Journal of Behavioral and Brain Science
Vol. 2 No. 1 (2012) , Article ID: 17715 , 4 pages DOI:10.4236/jbbs.2012.21001
The Time Course of D1 Agonist Induced Striatonigral ERK1/2 Signaling in a Rat Model of Parkinson’s Disease
Department of Pharmacology and Toxicology, School of Medicine-Northwest, Indiana University, Gary, USA
Email: *ssivam@iun.edu, cimoreno@iupui.edu
Received September 30, 2011; revised October 24, 2011; accepted November 11, 2011
Keywords: Natural Asset; Financial Value; Neural Network
ABSTRACT
Using a rat model of hemiparkinsonism, we examined the time-course of D1 agonist, SKF-38393-induced changes in extracellular signaling regulated kinases 1/2 (ERK1/2) phosphorylation in the striatum and substantia nigra (SN). We unilaterally lesioned the rat median forebrain bundle with 6-hydroxydopamine. Dopaminergic lesioned rats were administered with SKF-38393 and perfused at 15, 30, 60, or 120 minutes after the drug. Immunohistochemical analysis of striatum and SN revealed, as expected, a loss of tyrosine hydroxylase and a decrease of substance P in lesioned rats. SKF-38393 induced a robust increase in phospho-ERK1/2 levels in the lesioned striatum, which peaked at 15 min and substantially declined by 120 min. We report for the first time that similar changes were observed in the SN. The time-dependent ERK 1/2 activation in the striatonigral neurons may play a role in the therapeutic and/or side effects such as dyskinesias related to the dopamine agonist treatment for Parkinson’s disease.
1. Introduction
Dopamine (DA) is involved in many physiological functions including motor control, mood and reward pathways [1-7]. DA receptors constitute a subfamily of G-protein coupled receptors (GPCRs) and are classified into two main subtypes D1 and D2 [8,9]. The normal functioning of basal ganglia is dependent on the activity of the ‘direct’ (striatonigral) and ‘indirect’ (striatopallidal) output pathways subserved predominantly by the D1 and D2 receptor subtypes [10,11]. Degeneration of the nigrostriatal DA pathway results in an imbalance of the activity of the direct and indirect projection pathways and is thought to be responsible for the movement disorders associated with Parkinson’s disease [3,7,11]. DA agonists restore the normal functions of the direct and indirect pathways when DA is lost [10,11]. Though DA agonists alleviate Parkinsonism symptoms, chronic administration of these drugs leads to the development of debilitating side effects such as dyskinesias [12]. One effect of DA-depletion, which is not normalized by DA agonist treatment, is the supersensitive response of direct pathway neurons to D1 agonists [6,12-15].
MAP kinases (MAPKs) have been implicated in a wide variety of cellular process such as proliferation, differentiation, apoptic cell death, synaptic plasticity and memory [16,17]. Extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinase (JNK) and p38 mitogen-activated protein kinase (p38MAPK) comprise the three major classes of MAPKs that are involved in cell signaling [18,19]. The ERK-linked signaling pathways are stimulated by receptor tyrosine kinases and G-protein coupled receptors (GPCRs) and generally lead to proliferative and mitogenic responses [20]. Accumulated evidence indicates that GPCRs such as DA receptors influence ERK pathways [21,22]. ERK 1/2 phosphorylation has been implicated in many forms of synaptic plasticity in the brain [17,23]. Other evidence shows that changes in expression and/or function of brain proteins that are involved in signal transduction/gene transcription contribute to neuronal adaptations in a variety of disease models or drug treatments [24-29].
Several studies have shown that D1 agonists such as SKF-38393 induce increases in ERK1/2 expression in the 6-OHDA lesioned striatum [7,30-33]. However, little is known about the changes in ERK1/2 in the SN (SN) of dopaminergic denervated animals. Our preliminary study focused on the changes of phospho-ERK1/2 levels in unilaterally DA lesioned striatum and SN, 30 min following the administration of D1 receptor agonist SKF-38393 [34]. The aim of our present study is to investigate and compare the time course of SKF-38393 induced changes in ERK1/2 signaling in the striatum and SN of a rat model of Parkinson’s disease.
