Soft Nanoscience Letters
Vol.3 No.4A(2013), Article ID:40405,5 pages DOI:10.4236/snl.2013.34A001
Aptamers—A Promising Approach for Sensing of Biothreats Using Different Bioinformatics Tools
![]()
Department of CBRN Defence, Institute of Nuclear Medicine & Allied Sciences, Delhi, India.
Email: *rksharmadrl@yahoo.com
Copyright © 2013 Anamika Sharma, Rakesh Kumar Sharma. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received October 1st, 2013; revised November 1st, 2013; accepted November 8th, 2013
Keywords: Biosensors; Aptamers; SELEX; Insilco Designing
ABSTRACT
Intentional release of pathogens or biotoxin against humans, plants, or animals is an impending threat all over the world. Continuous monitoring of environment is required for their detection. These signals can help to distinguish whether the bioattack has occurred or not. Biosensors utilise biological response including different biochemical reactions, antigen antibody reactions, electrochemical reactions, aptameric reactions etc. The currently available biosensors have a limit of detection, specificity and less linearity which affect their sensitivity. Aptamers are single stranded oligonucleotides binding species which are capable of tightly binding to their distinguishing targets. They are evolved from random oligonucleotides pools by using different strategies. These are capable of conscientiously distinguishing their target ligands. They have high sensitivity and a wide range of detection limit. The versatility of nucleic acid based methods allowed for the design of specific aptamer sequences, typically on the order of 10 to 30 base pairs in length, identifying the different biothreat agents in the environment. By using different bioinformatics tools we can design RNA aptamers for toxins of lectin family.
1. Introduction
Biological warfare agents include use of certain bacteria, virus, and toxin of certain plants, animals, fungi and bacteria. Dedicated biosensors that have high sensitivity, linearity and less reaction time are required for their early detection [1]. Biosensors are analytical devices composed of some biological recognition including the use of biochemical reactions, antibody antigen reaction, electrochemical reactions [1,2]. These responses directly are interfaced to signal transducers which together relate the concentration of an analyte to a measurable response. Aptamers are synthetic single stranded oligonucelotides DNA/RNA which specifically bind to the target molecules, proteins, nucleic acids and even cells [3-5]. The etiology of the term aptamers is from the Latin word “aptum” meaning “to fit” [5]. It works on the basis of the lock and key between the aptamers and the targets molecule. This depends on the interactions between the DNA/RNA to their corresponding target and can be used for early detection and monitoring purpose [6]. The transducer will convert any physicochemical changes taking place in its proximity into visible signals [7,8]. These are based on electrochemical, optical, mass-sensitive, thermal and electronics principles. The biosensors based on the antibody antigen detection have several limitations i.e. antibodies against non-immunogenic or toxic substances cannot be obtained [9], irreversible denaturation after heat treatment. These limitations are inherent in the existing range of biosensors. But the biosensors based on the aptamers are not having these limitations [10]. So keeping this in our mind we attempt to identify the aptamers for the different targets by using the computational biology tools for early detection of the threats. We had chosen the two toxins of lectin family that are highly toxic and no antidote available to them only symptomatic and supportive treatments are available [11,12].
The detailed and precized study of aptamer-apatope interaction and evolutionary processes induced by the interplay lag behind the wide application of the SELEX products [13]. The binding property of aptamer is a function of both sequences and structure. Generally the product of last cycle of SELEX was taken and sequenced and examined for the possible sequence motifs using different motif binding programs [14,15]. Binding assays were allowed for the quantification of the binding strength considering the region as functionally active [16]. The different bioinformatics tools have opened the door to understand the interactions of aptamer and their corresponding target. So we had chosen those sequences that had minimum free energy (MFE). The binding energy of the target and the ligand should be as low as possible. In this present paper we attempt to isolate some sequences of ss DNA/ RNA aptamer and predict their secondary structure based on minimum free energy [17-20].
