDAG(
by calling HOM_RDAG( ) until there are no reduction pair left.
) until there are no reduction pair left.
Test if all boundaries are trivial. If not, then continue the reduction process with another method such as SNF.
4) (Return the Betti numbers) Assign the number of PROJECTED d-cells to .
.
BuildRDAG(Cell c) builds a reduction DAG from cell c.
1) (Initialization) Save c.nbUpdates into nbUpdatesLimit for later use.
2) (Find a reduction pair) Find a coface B of c such that B is NORMAL or VISITED. If a coface B is found, then call PairCells(c, B), otherwise set c to VISITED and exit BuildRDAG().
3) (Expand the reduction DAG) Expand the reduction DAG (repeat step 2) from all NORMAL faces of B that have nbUpdates £ nbUpdatesLimit. Proceed in a breadth first search approach.
PairCells(Cell c, Cell B) adds the reduction pair  to the sequence and updates the data structures accordingly.
to the sequence and updates the data structures accordingly.
1) (Update projCells) For all cofaces C of c different than B, if C is REDUCED, then add lc*C.projCells to
B.projCells. Otherwise, set C to PROJECTED and add lc*C to B.projCells, where .
.
2) (Update the nbUpdates variable) For all faces a of B different than c, add
|B.projCells|-1 to c.nbUpdates. For all faces of c, remove one to nbUpdates.
BuildSimplifiedComplex(Complex K) extracts the boundary and coboundary information from the data structures to build a simplified complex .
.
Note: In the preceding methods, the coboundary list of a cell is never modified. Thus, when referring to cofaces, it is about the cofaces in the complex K at a given recursion level before any reduction is performed.
1) (Update boundary) Proceed by increasing order of dimension. Let c be a PROJECTED d-cell of K. For all cofaces C of c, iterate through C. projCells. Let B be a  -cell in C. projCells and
-cell in C. projCells and  its associated coefficient. Add
its associated coefficient. Add  to B.boundary and update c.coboundary accordingly.
to B.boundary and update c.coboundary accordingly.
Finally, remove all REDUCED cells remaining in the boundary and coboundary of PROJECTED cells. Copy the pointers of all PROJECTED cells into K’.E.
5. Experimental Results
Because of its heuristic and recursive design, we had major difficulties to analyze the time complexity of our algorithm. Instead, we approached the problem by evaluating its performance on a wide range of experimentations. We compared its performance with the reduction algorithm 2 on different data series: d-balls, d-spheres, tori, Bing’s houses, 3D medical scans and randomly generated d-complexes.
The series of d-balls and d-spheres were created by juxtaposing d dimensional cubes along each dimension. A 3-ball of size 10 is the cube obtained by placing side by side 10 × 10 × 10 3-cubes and making the corresponding identifications of lower dimensional cells. A d-sphere is a d -ball quotiented by its boundary. We also constructed the tori and Bing’s houses from 3-cubes, although they are both 2 dimensional complexes. This construction does not affect the homology. We created two data series from 3D medical scans: brain and heart. Each of them is created from 3D images by selecting the pixels whose values are higher than a threshold. Lastly, we randomly generated d-complexes by choosing  random numbers referring to the number of generators. We created a d-cell for each d-generator and randomly linked the d-cells to the
random numbers referring to the number of generators. We created a d-cell for each d-generator and randomly linked the d-cells to the  -cells. By subdivision, we obtained complexes of various data sizes.
-cells. By subdivision, we obtained complexes of various data sizes.
In the experimentation, we use the following terminology. A data series refers to one of d-balls, d-spheres, tori, Bing’s houses, 3D medical scans or randomly generated d-complexes. Each data series is constituted of many datasets. For example, d-balls contain the 2-ball, 3-ball, ..., 6-ball. Each dataset contains many files of complexes of various data sizes. For example, there are 15 files in the 2-ball data set for which the data size vary from 2000 up to 10,000,000. The size of a complex is measured as the number of cells plus the number of links (entries in boundary lists). A test is an execution of the algorithm on a file of a specified data set.
