Journal of Behavioral and Brain Science
Vol.06 No.09(2016), Article ID:69725,7 pages
10.4236/jbbs.2016.69034
Alterations in Caspase-3 in Juvenile Rats Treated Neonatally with Domoic Acid
Mark A. Robbins1, Catherine L. Ryan2, Tracy A. Doucette2
1Department of Biology, University of Prince Edward Island, Charlottetown, Canada
2Department of Psychology, University of Prince Edward Island, Charlottetown, Canada

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 8 March 2016; accepted 12 August 2016; published 15 August 2016
ABSTRACT
The clinical presentation of schizophrenia involves a variety of symptoms, which in many cases include hallucinations and delusions. Experimentally revealed alterations in both pre-pulse inhibition (PPI) and latent inhibition (LI) are also apparent in individuals afflicted with this disorder. Many have speculated that altered synaptic connections are, in part, responsible for this subset of behavioral abnormalities. We have previously reported that neonatal chronic low-dose injections of domoic acid (DOM) produce adult rats with deficits in PPI and LI. The current study was conducted to determine whether this toxin-treatment would alter the degree of apoptosis occurring in the developing brain. Results revealed significant decreases in caspase-3 within the right prelimbic cortex (PrL) in both male and female DOM-treated rats suggesting that even modest alterations in glutamate (Glu) signaling during critical periods of central nervous system (CNS) maturation will modify ontogenetic processes in the prefrontal cortex (PFC) of the juvenile rat.
Keywords:
Neonatal Rat, Domoic Acid, Prefrontal Cortex, Apoptosis, Caspase-3

1. Introduction
Apoptosis is a form of programmed cellular death which, unlike necrosis, does not involve the release of harmful substances into the extra-cellular environment. Three main pathways are involved in apoptotic signalling: the extrinsic pathway, the intrinsic pathway, and the perforin/granzyme. Each pathway leads to the activation of the so termed “execution pathway”; beginning with caspase-3 activation, and involving various mechanisms of cellular death (e.g. chromosomal degradation, cytoskeletal degradation, phagocytic uptake of apoptotic bodies) (reviewed in [1] ). Consequently, the quantification of activated capase-3 is a common marker for determining apoptotic cell death (reviewed in [2] [3] ).
High levels of apoptosis occur during development as a consequence of an initial overproduction of neurons. Estimates are that up to half of the original neuronal population is later pruned through apoptosis [4] [5] . Neurons receiving adequate stimulation through trophic support from glial cells, presynaptic cells, and steroid hormones are retained, while neurons not receiving proper stimulation are pruned [6] - [8] . This process is tightly regulated, as excessive levels can result in over-pruning of synaptic connections while insufficient levels can result in under-pruning of synaptic connections, with either scenario being sub-optimal for and detrimental to normal CNS maturation.
Although appropriate cell death via apoptosis is normal and necessary, these same apoptotic processes can be inappropriately commandeered, and are indeed implicated in cellular death which is innate to various neuropathological states/neurodegenerative disorders [9] - [11] . Inappropriate cell death also appears to be part of the underlying pathophysiology of schizophrenia (as reviewed in [12] ), with reduced neuropil [13] [14] , reductions in presynaptic markers such as synaptophysin and synaptosomal-associated protein 25 [15] , increased prefrontal neuronal apoptosis inhibitor protein [16] , abnormalities in prefrontal synaptic proteins such as N-ethylmaleimide sensitive factor fusion protein and synapsin II [17] and decreased prefrontal dendritic spine density [13] being reported in the cortex of those with schizophrenia. Additionally, others report decreased prefrontal white mater [18] layer-specific reductions in interneurons in layer II of prefrontal cortex and in layers IIBVI of the anterior cingulate cortex [19] and pyramidal neurons in layer IV of anterior cingulate cortex [20] , reductions in neuronal number in the nucleus accumbens (NAcc) [21] and mediodorsal thalamus [22] , and reductions in oligodendrocytes and glial cells in general in the PFC [23] [24] in those with schizophrenia. Collectively these data suggest that either increased cellular apoptosis or increased synaptic apoptosis, the latter referring to a form of apoptosis that targets synapses and/or distal neurites without affecting the remaining portion of a neuron [25] , occurs in individuals with schizophrenia.