2. Materials and Methods
2.1. SELEX Libraries
We had created a randomized library of 108 - 109 DNA/ RNA [18,19] then sequences were chosen and their structure were predicted by using the different tools available. We had used RNAfold (http://rna.tbi.univie.ac.at) web server and 3D-DART for prediction of the ss DNA/RNA molecules. RNAfold webserver [21,22] is an online tool that predicts the structure of the single stranded nucleotides which based on the minimum free energy (MFE). We had got the detailed thermodynamic description (loop free-energy decomposition) of our ss DNA/RNA structures. We had calculated the thermodynamic ensemble prediction which was based on the free energy of the thermodynamic ensemble (kcal/mol), frequency of the MFE structure (%) and ensemble diversity of the ss DNA/RNA [22,23]. The 3D-DART webserver (3DNADriven DNA Analysis and Rebuilding Tool) provides a convenient means of generating custom 3D structural models of DNA. 3D-DART uses the DNA rebuild functionality of the well-known software package 3DNA [24,25]. The sequences obtained from the analysis were chosen for the docking proposes and the using two webserver were compared and then by using the HADDOCK [26,27] we had chosen those sequences that were having a low binding energy. HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an online software that provide information-driven flexible docking approach for the modelling of biomolecular complexes [23,24]. We had used toxin ricin and abrin as the target and the ss DNA/ RNA sequences as the ligand molecules for getting the sequences of lower entropy units of aptamer and apatope.
2.2. Ricin and Abrin
Ricin from Ricinus communis, is a highly toxic, naturally occurring protein. Abrin is a toxalbumin that is found in the seeds of a plant called the lucky bean, rosary pea or jequirity pea Abrus precatorius. Abrin is similar to ricin but far more deadly than ricin, the toxin found in the seeds of the castor oil plant [11,12]. The different protein database bank (PDB) has enough information related to the toxic proteins. We had taken PDB files of these proteins from the database and then use them as the target.
2.3. Selection of Aptamers
Aptamers are generally evolved by a combinatorial biochemistry technique in which “Systematic Evolution of Ligands by Exponential enrichment (SELEX)” take place [19,21] These molecules are designed to bind with high affinity and high specificity to wide range of molecular targets (apatopes), small organic molecules [9,10] protein molecules and to entire cell [12] (Figure 1). Due to their structural ability and diverse functionality in a wide range of pH and temperature make them ideal candidate for a broad spectrum of different in vivo tools [13-15]. The experimental design of SELEX is based on the assumption that a large pool of sequences is likely to contain nucleotide strands that are capable of binding to any target molecule [21,23]. In the traditional SELEX protocol there are five steps in one selection cycle: incubation, binding, partitioning and washing, target bound elution and amplification [15,16]. The nucleotide species in the pool potentially bind the target of interest which depends on the specificity sequence. In the last of every cycle the non binding species removed and bound species were eluted and amplified, forming the input for the next round. The SELEX methods started with the random sequence library of ss nucleic acid DNA/RNA. Generally these contain 20 - 100 nucleotides in length [25]. The more the randomization of ss nucleic acid sequences provides a wide range of diversity 4n where n refers for the number of nucleotides that were used for the selection of aptamers [2,6,17]. Libraries containing these nucleotides were flanked by the constant sequences for priming or capturing. Then individual aptamers were clones can be generated and tested for target binding affinity and specificity.
3. Results and Discussion
The approach which based on the assumption that if we predicted the aptamer structure by using different bioinformatics tools. Then if we calculate the energy of binding of aptamer and apatope we can easily isolate different the single stranded ss DNA/RNA sequences that were based on the difference in the energy of the target and ligand. The sequences which can be used as aptamer chosen from the fact that lowers the entropy of the system higher the stabilization of the system [18-20]. The sequences for ricin and abrin which had the lowest values of the MFE had shown a wide range of the binding coefficient [18]. The major result of secondary structures shows promising values for the binding of the two proteins and their different parameter given in the Table 1. In the process of selection of the different ss DNA/RNA we had identified some sequences that were showing a high value in the in silico studies. We had isolated the sequences from a randomised library of 108 - 109 to find out the ss DNA/RNA sequences by the use of online tools RNA fold and 3D-DART. Analysis of the docking result had shown the values of binding energy in rage of −6.56 kcal/mol to −10.68 kcal/mol. Molecular dynamics study we had found out the least energy binding sequences. The sequences obtained from the RNA fold webserver is shown in the Figure 2. The sesquence shown in the (a) was found to be having least binding energy −9.8 kcal/mol for ricin molecule while the sequence (b) and (c) had −6.56 and −10.8 kcal/mol. The secondary sequences which had the higher MFE and had pseudoknots were not found to be promising candidate for aptamer. We had also search for the sequences that show homology to our sequences running BLAST (http://blast.ncbi.nlm.nih.gov) at NCBI but there is no significant correlation found by this similarity-searching tool.