2. Materials and Methods
2.1. Animals
Female Harlan Sprague Dawley rats (Sprague-Dawley, Haran Sprague-Dawley Inc., Indianapolis, IN) weighing 250 - 350 g were used. All animals were maintained on a 12/12 h light/dark cycle at 22˚C ± 2˚C and 50% ± 10% humidity. The animals were housed two per cage and had continuous access to Wayne Lab Box chow and water. Animals were used in accordance with the NIH Guide to the Care and Use of Laboratory Animals and approval by Institutional Animal Care and Use Committee of Indiana University School of Medicine.
2.2. Unilateral Dopaminergic Lesion with 6-Hydroxydopamine (6-OHDA)
Unilateral lesions of the nigrostriatal DA pathway at the level of median forebrain bundle (MFB) were made in rats anesthetized with ketamine HCl/xylazine HCl solution (80/10 mg/kg, i.p., Sigma-Aldrich). Denervation was achieved by infusion of 9 µg of the free base of 6OHDA in 4 µl of vehicle (0.1% ascorbic acid in normal saline) into the MFB according to the atlas of [35]. The coordinates relative to bregma were: A: 4.4, L: 1.5 and V: 8.7. Desipramine HCl (15mg/kg, i.p., Sigma-Aldrich), was administered 60 min prior to 6OHDA infusion to protect noradrenergic neurons [36]. Animals were allowed to recover from the anesthesia and were put back into their homes cages, where they were given access to food and water ad libitum. For two consecutive days post-surgery, animals were given meloxicam (Sigma-Aldrich) once a day at 1 mg/kg, s.c., for pain relief.
2.3. Apomorphine-Induced Rotation Test
Rats were screened to test the extent of the 6-OHDA lesion by apomorphine-induced rotation test. Fourteen days post-surgery, animals were challenged with apomorphine HCl (0.1 mg/kg, i.p., Sigma-Aldrich) in sodium bisulphite solution (0.1% in saline). Each animal was placed in a large hemispherical bowl (14 inch diameter) enclosed by transparent Plexiglas cylinder and the animals were observed for contralateral rotation. The number of contralateral rotations in 5 minutes at 15, 30, and 45 minute intervals after the apomorphine injection was recorded. Animals averaging 6 or more rotations/min were determined to have greater than 90% DA depletion and were selected for further drug treatment.
2.4. SKF-38393 Treatments
Selected 6-OHDA-lesioned animals were treated with the partial D1 agonist, SKF-38393 (6 mg/kg, i.p., in saline). A separate group of 6-OHDA lesioned animals were administered saline to serve as controls. The experimental design allowed us to compare relative changes from the intact versus lesioned side in the same animal as well as to the independent control group. Ten min after injection of SKF-3839, rats were observed for 5 min to verify rotational responsiveness to the agonist. Animals were killed 15, 30, 60 or 120 min after SKF-38393 administration by an overdose of sodium pentobarbital (100 mg/kg i.p., Sigma-Aldrich).
2.5. Immunohistochemistry
Deeply anesthetized animals were perfused transcardially for 4 min with saline (50 mL) followed by 4% phosphate buffered paraformaldehyde (0.1 M sodium phosphate). The brains were removed, stored in the fixative solution overnight and then stored in a 30% sucrose - 0.9% saline solution for 24 hr at 4˚C. After the brains had sunken in the sucrose solution, they were removed and frozen, and coronal sections through the striatum and SN were cut (30 µm) on a sliding microtome. Sections were collected into 30% sucrose—0.9% saline solution for immunohistochemistry following the general procedures of [33]. A brief description of the immunohistochemistry procedure is given below.
After rinsing in fresh PBS (0.15 M NaCl, 0.1M sodium phosphate, pH 7.4) three times (5 min each), freefloating tissue sections were soaked in 0.3% H2O2 for 30 min, rinsed three times in PBS—0.3% Triton X-100 (5 min each), and blocked in 10% normal goat serum and 0.2% Triton X-100 in PBS for 90 min. Affinity-purified monoclonal TH (Affinity Bioreagents, Golden, CO), phospho-ERK1/2 [phospho-p44/42 MAP kinase (thr 202/ tyr204); Cell Signaling Technology, Beverly, MA], and substance P (SP, [37]) were used to detect immunoreactivity.