Although the literature clearly suggests that changes in apoptosis occur as part of the pathogenesis of schizophrenia, paradoxically, not all findings report an increase in apoptotic processes. In fact, in a review by Jarskog (2006) [26] , he summarizes a number of studies that report a decrease in apoptotic markers in schizophrenia, particularly in cases of chronic schizophrenia. He speculates that this can reflect some one, or combination of, factors that result from an inappropriate response to an apoptotic insult or may reflect some compensatory response to earlier apoptotic processes.
In summary, alterations in programmed cell death consistently occur as a correlate in schizophrenia. Whether apoptosis is up-regulated or down-regulated may depend on the timing of the analysis or on the chronicity of the disorder, but clearly alterations in apoptosis reflect alterations in neuronal survival/circuitry that likely play a significant role in the behavioral manifestations of the disorder.
We have previously reported that, as adults, rats that were chronically exposed to very low doses of DOM during critical periods of CNS maturation, demonstrate persistent behavioural abnormalities that are consistent with those observed within the clinical population and that are homologous with those reported in other animal models of this disorder (reviewed in [27] ). These behavioural abnormalities include deficits in PPI [28] and LI [29] memory dysfunction [30] [31] and social withdrawal [32] . Therefore, the current study was conducted in order to determine whether early exposure to domoate would alter apoptotic processes in the medial prefrontal cortext (mPFC) in pre-weanling rats, thus altering the normal course of CNS maturation.
2. Materials and Methods
2.1. Experimental Animals and Injection Procedure
All experiments were conducted on the offspring of untimed pregnant Sprague-Dawley rats (Charles River Laboratories, St Constant, QC). Dams were left undisturbed until the day of birth, which was designated as postnatal day (PND) 0. Within 24 hours of birth, litters were culled to 10 pups (5 male and 5 female, where possible) and, using stratified-random sampling, pups from each litter were pseudo-randomly assigned to saline- or DOM- treatments.
Domoic acid, obtained from Diagnostic Chemicals Ltd. (Charlottetown, PE), was dissolved in sterile saline with injections (s.c.) administered in a volume of 10 ml/kg. From PND 8 - 14, pups were weighed, marked for identification (i.e., ear-notched), and given a single daily injection of 20 μg/kg DOM (i.e. a dose which has been previously shown to be physiologically relevant, and at which produces no overt signs of behavioral toxicity) [33] or an equal volume of saline. Litters were housed in a colony room maintained at approximately 22˚C and were housed under a 12 hour reverse light cycle (lights on at 07:00 h). Food and water (Purina Lab Chow) were administered ad libitum and all procedures were conducted according to the guidelines established by the Canadian Council on Animal Care and in accordance with the Animal Care Committee at the University of Prince Edward Island.
2.2. Tissue Collection and Sectioning
On PND 21, rats (n = 20; 5 of each sex × treatment with only one pup per each sex × treatment obtained from each litter) were deeply anesthetized with sodium pentobarbitol and decapitated. Brains were dissected from the skull, stored in 4% paraformaldehyde for 10 days, and then transferred to 0.08% sodium azide in phosphate- buffered saline (PBS) until sectioning. Coronal sections (50 µm) were produced in the area of the PrL (3.7 to 2.2 relative to bregma) [34] , and tissue was stored in 24-well plates containing 0.08% sodium azide in PBS until processed.
2.3. Immunohistochemical Analyses
Immunohistochemical procedures were adapted from [35] and began with a series of rinses (3 × 5 min in PBS unless otherwise stated) followed by a 30 minute incubation in 1% hydrogen peroxide in PBS. Sections were re-rinsed and incubated for 30 minutes in 1% sodium borohydride in PBS. Following re-rinsing, sections were incubated in blocking solution for 30 min (5% NHS, 2% BSA, 0.2% milk, 0.5% Triton X-100 in TBS) and were then treated with the primary antibody to active caspase-3 (rabbit anti-cleaved caspase-3; polyclonal) (Millipore, Billerica MA, USA) at a working dilution of 1:1000 in PBS for approximately 60 hours at 4EC. Rinses (5 × 5 min) in TBS were then performed prior to incubation for 2 hours in biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame CA, USA) in TBS at a dilution of 1:100. Tissue was rinsed in TBS and incubated with avidin-biotin complex (Vector Laboratories, Burlingame CA, USA) for 2 hrs. A final set of rinses in TBS was performed, sections were floated onto slides, and the peroxidase reaction was developed using 0.2% hydrogen peroxide in DAB for 6 minutes. Sections were then rinsed with approximately 2 mL of both PBS and distilled water and were left to dry overnight. The following day, sections were dehydrated (3 minute rinses in 70% EtOH, 95% EtOH, 100% EtOH, 100% EtOH, xylene, xylene before a final 45 minute rinse in xylene) and cover-slipped using Permount.