4. Conclusion
The single stranded sequences were binding to their target with high specificity and they work as the monoclonal antibodies do. For isolation of the appropriate sequences binding to the target is generated from the library that had a random large pool of the nucleotide sequences. The sequences which have the lower MFE and have low values of the frequency of the ensemble diver-
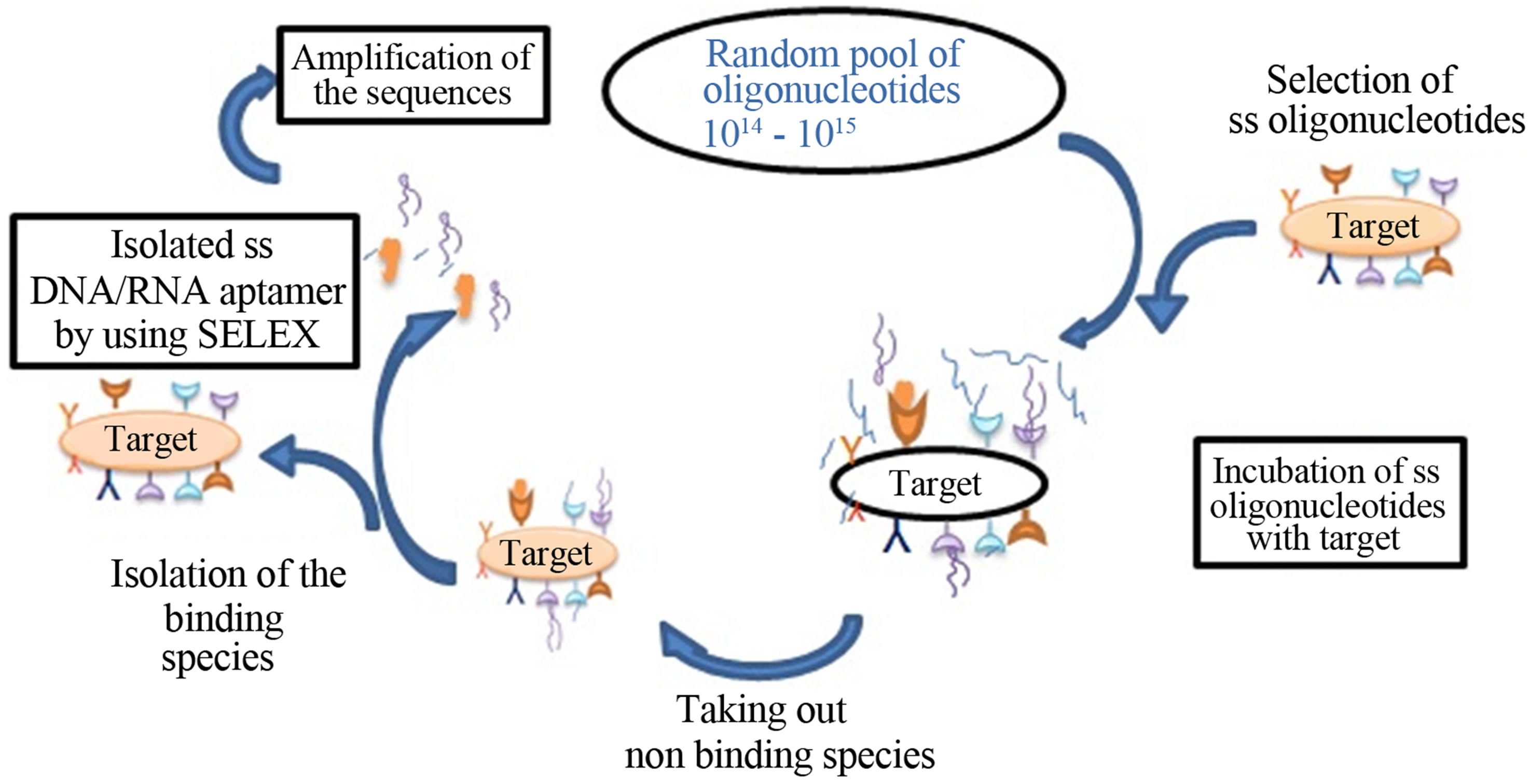
Figure 1. Schematic steps in selection of aptamers by using the SELEX techniques.