All sections were incubated in 3% normal serum, 0.2% Triton X-100 and the primary antibodies for 24 hrs at 4˚C with agitation. Following incubation in the primary antibodies, sections were rinsed 3 times in PBS—0.3% Triton-X 100 (5 min each) and incubated in affinity-purified biotinylated anti-mouse IgG for the detection of TH and anti-rabbit IgG (Vector Laboratories, Burlingame, CA) for the detection of SP and phospho-ERK1/2. Tissue sections were further processed using Ready-to-use Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) with detection using nickel-cobalt intensification of the diaminobenzidine reaction product. Sections were rinsed, transferred to slides, and put through a series of graded ethanol rinses followed by a final rinse in xylene, reagent ACS (Acros, Morris Plains, and NJ). All sections were cover slipped with Permount (Fisher Scientific, Fair Lawn, NJ).
2.6. Drugs and Chemicals
6-OHDA HBr were obtained from Research Biochemicals Inc., Wayland, MA. SKF-38383, desipramine HCl, apomorphine HCl and meloxicam sodium salt hydrate were purchased from Sigma-Aldrich, St. Louis, MO. Other chemicals and reagents were purchased from commercial sources.
2.7. Quantification and Statistical Analysis
The intensity of immunostaining for TH, SP and phospho-ERK1/2 in the striatum and SN of the lesioned and intact sides were determined using ImageJ (NIH) and quantified with QuantiScan software (Biosoft, Ferguson, MO). The values for the lesioned side were expressed as percent change from the intact side. Changes in the intact versus lesioned side was assessed by Student’s t-test. For time course changes, the percent change on the lesioned side following SKF-38393 treatment at different times was compared to the changes in the control group. All data are presented as mean ± standard error of the mean. The data were subjected to one way analysis of variance followed by Newman-Keuls multiple range test for the comparison of group means using SigmaStat (Systat Software, Point Richmond, CA). P < 0.05 was considered significant.
3. Results
3.1. Assessment of TH and SP Immunoreactivity in the Striatum and SN of the Unilaterally 6-OHDA Lesioned Animals
TH immunoreactivity was determined for all unilaterally lesioned animals in order to verify that 6-OHDA induced an adequate loss of dopaminergic neurons within the lesioned striatum and SN. A greater than 90% decreases in TH immunoreactivity in the lesioned striatum and SN were observed for saline as well as SKF-38393 treatment groups. A representative micrograph showing depletion of TH immunoreactivity only on the lesioned side as compared to intact side is presented in Figure 1, upper panel. SP immunoreactivity was assessed as an indirect marker for DA depletion because near total striatal DA denervation results in a decrease in substance P in the striatum and SN [38,39]. The lesioned striatum and SN of control or SKF-38393treated animals demonstrated a significant loss of SP immunoreactivity. A representative micrograph showing the decrease of SP immunoreactivity on the lesioned side as compared to the intact side is presented in Figure 1, lower panel. D1 agonist treatment did not alter the basal loss of SP or TH immunoreactivity in striatum and SN (Figure 4).
3.2. Time-Course of Effects of SKF-38393 on Phospo-ERK1/2 Levels in the Striatum of Unilaterally 6-OHDA Lesioned Rats
Phospho-ERK was evaluated in the striatum of unilaterally lesioned rats at various time points after SKF-38393 administration. In the intact striatum, phospho-ERK1/2 immunoreactivity was not apparent (Figure 2). In rats with a unilateral 6-OHDA lesion of the nigrostriatal DA system, SKF-38393 treatment resulted in the robust activation of phospho-ERK1/2 in striatum that peaked as early as 15 min and declined thereafter at 30, 60 and 120 min (Figures 2 and 4). Phospho-ERK immunoreactivity remained significantly elevated at 120 min after SKF- 38393.