2.4. Quantification of Staining
Equivalent sections, from separate subjects, were identified using the forceps minor of the corpus callosum and lateral ventricle as landmarks. The PrL of the mPFC was identified as being located at the midline of the brain and exactly equidistant from the most dorsal and ventral regions of each section [34] [35] . Photographs (1000× magnification) were taken of the left and right hemispheres of representative sections of the PrL using a digital Cannon Rebel EOS xSi camera (Canon Canada Inc., ON, Canada) mounted on a MEIJI trinocularML5000 series microscope (MEIJI Techno America, CA, USA). Instances of immunoreactivity in the PrL, defined as dark, punctuated somata which represent sites of caspase-3, were both manually counted and digitally quantified for optical density measurements using ImageJ software (1.41d, NIH, USA). All quantification procedures were conducted with the experimenter blind to the treatment of each specimen. Data obtained from the left and right hemispheres of male and female juvenile rats were analysed using planned orthogonal comparisons (PASW Statistics 18). A result of p ≤ 0.05 indicated statistical significance.
3. Results
Results revealed statistically significant treatment effects with DOM-treated males demonstrating decreased optical density measurements in the right PrL [t(8) = −2.838, p = 0.022] (Figure 1 and Figure 2) and with DOM- treated females demonstrating decreased immunopositive cell counts in the right PrL [t(8) = −2.307, p = 0.050] (Figure 3).
In addition to these statistically significant findings, a consistent tendency for decreased caspase-3 staining noted in DOM-treated rats compared to their saline counterparts. This pattern held true for all data―whether quantified using cell counting techniques, or when assessed using optical density methods―a pattern which was notable bilaterally and for both male and female rats (Table 1).

 (a) (b)
(a) (b)
Figure 1. Caspase-3 immunopositive staining in the right PRL of representative male (a) SAL-treated and (b) DOM-treated subjects.
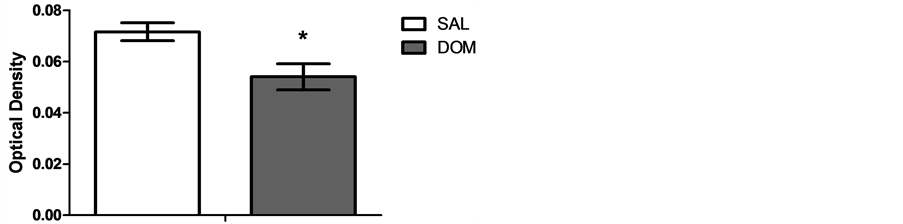
Figure 2. Mean ± SEM for the intensity of caspase-3 staining in the right PRL or DOM- and SAL-treated male subjects. Scores can range from 0 (lightest staining) to 1 (darkest staining). The asterisk indicates a statistically significant difference from controls (p ≤ 0.05).

Figure 3. Mean ± SEM cell count measurement of caspase-3 staining in the right PRL or DOM- and SAL-treated female subjects. The asterisk indicates a statistically significant difference from controls (p ≤ 0.05).

Table 1. Mean ± SEM for caspase-3 staining intensity, as quantified by optical density, and cell counts in the PRL of rats treated neonatally with DOM and SAL. Asterisk indicates a statistically significant difference (p ≤ 0.05).
4. Discussion
Data from the current study indicate that neonatal treatment with DOM from PND 8 - 14 (i.e. a critical period of brain development which is approximately equivalent to the third trimester of human development) [36] , resulted in decreased caspase-3 staining in the right PrL in both DOM-treated male (as demonstrated by optical density measurements) and female (as demonstrated by cell count measurements) juvenile rats.