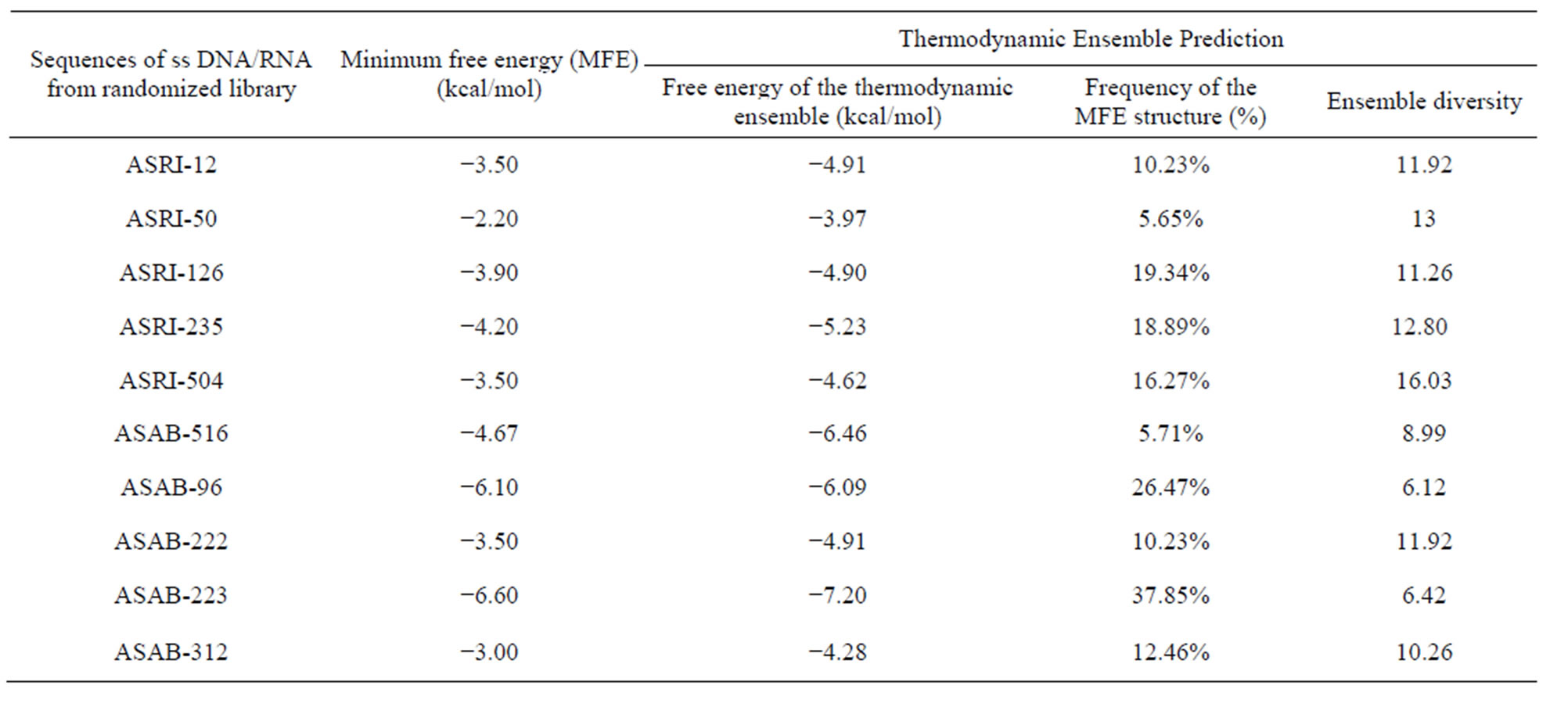
Table 1. Values of different parameters for different aptamer sequences.