Initially, we observed that a test could be misleading, especially when dealing with cubical data sets. The reason is because we create the complexes in an orderly fashion. Thus, during its execution, the algorithm is most probably benefiting from a favorable choice of reduction pairs due to the order inherent in the data. Consequently, to avoid misreporting on the algorithm’s performance, we took care to randomize the boundary and coboundary lists of all cells in the complex before each test.
We performed 30 tests for each file and computed the average, maximum and standard deviation of the time used for calculating the homology, the generators and sorting. We measured the sorting time because it has a time complexity of  and we wanted to verify that sorting would not monopolize the computation time. Also, we saved statistics on the number of recursions to see its influence on the global performance. Here we grouped and summarized the results per dataset.
and we wanted to verify that sorting would not monopolize the computation time. Also, we saved statistics on the number of recursions to see its influence on the global performance. Here we grouped and summarized the results per dataset.
We begin with our most interesting results (see Table 1) which compare the performance of the reduction DAGs algorithm with the classical reduction method on different datasets. We can observe a significant improvement between the two methods. Knowing that the time complexity of the reduction method is quadratic (using ordered lists), those results suggest a subquadratic time complexity for the reduction DAGs algorithm which is what we measured in Table 2.

Table 1. Comparison of the reduction DAGs algorithm versus the classical reduction method.
Table 2. Approximated performance with respect to dataset, data size and dimension.
In Table 2 we report the approximative time that our algorithm uses to calculate homology on various datasets. The second column (N) gives the number of files within the dataset. The third and fourth columns show the parameters  and
and  that best-fit the equation “
that best-fit the equation “ “, where time is expressed in seconds. We obtained the best-fits from the times measured on the individual files of each dataset. In the last four columns, we present the times in milliseconds that we get from the best-fit equation for different data sizes. Those times approximate the real times that we measured in real experiments. For example, on 30 tests, we measured an average time of 4992.24 ms on a torus of size 10944304. The best-fit equation approximates a time of 4573.64 ms for a torus of size 10000000.
“, where time is expressed in seconds. We obtained the best-fits from the times measured on the individual files of each dataset. In the last four columns, we present the times in milliseconds that we get from the best-fit equation for different data sizes. Those times approximate the real times that we measured in real experiments. For example, on 30 tests, we measured an average time of 4992.24 ms on a torus of size 10944304. The best-fit equation approximates a time of 4573.64 ms for a torus of size 10000000.
From Table 2, we observe that  and
and  for d-balls and d-spheres without regard of the dimension. Except for few values,
for d-balls and d-spheres without regard of the dimension. Except for few values,  is relatively constant while
is relatively constant while  gradually decreases as the dimension increases. We explain this by the fact that the ratio of exterior face reductions versus interior face reductions increases with the dimension. An exterior face reduction denotes a reduction pair
gradually decreases as the dimension increases. We explain this by the fact that the ratio of exterior face reductions versus interior face reductions increases with the dimension. An exterior face reduction denotes a reduction pair  for which a has only the coface B in its coboundary list. On the other hand, we use the term interior face reduction when a is shared by more than one coface. Algorithmically speaking, an exterior face reduction is more advantageous because it does not cost any update to the projection lists, while an interior face reduction costs at least an update.
for which a has only the coface B in its coboundary list. On the other hand, we use the term interior face reduction when a is shared by more than one coface. Algorithmically speaking, an exterior face reduction is more advantageous because it does not cost any update to the projection lists, while an interior face reduction costs at least an update.
We observe that the approximation times and the parameters  and
and  for the heart and brain datasets, remain in the same order as those of d-balls and d-spheres despite the high numbers of generators as shown in Table 3. For example, the file heart96 had 30 connected components, 107 holes and 19 voids. For these two datasets, the number of generators was not a relevant factor for the performance of the algorithm. We think it is because those two datasets contain a high ratio of exterior face reductions, which cost near to nothing. If we compare with the random d-complexes then the topology is an important factor. Indeed, as the data size increased we observed bigger differences in the time performance.
for the heart and brain datasets, remain in the same order as those of d-balls and d-spheres despite the high numbers of generators as shown in Table 3. For example, the file heart96 had 30 connected components, 107 holes and 19 voids. For these two datasets, the number of generators was not a relevant factor for the performance of the algorithm. We think it is because those two datasets contain a high ratio of exterior face reductions, which cost near to nothing. If we compare with the random d-complexes then the topology is an important factor. Indeed, as the data size increased we observed bigger differences in the time performance.