We believe that these findings, while modest, could serve as an important first step in mechanistically linking our earlier reported behavioral abnormalities to their more proximal cause. Our previous reports which have shown altered PPI [28] , abnormal LI [29] , social withdrawal [32] , changes in emotionality [37] , altered responses to novelty and drug reinforcement [38] and deficits in learning and memory [30] [31] [39] following this early toxin-treatment regime could indeed be consequent to altered apoptotic processes. For instance, decreases in caspase-3 immunoreactivity could reflect lower levels of apoptosis during the time period assessed (i.e. 1 week following toxin treatment). It may be that low doses of DOM provide survival signals via glutamate receptor activation [40] . However, it is also possible that the opposite is true; that our results indicate an earlier-than-normal period of increased cell death (i.e. during the treatment regime), perhaps as a result of glutamate hyperactivity/excitotoxicity [41] [42] . Either scenario would most likely alter PFC circuitry and, in turn, could result in the behavioural anomalies we have reported on previously.
Although much remains to be discerned with respect to the proximal cause(s) and more distal consequence(s) of the results reported herein, we have demonstrated alterations tocaspase-3 expression in the PrL of pre- weanling rats. As such, these data suggest that as a consequence of this early low-dose toxin treatment, the establishment of appropriate prefrontal connections (e.g. synaptic structure and function) may be persistently altered.
Etiological validity involves recapitulating the clinical etiology of the disorder in question [43] . It is our contention that, in addition to face validity (e.g. symptom similarities), our treatment regime of neonatal chronic low-dose DOM-exposure has inherent etiological validity as an animal model of schizophrenia due to its neurodevelopmental nature. Moreover, we believe that this validity is further strengthened by the results reported herein.
5. Conclusion
In summary, altered caspase-3 in the PrL suggests that early domoate-treatment may result in persistent abnormalities in synaptic structure and function, a finding that is relevant in terms of improving the etiological validity of our early domoate-treatment regime as a neurodevelopmental model which may have application in schizophrenia research. These findings provide a contextual frame-work for understanding the cognitive abnormalities that are reliably observed in adult animals that are treated neonatally with very low doses of domoic acid during critical periods of CNS maturation.
Acknowledgements
Funding for this study was provided by a grant from the Natural Science and Engineering Research Council of Canada (NSERC) awarded to TA Doucette. MAR was supported by NSERC and Hooper/Klarenbach Research Post-Graduate Scholarships.
Cite this paper
Mark A. Robbins,Catherine L. Ryan,Tracy A. Doucette, (2016) Alterations in Caspase-3 in Juvenile Rats Treated Neonatally with Domoic Acid. Journal of Behavioral and Brain Science,06,357-363. doi: 10.4236/jbbs.2016.69034
References
- 1. Jin, Z. and El-Deiry, W.S. (2005) Overview of Cell Death Signaling Pathways. Cancer Biology & Therapy, 4, 139-163.
http://dx.doi.org/10.4161/cbt.4.2.1508 - 2. Jakob, S., Corazza, N., Diamantis, E., Kappeler, A. and Brunner, T. (2008) Detection of Apoptosis in Vivo Using Antibodies against Caspase-Indcued Neo-Epitopes. Methods, 44, 255-261.
http://dx.doi.org/10.1016/j.ymeth.2007.11.004 - 3. Resendes, A.R., Majo, N., Segales, J., Espadamala, J., Mateu, E., Chianini, F., Nofrarias, M., et al. (2004) Apoptosis in Normal Lymphoid Organs from Healthy Normal, Conventional Pigs at Different Ages Detected by TUNEL and Cleaved Caspase-3 Immunohistochemistry in Paraffin-Embedded Tissues. Veterinary Immunology and Immunopathology, 99, 203-213.
http://dx.doi.org/10.1016/j.vetimm.2004.02.001 - 4. Burek, M. and Oppenheim, R. (1999) Cellular Interactions That Regulate Programmed Cell Death in the Developing Vertebrate Nervous System. In: Koliatsos, V. and Ratan, R., Eds., Cell Death and Disease of the Nervous System, Vol. 1, Humana, Totowa, 145B80.
http://dx.doi.org/10.1007/978-1-4612-1602-5_8 - 5. Oppenheim, R.W. (1981) Cell Death of Motoneurons in the Chick Embryo Spinal Cord: Evidence on the Role of Cell Death and Neuromuscular Function in the Formation of Specific Peripheral Connections. Journal of Neuroscience, 1, 141B51.