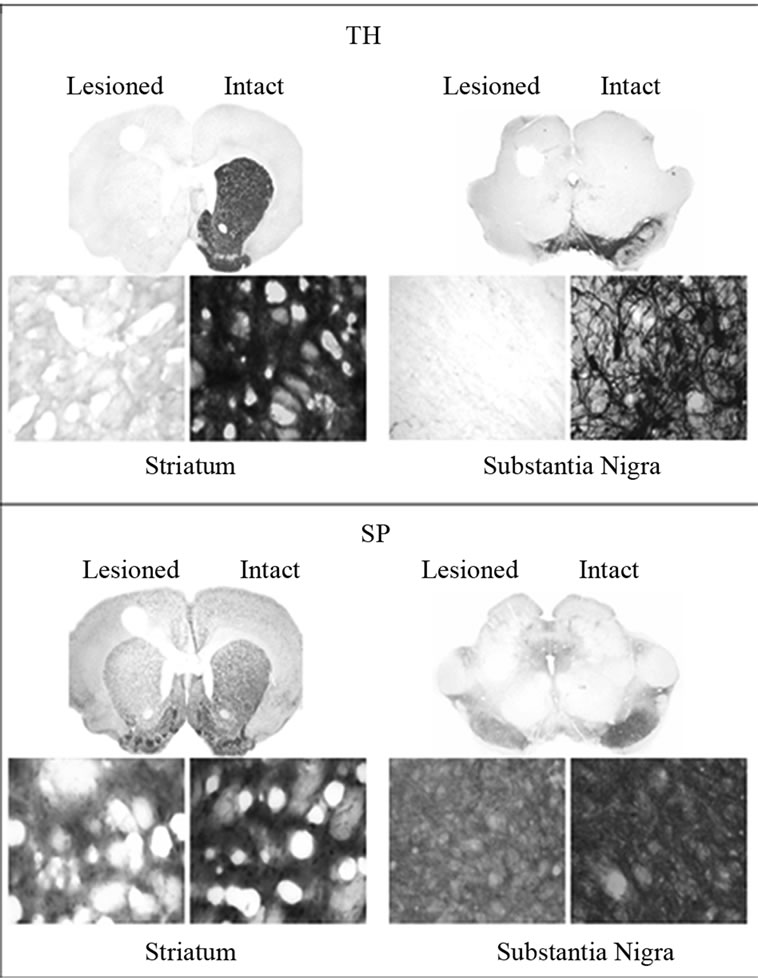
Figure 1. Upper Panel: Immunohistochemistry for tyrosine hydroxylase (TH) in coronal sections representing the striatum (left) and SN (right) of in unilaterally 6-OHDA lesioned rats. Representative micrographs showing the depletion of DA as evidenced from the loss of TH in the striatum on the lesioned side as compared to the intact side. The saline control served as a basal independent control. The loss of TH immunoreactivity in the SKF-38393 treatment groups at 15, 30, 60 and 120 min after the drug was similar to the saline control group. Lower Panel: Immunohistochemistry for substance P (SP) in coronal sections representing the striatum and SN of in unilaterally 6-OHDA lesioned rats. Representative micrographs show the modest loss of SP in the striatum SN on the lesioned side as compared to the intact side. The saline control served as a basal independent control. The loss of SP immunoreactivity in the SKF-38393 treatment groups at 15, 30, 60 and 120 min after the drug was similar to the saline control group. The coronal slices represent the magnification at 1.25× and the bottom panels show magnification at 400×.
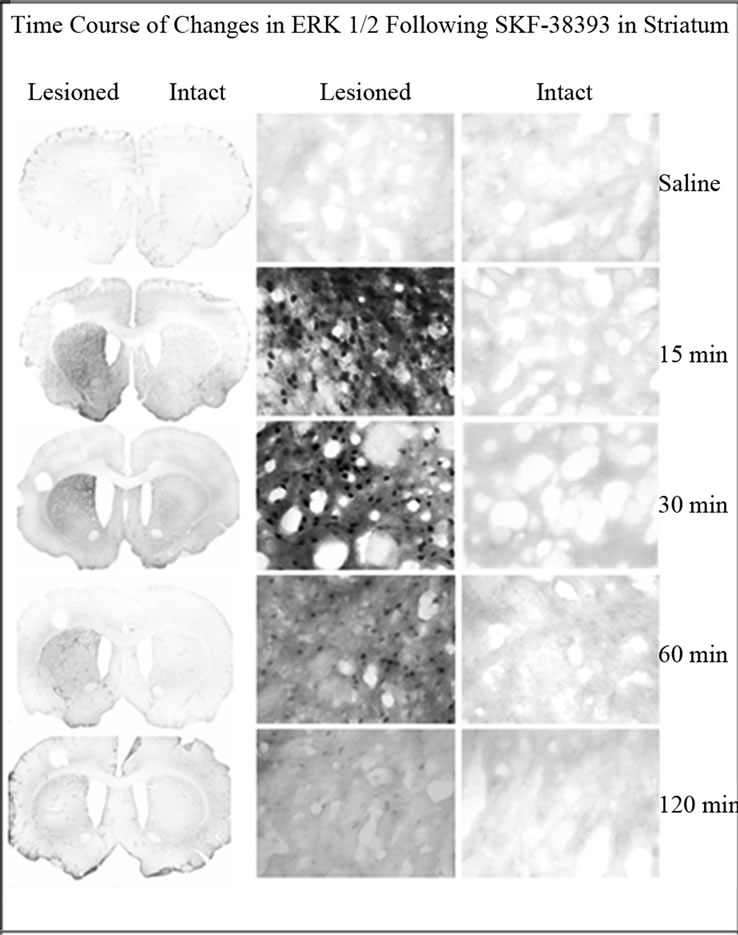
Figure 2. Immunohistochemistry for phospho-ERK1/2 in coronal sections representing the striatum of in unilaterally 6-OHDA lesioned rats. Representative micrographs showing the time course of ERK1/2 in the striatum on the lesioned side as compared to the intact side. The saline control served as a basal independent control. There was an increase of phospho-ERK1/2 immunoreactivity in the SKF- 38393 treatment groups at 15, 30, 60 and 120 min after the drug.