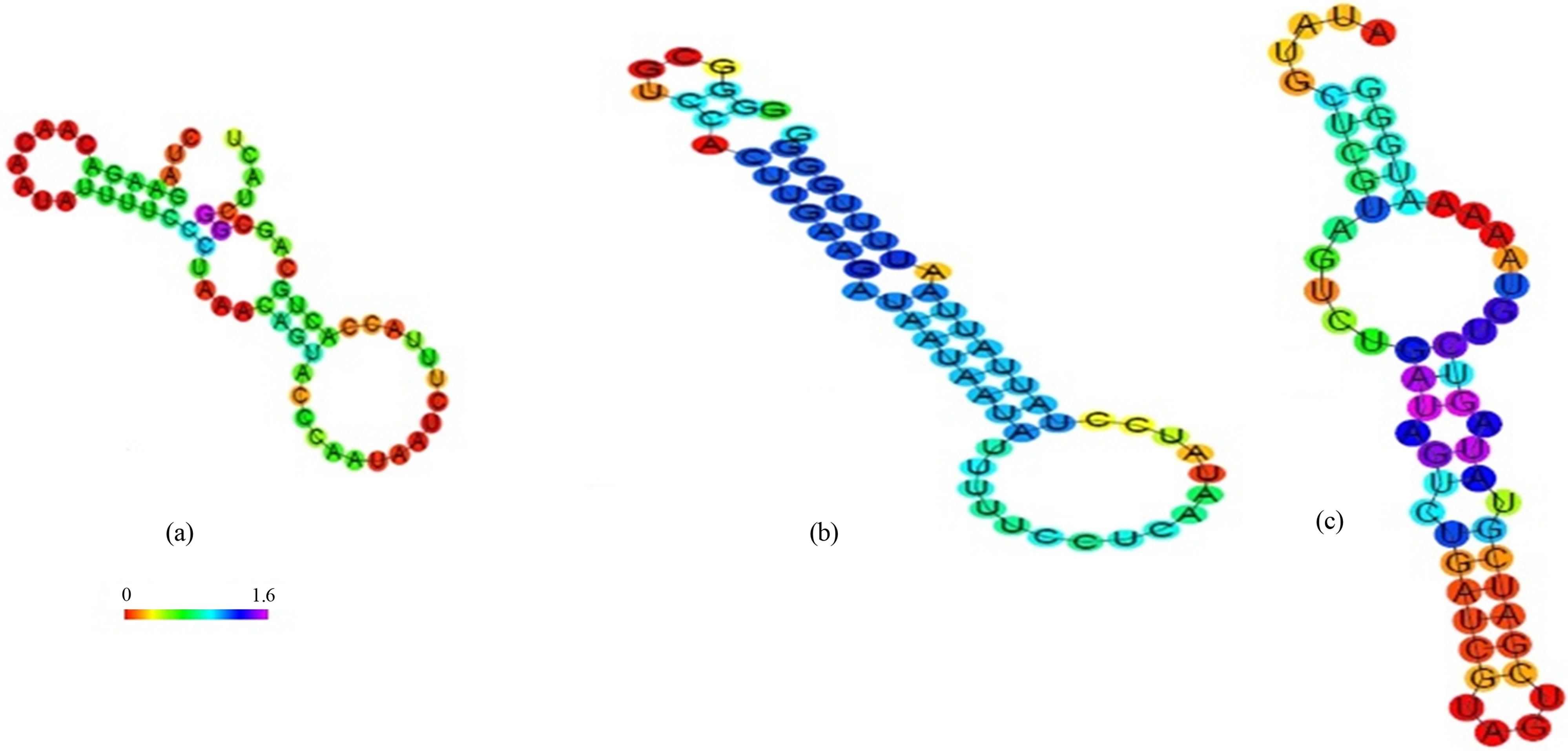
Figure 2. The structure of the top three ss of DNA/RNA sequences based on RNA fold. (a) sequence has higher probability binding to the ricin molecule (b) and (c) sequences have higher probability binding to the abrin molecule.
sity have appeared promising candidate for aptamer for the ricin molecules. The bioinformatics tools play important roles in the selection of the aptamers. The predicted ss DNA/RNA which is based on the online tools plays a important role in selection of the aptamer sequence. The molecular dynamics studies using HADDOCK had shown the interaction of the aptamer with it; apatope had shown the most promising sequences. We had isolated the sequences based on the molecular dynamics and secondary structure of the ss sequences of the aptamers.