- 6. Nordeen, E.J., Nordeen, K.W., Sengelaub, D.R. and Arnold, A.P. (1985) Androgens Prevent Normally Occurring Cell Death in a Sexually Dimorphic Spinal Nucleus. Science, 229, 671B73.
- 7. Okado, N. and Oppenheim, R.W. (1984) Cell Death of Motoneurons in the Chick Embryo Spinal Cord: The Loss of Motoneurons Following Removal of Afferent Inputs. Journal of Neuroscience, 4, 1639B52.
- 8. Lindsay, R.M. (1979) Adult Rat Brain Astrocytes Support Survival of Both NGF Dependent and NGF Insensitive Neurones. Nature, 282, 80B82.
http://dx.doi.org/10.1038/282080a0 - 9. Hickey, M.A. and Chesselet, M.F. (2003) Apoptosis in Huntington’s Disease. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 27, 255-265.
http://dx.doi.org/10.1016/S0278-5846(03)00021-6 - 10. Martin, L.J. (1999) Neuronal Death in Amyotrophic Lateral Sclerosis Is Apoptosis: Possible Contribution of a Programmed Cell Death Mechanism. Journal of Neuropathology & Experimental Neurology, 58, 459-471.
http://dx.doi.org/10.1097/00005072-199905000-00005 - 11. Su, J.H., Anderson, A.J., Cummings, B.J. and Cotman, C.W. (1994) Immunohistochemical Evidence for Apoptosis in Alzheimer’s Disease. Neuroreport, 5, 2529-2533.
http://dx.doi.org/10.1097/00001756-199412000-00031 - 12. Jarskog, L.F., Glantz, L.A., Gilmore, J.H. and Lieberman, J.A. (2005) Apoptotic Mechanisms in the Pathophysiology of Schizophrenia. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 29, 846-858.
http://dx.doi.org/10.1016/j.pnpbp.2005.03.010 - 13. Garey, L., Ong, W., Patel, T., Kanani, M., Davis, A., Mortimer, A., Barnes, T. and Hirsch, S. (1998) Reduced Dendritic Spine Density on Cerebral Cortical Pyramidal Neurons in Schizophrenia. Journal of Neurology, Neurosurgery & Psychiatry, 65, 446-453.
http://dx.doi.org/10.1136/jnnp.65.4.446 - 14. Glantz, L.A. and Lewis, D.A. (2000) Decreased Dendritic Spine Density on Pre-frontal Cortical Pyramidal Neurons in Schizophrenia. Archives of General Psychiatry, 57, 65B73.
- 15. Karson, C.N., Mrak, R.E., Schluterman, K.O., Sturner, W.Q., Sheng, J.G. and Griffin, W.S. (1999) Alterations in Synaptic Proteins and Their Encoding mRNAs in Prefrontal Cortex in Schizophrenia: A Possible Neurochemical Basis for Hypofrontality. Molecular Psychiatry, 4, 39B45.
http://dx.doi.org/10.1038/sj.mp.4000459 - 16. Vawter, M.P., Crook, J.M., Hyde, T.M., Kleinman, J.E., Weinberger, D.R., Becker, K.G. and Freed, W.J. (2002) Microarray Analysis of Gene Expression in the Prefrontal Cortex in Schizophrenia: A Preliminary Study. Schizophrenia Research, 58, 11-20.
http://dx.doi.org/10.1016/S0920-9964(01)00377-2 - 17. Mirnics, K., Middleton, F.A., Marquez, A., Lewis, D.A. and Levitt, P. (2000) Molecular Characterization of Schizophrenia Viewed by Microarray Analysis of Gene Expression in Prefrontal Cortex. Neuron, 28, 53-67.