In Figure 16, we also reproduced the algorithm’s performance on the files of each dataset. We used a logarithmic scale on both axes to better distinguish the trends. In Figure 16(d), each file in a d-complex has a distinct topology. It explains why we see only a weak trend in opposition to the other datasets where the trends are strong.
Another interesting aspect of the algorithm to consider is the number of recursions which is expressed as the number of reduction calls in Table 4. Interestingly, one reduction call was enough to calculate the homology of all d-balls, d-spheres and Bing’s houses. It means that the algorithm did not reconstruct an intermediate simplified complex to obtain the homology. For other datasets, the algorithm had to reconstruct simplified complexes to carry out the computation. As an example, a value of n means that, on average, the algorithm constructed  simplified complexes. The Max column shows the average of the maximum number of calls and the last column refers to the average standard deviation.
simplified complexes. The Max column shows the average of the maximum number of calls and the last column refers to the average standard deviation.
Table 3. Number of generators of the files in the heart and brain datasets.
 (a)
(a) (b)
(b) (c)
(c) (d)
(d)
Figure 16. Performance of the algorithm on various data series: (a) d-balls, (b) d-spheres, (c) heart, brain, Bing’s house and torus, (d) random d-complexes.
Finally, in Table 5 we show how much time the algorithm spent on sorting and calculating the generators. Regarding sorting, the issue is that the heuristic uses sorting in attempt to choose reduction pairs that should minimize the number of lists updates. Consequently, we wanted to verify that sorting did not monopolize the computation time. Otherwise, the heuristic would hinder more rather than help the computation. So we measured the average time spent on sorting relative to the total time of a computation and expressed the results in percentage terms. Also, we give the average maximum and average standard deviation. We observe that, on average, the algorithm spent around 5% - 10% of the time on sorting and the extrema are almost always less than 15%. In addition, the algorithm performed much slower when we disabled the heuristic.
Table 4. Number of reduction calls per dataset.
Table 5. Cost for computing generators and sorting.
Regarding the computation of generators, our algorithm allows to obtain them for an additional cost of about 10% or less on average. From Table 5 we see that most average times fluctuate around 5% and the maximums fall into the 15% - 30% range. Moreover, the algorithm performed as strongly on datasets with a large amount of generators than on those with a small amount.
6. Conclusions
We developed in this paper a novel and efficient algorithm for the computation of homology groups and homology generators that works at the level of chain complexes, and hence allows handling a variety of geometric complexes. The main idea is based on organizing sequences of cell reductions into a structure of directed acyclic graphs, which makes it possible to derive global projection formulas and perform large collections of reductions at once. In this method, the boundary operators of the complex are not explicitly updated after each reduction step. Instead, this information is always preserved implicitly within the data structures and processed globally for each reduction graph allowing reducing considerably the computational time.
Our experimentations show that this algorithm performs significantly faster than the classical reduction method. In addition, for all the datasets that we tested, which cover a wide range of types of data, their global performance indicated a subquadratic time complexity. Moreover, it allows calculating the homology generators at a small additional time cost. Interestingly enough, in all the datasets we dealt with, the algorithm needed only to construct few intermediate complexes to obtain the homology of the original complex. Our feeling is that if one encounters a specially designed complex in which the number of recursions is substantially high then the algorithm may underperform. However, it is not yet clear how to construct such a complex.