3.3. Effects of SKF-38393 on Phospo-ERK1/2 Levels in the SN of Unilaterally 6-OHDA Lesioned Rats
SKF-38393 induced robust increase in phospho-ERK1/2 on the lesioned side as compared to the intact side (Figure 3 and 4). Results similar to those seen in the striatum were observed within the SN with regard to time course changes in phopho-ERK1/2 following SKF-38393 treatment.
4. Discussion
The present study demonstrates that SKF-38393 induced a robust increase in phospho-ERK1/2 levels only in the lesioned striatum and SN as compared to the intact side. The results confirm and extend previous reports that DA agonists increase phosphorylation of ERK1/2 in the DAdepleted striatum. The present study reveals for the first time that increased phosphorylation of ERK1/2 also occurs in the SN, suggesting an involvement of the entire striatonigral pathway following DA agonist administra-
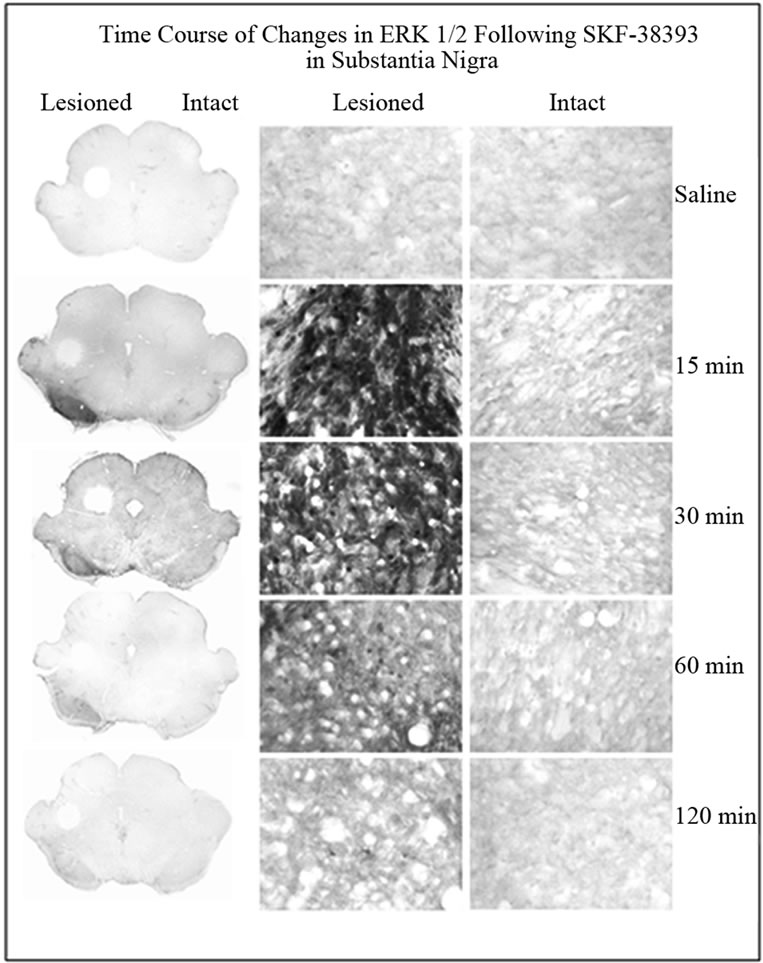
Figure 3. Immunohistochemistry for phospho-ERK1/2 in coronal sections representing the SN of in unilaterally 6- OHDA lesioned rats. Representative micrographs showing the time course of ERK1/2 in the striatum on the lesioned side as compared to the intact side. The saline control served as a basal independent control. There was an increase of phospho-ERK1/2 immunoreactivity in the SKF- 38393 treatment groups at 15, 30 and 60 min after the drug.