http://dx.doi.org/10.1016/S0896-6273(00)00085-4 - 18. Breier, A., Buchanan, R.W., Elkashef, A., Munson, R.C., Kirkpatrick, B. and Gellad, F. (1992) Brain Morphology and Schizophrenia: A Magnetic Resonance Imaging Study of Limbic, Prefrontal Cortex, and Caudate Structures. Archives of General Psychiatry, 49, 921-926.
http://dx.doi.org/10.1001/archpsyc.1992.01820120009003 - 19. Benes, F.M., McSparren, J., Bird, E.D., SanGiovanni, J.P. and Vincent, S.L. (1991) Deficits in Small Interneurons in Prefrontal and Cingulate Cortices of Schizophrenic and Schizoaffective Patients. Archives of General Psychiatry, 48, 996-1001.
http://dx.doi.org/10.1001/archpsyc.1991.01810350036005 - 20. Benes, F.M., Vincent, S.L. and Todtenkopf, M. (2001) The Density of Pyramidal and Nonpyramidal Neurons in Anterior Cingulate Cortex of Schizophrenic and Bipolar Subjects. Biological Psychiatry, 50, 395-406.
http://dx.doi.org/10.1016/S0006-3223(01)01084-8 - 21. Pakkenberg, B. (1990) Pronounced Reduction of Total Neuron Number in Mediodorsal Thalamic Nucleus and Nucleus Accumbens in Schizophrenics. Archives of General Psychiatry, 47, 1023-1028.
http://dx.doi.org/10.1001/archpsyc.1990.01810230039007 - 22. Pakkenberg, B. (1987) Post Mortem Study of Chronic Schizophrenic Brains. The British Journal of Psychiatry, 151, 744-752.
http://dx.doi.org/10.1192/bjp.151.6.744 - 23. Cotter, D., Mackay, D., Chana, G., Beasley, C., Landau, S. and Everall, I.P. (2002) Reduced Neuronal Size and Glial Cell Density in Area 9 of the Dorsolateral Prefrontal Cortex in Subjects with Major Depressive Disorder. Cerebral Cortex, 12, 386-394.
http://dx.doi.org/10.1093/cercor/12.4.386 - 24. Hof, P.R., Haroutunian, V., Friedrich Jr, V.L., Byne, W., Buitron, C., Perl, D.P. and Davis, K.L. (2003) Loss and Altered Spatial Distribution of Oligodendrocytes in the Superior Frontal Gyrus in Schizophrenia. Biological Psychiatry, 53, 1075-1085.
http://dx.doi.org/10.1016/S0006-3223(03)00237-3 - 25. Mattson, M.P., Keller, J.N. and Begley, J.G. (1998) Evidence for Synaptic Apoptosis. Experimental Neurology, 153, 35-48.
http://dx.doi.org/10.1006/exnr.1998.6863 - 26. Jarskog, L.F. (2006) Apoptosis in Schizophrenia: Pathophysiologic and Therapeutic Considerations. Current Opinion in Psychiatry, 19, 307-312.
http://dx.doi.org/10.1097/01.yco.0000218603.25346.8f - 27. Jones, C.A., Watson, D.J.G. and Fone, K.C.F. (2011) Review: Animal Models of Schizophrenia. British Journal of Pharmacology, 164, 1162-1194.
http://dx.doi.org/10.1111/j.1476-5381.2011.01386.x - 28. Adams, A.L., Doucette, T.A. and Ryan, C.L. (2008) Altered Pre-Pulse Inhibition in Adult Rats Treated Neonatally with Domoic Acid. Amino Acids, 35, 157-160.
http://dx.doi.org/10.1007/s00726-007-0603-3 - 29. Adams-Marriott, A.L., Ryan, C.L. and Doucette, T.A. (2012) Neonatal Domoic Acid Treatment Produces Alterations to Prepulse Inhibition and Latent Inhibition in Adult Rats. Pharmacology Biochemistry and Behavior, 103, 338-344.
http://dx.doi.org/10.1016/j.pbb.2012.08.022 - 30. Robbins, M.A., Ryan, C.L., Adams-Marriott, A.L. and Doucette, T.A. (2013) Temporal Memory Dysfunction and Alterations in Tyrosine Hydroxylase Immunoreactivity in Adult Rats Following Neonatal Exposure to Domoic Acid. Neuroscience and Medicine, 4, 29-35.