5. Acknowledgements
The authors are thankful to the Director of INMAS, for his kind cooperation and providing the necessary facilities for carrying the research work. AS is thankful to DST INSPIRE fellowship, Government of India for financial support.
REFERENCES
- D. R. Thevenot, K. Toth, R. A. Durst and G. S. Wilson, “Electrochemical Biosensors: Recommended Definitions and Classification,” Pure and Applied Chemistry, Vol. 71, No. 12, 1999, pp. 2333-2348. http://dx.doi.org/10.1351/pac199971122333
- H. Park and I. R. Paeng, “Development of Direct Competitive Enzyme-Linked Aptamer Assay for Determination of Dopamine in Serum,” Analytica Chimica Acta, Vol. 685, No. 1, 2011, pp. 65-73. http://dx.doi.org/10.1016/j.aca.2010.11.010
- C. Tuerk and L. Gold, “Systematic Evolution of Ligands by Exponential Enrichment: RNA Ligands to Bacteriophage T4 DNA Polymerase,” Science, Vol. 24, No. 9, 1990, pp. 505-510. http://dx.doi.org/10.1126/science.2200121
- A. D. Ellington and J. W. Szostak, “In Vitro Selection of RNA Molecules That Bind Specific Ligands,” Nature, Vol. 3, No. 46, 1990, pp. 818-822. http://dx.doi.org/10.1038/346818a0
- L. Wang, C. Zhu, L. Han, L. Jin and S. Dong, “Label Free Regenerative and Sensitive Surface Plasmon Resonance and Electrochemical Aptasensors Based on Grahene,” Chemical Communications, Vol. 47, No. 27, 2011 pp. 7794-7796. http://dx.doi.org/10.1039/c1cc11373a
- A. D. Keefe, S. Pai and A. D. Ellingtion, “Aptmers as Therapeutics,” Nature Reviews Drug Discovery, Vol. 9, No. 7, 2010, pp. 537-550 http://dx.doi.org/10.1038/nrd3141
- M. Rajendran and A. D. Ellington, “Selection of Fluorescent Aptamer Beacons That Light up in the Presence of Zinc,” Analytical and Bioanalytical Chemistry, Vol. 390, No. 4, 2008, pp. 1067-1075. http://dx.doi.org/10.1007/s00216-007-1735-8
- J. E. Barrick and R. R. Breaker, “The Distributions, Mechanisms, and Structures of Metabolite-Binding Riboswitches,” Genome Biology, Vol. 8, 2007, p. 239. http://dx.doi.org/10.1186/gb-2007-8-11-r239
- C. Lozupone, et al., “Selection of the Simplest RNA That Binds Isoleucine,” RNA, Vol. 9, 2003, pp. 1315-1322. http://dx.doi.org/10.1261/rna.5114503
- Y. H. Kim, et al., “An RNA Aptamer That Specifically Binds Pancreatic Adenocarcinoma Up-Regulated Factor Inhibits Migration and Growth of Pancreatic Cancer Cells,” Cancer Letters, Vol. 313, No. 1, 2011, pp. 76-83. http://dx.doi.org/10.1016/j.canlet.2011.08.027
- 11J. Audi, M. belson, M. Patel, J. Schier and J. Osterloh, “Ricin Posining: A Comphersive Review,” JAMA, Vol. 294, No. 18, 2005, pp. 2342-2351. http://dx.doi.org/10.1001/jama.294.18.2342
- K. J. Dickers, S. M. Bradberry, P. Rice, G. D. Griffiths and J. A. Vale, “Abrin Poisoning,” Toxicological Reviews, Vol. 22, No. 3, 2003, pp. 137-142. http://dx.doi.org/10.2165/00139709-200322030-00002
- M. C. Frith, N. F. W. Saunders, B. Kobe and T. L. Bailey, “Discovering Sequence Motifs with Arbitrary Insertions and Deletions,” PLOS Computational Biology, Vol. 4, No. 5, 2008, Article ID: e1000071. http://dx.doi.org/10.1371/journal.pcbi.1000071
- C. L. Esposito, et al., “A Neutralizing RNA Aptamer against EGFR Causes Selective Apoptotic Cell Death,” PloS One, Vol. 6, No. 9, 2011, Article ID: e24071. http://dx.doi.org/10.1371/journal.pone.0024071