tion. In addition, our pilot experiments revealed that pretreatment with D1 antagonist SCH-23390 (1 mg/kg, i.p.) blocked the SKF-38393 induced increase in ERK1/2 levels both in the striatum and SN (unpublished observations).
The present results showing that there is an increase in striatal ERK1/2 following DA agonist administration in DA denervated rat striatum are in accordance with previous studies [31,33,34,40,41].
DA agonists also induce ERK1/2 in DA deficient mouse striatum in which the TH gene is specifically inactivated in dopaminergic neurons [32]. Gerfen et al. (2002) using the unilateral lesion model and Western blot technique reported that 2 mg/kg of SKF-38393 increased ERK1/2 levels in the lesioned but not in the intact striatum. Papadeas et al. (2004) reported that SKF-38393 (3 mg/kg) increased the immunoreactivity of ERK1/2 in the striatum of neonatally dopaminergic denervated rats tested as adults (bilateral lesion). In the present study with the unilateral adult lesion model, we used a dose of 6 mg/kg SKF-38393 and found that the increase in striatal ERK1/2 levels peaked at 15 min and declined thereafter but remained significantly elevated at 120 min. These studies taken together suggest dose dependency of dopamine agonist treatment for the induction of ERK1/2 in the striatum.
Several lines of evidence suggest enhanced ERK1/2 activation drives immediate early genes in the striatum resulting in transcription of a variety of genes [6,31]. However, sustained ERK1/2 activation (>60 min) is necessary for translocation of ERK1/2 from the cytoplasm to the nucleus to induce gene transcription that can lead to enduring plastic changes [42,43]. Such changes may influence the outcome of therapy or side effects of DA agonist treatments for Parkinson’s disease [44].
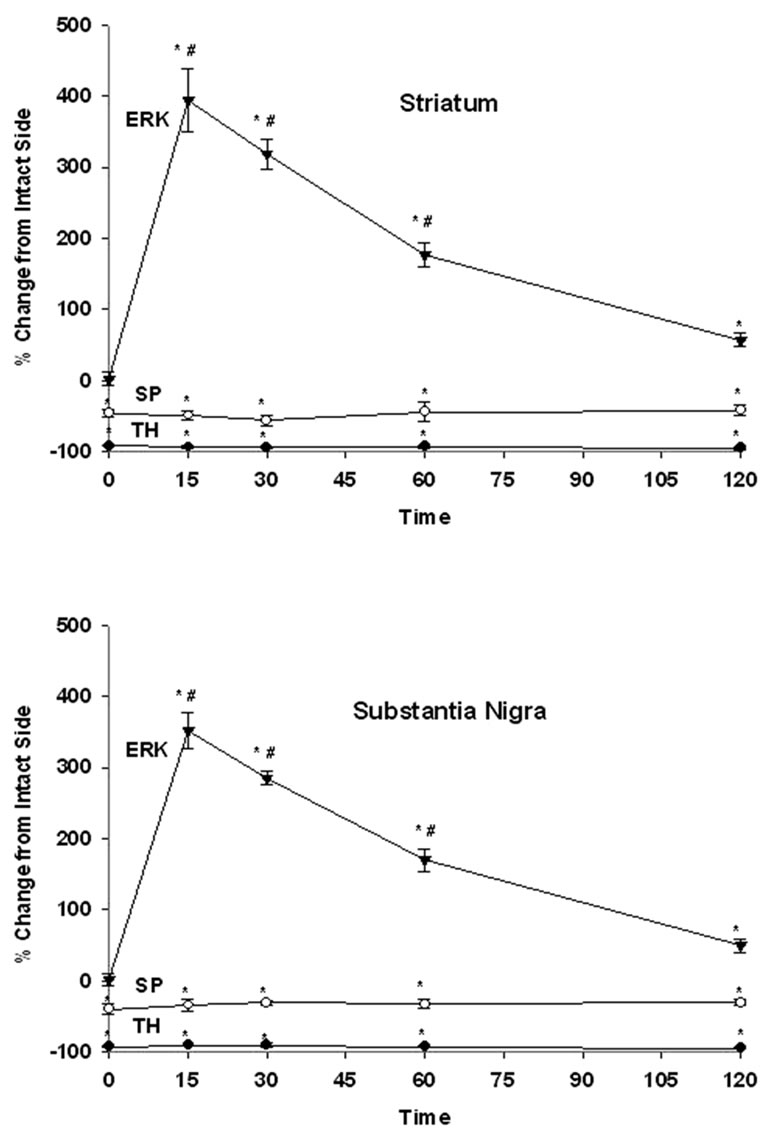
Figure 4. The time course of effects of SKF-3893 treatment on TH, SP and ERK1/2 in the striatum (upper panel) and SN (lower panel) of unilaterally dopaminergic lesioned rats. Rats received SKF-38393, 6 mg/kg, i.p and were perfused at 15, 30, 60 or 120 min later. TH, SP and ERK1/2 levels were determined by immunohistochemistry as described in Methods. Values represent mean ± S.E.M. Data were expressed as percent control of the intact side. The numbers of animals used were: 6, 4, 4, 4, 4 for control, 15, 30, 60, 120 min after SKF-39393 treatment, respectively. * P < 0.05 as compared to the intact side; # < 0.05 as compared to the control.