http://dx.doi.org/10.4236/nm.2013.41005 - 31. Adams, A.L., Doucette, T.A., James, R. and Ryan, C.L. (2009) Persistent Changes in Learning and Memory in Rats Following Neonatal Treatment with Domoic Acid. Physiology & Behavior, 96, 505-512.
http://dx.doi.org/10.1016/j.physbeh.2008.11.019 - 32. Ryan, C.L., Robbins, M.A., Smith, M.T., Gallant, I.C., Adams, A.L. and Doucette, T.A. (2011) Altered Social Interaction in Adult Rats Following Neonatal Treatment with Domoic Acid. Physiology & Behavior, 102, 291-295.
http://dx.doi.org/10.1016/j.physbeh.2010.11.020 - 33. Doucette, T.A., Bernard, P.B., Yuill, P.C., Tasker, R.A. and Ryan, C.L. (2003) Low Doses of Non NMDA Glutamate Receptor Agonists Alterneuro Behavioural Development in the Rat. Neurotoxicology and Teratology, 25, 473-479.
http://dx.doi.org/10.1016/S0892-0362(03)00034-5 - 34. Ramachandra, R. and Subramanian, T. (2011) Atlas of the Neonatal Rat Brain. CRC Press, Boca Raton.
http://dx.doi.org/10.1201/b10500 - 35. Lanshakov, D.A., Bulygina, V.V., Romanova, I.V. and Dygalo, N.N. (2009) Immuno-histochemical Analysis of Active Caspase-3 Expression in Structures of Neonatal Brain. Bulletin of Experimental Biology and Medicine, 147, 635-638.
http://dx.doi.org/10.1007/s10517-009-0563-6 - 36. Clancy, B., Finlay, B.L., Darlington, R.B. and Anand, K.J.S. (2007) Extrapolating Brain Development from Experimental Species to Humans. Neurotoxicology, 28, 931-937.
http://dx.doi.org/10.1016/j.neuro.2007.01.014 - 37. Doucette, T.A., Ryan, C.L. and Tasker, R.A. (2007) Gender Based Changes in Cognition and Emotionality in a New Rat Model of Epilepsy. Amino Acids, 32, 317-322.
http://dx.doi.org/10.1007/s00726-006-0418-7 - 38. Burt, M.A., Ryan, C.L. and Doucette, T.A. (2008) Altered Responses to Novelty and Drug Reinforcement in Adult Rats Treated Neonatally with Domoic Acid. Physiology & Behavior, 93, 327-336.
http://dx.doi.org/10.1016/j.physbeh.2007.09.003 - 39. Tasker, R.A., Perry, M.A., Doucette, T.A. and Ryan, C.L. (2005) NMDA Receptor Involvement in the Effects of Low Dose Domoic Acid in Neonatal Rats. Amino Acids, 28, 193-196.
http://dx.doi.org/10.1007/s00726-005-0167-z - 40. Bahr, B.A., Bendiske, J., Brown, Q.B., Munirathinam, S., Caba, E., Rudin, M., Urwyler, S., Sauter, A. and Rogers, G. (2002) Survival Signaling and Selective Neuroprotection through Glutamateric Transmission. Experimental Neurology, 174, 37-47.
http://dx.doi.org/10.1006/exnr.2001.7852 - 41. Sattler, R. and Tymianski, M. (2001) Molecular Mechanisms of Glutamate Receptor-Mediated Excitotoxic Neuronal Cell Death. Molecular Neurobiology, 24, 107-129.
http://dx.doi.org/10.1385/MN:24:1-3:107 - 42. McDonald, J.W. and Johnston, M.V. (1990) Physiological and Pathophysiological Roles of Excitatory Amino Acids during Central Nervous System Development. Brain Research Reviews, 15, 41-70.
http://dx.doi.org/10.1016/0165-0173(90)90011-C - 43. Belzung, C. and Lemoine, M. (2011) Criteria of Validity for Animal Models of Psychiatric Disorders: Focus on Anxiety Disorders and Depression. Biology of Mood & Anxiety Disorders, 1, 9.
http://dx.doi.org/10.1186/2045-5380-1-9