- S. C. Walker, et al., “The Dual Use of RNA Aptamer Sequences for Affinity Purification and Localization Studies of RNAs and RNA-Protein Complexes,” Methods in Molecular Biology, Vol. 714, 2011, pp. 423-444. http://dx.doi.org/10.1007/978-1-61779-005-8_26
- P. Wang, et al., “Aptamers as Therapeutics in Cardiovascular Diseases,” Current Medicinal Chemistry, Vol. 18, No. 27, 2011, pp. 4169-4174. http://dx.doi.org/10.2174/092986711797189673
- J. Hoinka, E. Zotenk, A. Friedman, Z. E. Sauna and T. M. Przytycka, “Identification of Sequence Structure RNA Binding Motifs for SELEX-Derived Aptamers,” Bioinformatics, Vol. 28, No. 12, 2012, pp. i215-i223. http://dx.doi.org/10.1093/bioinformatics/bts210
- R. Stoltenburg, C. Reinemann and B. Strehlitz, “SELEX —A (r) Evolutionary Method to Generate High-Affinity Nucleic Acid Ligands,” Biomolecular Engineering, Vol. 24, No. 4, 2007, pp. 381-403. http://dx.doi.org/10.1016/j.bioeng.2007.06.001
- A. K. Dubey, C. S. Baker, T. Romeo and P. Babtizke “RNA Sequence and Secondary Structure Participate in High Affinity CsrA-RNA Interaction,” RNA, Vol. 11, 2005, pp. 1579-1587. http://dx.doi.org/10.1261/rna.2990205
- H. Li, et al., “Aptamer Selection for the Detection of Escherichia coli K88,” Canadian Journal of Microbiology, Vol. 57, No. 6, 2011, pp. 453-459. http://dx.doi.org/10.1139/w11-030
- S. E. Osbrone and A. D. Ellington, “Nucleic Acid Selection and the Challenge of Combinatorial Chemistry,” Chemical Reviews, Vol. 97, No. 2, 1997, pp. 349-370. http://dx.doi.org/10.1021/cr960009c
- D. H. Mathews, M. D. Disney, J. L. Childs, S. J. Schroeder, M. Zuker and D. H. Turner, “Incorporating Chemical Modification Constraints into a Dynamic Programming Algorithm for Prediction of RNA Secondary Structure,” Proceedings of the National Academy of Sciences, Vol. 101, No. 19, 2004, pp. 7287-7292. http://dx.doi.org/10.1073/pnas.0401799101
- R. Lorenz, S. H. Bernhart, H. zu Siederdissen, H. Tafer, C. Flamm, P. F. Stadler and I. L. Hofacker, “Vienna RNA Package 2.0,” Algorithms for Molecular Biology, Vol. 6 No. 1, 2011, p. 26. http://dx.doi.org/10.1186/1748-7188-6-26
- X. J. Lu and W. K. Olson, “3DNA: A Software Package for the Analysis Rebuilding and Visualization of ThreeDimensional Nucleic Acid Structures,” Nucleic Acids Research, Vol. 31, No. 17, 2003, pp. 5108-5121. http://dx.doi.org/10.1093/nar/gkg680
- R. Lavery and H. Sklenar “Defining the Structure of Irregular Nucleic Acids: Conventions and Principles,” Journal of Biomolecular Structure and Dynamics, Vol. No. 6, 1989, pp. 655-667. http://dx.doi.org/10.1080/07391102.1989.10507728
- S. J. de Vries, M. van Dijk and A. M. J. J. Bonvin, “The HADDOCK Web Server for Data-Driven Biomolecular Docking,” Nature Protocols, Vol. 5, 2010, pp. 883-897. http://dx.doi.org/10.1038/nprot.2010.32
- M. V. Dijk, A. D. van Dijk, V. Hsu, R. Boelens and A. M. Bonvin, “Information-Driven Protein-DNA Docking Using HADDOCK: It Is A Matter of Flexibility,” Nucleic Acids Research, Vol. 34, No. 11, 2006, pp. 3317-3325. http://dx.doi.org/10.1093/nar/gkl412
NOTES
*Corresponding author.