Cite this paper
MadjidAllili,DavidCorriveau, (2016) A Global Reduction Based Algorithm for Computing Homology of Chain Complexes. Advances in Pure Mathematics,06,113-137. doi: 10.4236/apm.2016.63010
References
- 1. Allili, M. and Corriveau, D. (2007) Topological Analysis of Shapes using Morse Theory. Computer Vision and Image Understanding, 105, 188-199.
http://dx.doi.org/10.1016/j.cviu.2006.10.004 - 2. Allili, M., Corriveau, D. and Ziou, D. (2004) Morse Homology Descriptor for Shape Characterization. Proceedings of the 17th International Conference on Pattern Recognition, Vol. 4, 27-30. http://dx.doi.org/10.1109/icpr.2004.1333697
- 3. Allili, M., Corriveau, D., Derivière, S., Kaczynski, T. and Trahan, A. (2007) Discrete Dynamical System Framework for Construction of Connections between Critical Regions in Lattice Height Data. Journal of Mathematical Imaging and Vision, 28, 99-111.
http://dx.doi.org/10.1007/s10851-007-0010-0 - 4. Collins, A., Zomorodian, A., Carlsson, G. and Guibas, L. (2004) A Barcode Shape Descriptor for Curve Point Cloud Data. Computers and Graphics, 28, 881-894.
http://dx.doi.org/10.1016/j.cag.2004.08.015 - 5. Kaczynski, T., Mischaikow, K. and Mrozek, M. (2004) Computational Homology. Applied Mathematical Sciences Series 157, Springer-Verlag, New York.
- 6. Munkres, J.R. (1984) Elements of Algebraic Topology. Addison-Wesley.
- 7. Kannan, R. and Bachem, A. (1979) Polynomial Algorithms for Computing the Smith and Hermite Normal Forms of an Integer Matrix. SIAM Journal on Computing, 8, 499-507.
http://dx.doi.org/10.1137/0208040 - 8. Chou, T.W. and Collins, G.E. (1982) Algorithms for the Solutions of Systems of Linear Diophantine Equations. SIAM Journal on Computing, 11, 687-708.
http://dx.doi.org/10.1137/0211057 - 9. Iliopoulos, C.S. (1989) Worst-Case Complexity Bounds on Algorithms for Computing the Canonical Structure of Finite Abelian Groups and the Hermite and Smith Normal Forms of an Integer Matrix. SIAM Journal on Computing, 18, 658-669.
http://dx.doi.org/10.1137/0218045 - 10. Storjohann, A. (1996) Near Optimal Algorithms for Computing Smith Normal Forms of Integer Matrices. Proceedings of 1996 International Symposium on Symbolic and Algebraic Computation, ISSAC’96, Zurich, 24-26 July 1996, 267-274.
http://dx.doi.org/10.1145/236869.237084 - 11. Kaczynski, T., Mrozek, M. and Slusarek, M. (1998) Homology Computation by Reduction of Chain Complexes. Computers and Mathematics with Applications, 35, 59-70.
- 12. Mrozek, M., Pilarczyk, P. and Zelazna, N. (2008) Homology Algorithm Based on Acyclic Subspace. Computers and Mathematics with Applications, 55, 2395-2412.
http://dx.doi.org/10.1016/j.camwa.2007.08.044 - 13. Delfinado, C.J.A. and Edelsbrunner, H. (1995) An Incremental Algorithm for Betti Numbers of Simplicial Complexes on the 3-Sphere. Computer Aided Geometric Design, 12, 771-784.
http://dx.doi.org/10.1016/0167-8396(95)00016-Y - 14. Mrozek, M. and Batko, B. (2009) Coreduction Homology Algorithm. Discrete and Computational Geometry, 41, 96-118.
http://dx.doi.org/10.1007/s00454-008-9073-y - 15. Corriveau, D. and Allili, M. (2009) Computing Homology: A Global Reduction Approach. In: Brlek, S., Reutenauer, C. and Provençal, X., Eds., Discrete Geometry for Computer Imagery: Proceedings of 15th IAPR International Conference, DGCI 2009, Springer, Berlin Heidelberg, 313-324.
http://dx.doi.org/10.1007/978-3-642-04397-0_27