The normal functioning of basal ganglia is dependent on the activity of the “direct” (striatonigral) and “indirect” (striatopallidal) output pathways subserved predominantly by the D1 and D2 receptor subtypes [10,11]. Degeneration of the nigrostriatal DA pathway results in an imbalance of the activity of the direct and indirect projecttion pathways and is thought to be responsible for the movement disorders associated with Parkinson’s disease [3,7,11]. There are approximately an equal number of direct and indirect striatal projection neurons, which together constitute >90% of the neuron populations in the striatum [45]. Both direct and indirect striatal projection neurons receive nigrostriatal dopaminergic and corticostriatal glutamatergic inputs [46]. Previous studies indicated that in the intact striatum the ERK1/2 signaling pathway is normally used by indirect but not direct striatal projection neurons [7]. After nigrostriatal dopaminergic lesion, the induction of ERK1/2 signaling is activated through the supersensitive D1 receptors in direct pathway neurons [7,31,32]. However, recent studies have indicated other mechanisms that may also be involved in the regulation of ERK1/2 in the striatum. These include, among others, glutamate receptors [46,47], PKA-dependent phosphorylation of DARPP-32 [48], inhibition of protein phosphatase-1 [29], and growth factors such as glial cell line-derived neurotrophic factor (GDNF) [49].
Though there are a number of studies that elucidated the role of ERK1/2 in the striatum, there is a paucity of information in the literature regarding the influence of DA agonist on the induction of ERK1/2 in the SN. To our knowledge, this is first study to report that the SN exhibited a similar pattern of changes in ERK1/2 expression as that observed in the striatum following SKF- 38393 treatment in unilaterally 6OHDA lesioned animals. Based on literature search, we speculate that GABA, glutamate, GDNF, and/or cAMP/DARP-32 pathway may play a role in the induction of ERK1/2 in SN following DA agonist treatment.
DA cells are located in SN pars compacta (SNc) with their dendrites extending ventrally into the SN parts reticulata (SNr) which is composed primarily GABA projection neurons [50]. SNr neurons lack postsynaptic DA receptors [51]. D1 receptors were identified on the GABA [29,52] and glutamate [53] terminals synapsing on SNr neurons. The action of DA on SNr neurons is indirect, involving the presynaptic D1 receptors on GABA and/or glutamate afferents [54]. Therefore, it is possible that SKF-38393 can mediate its effects in the SNr neurons through activation of the D1 receptors on GABA and/or glutamate terminals which in turn may influence the induction of ERK1/2.
Reports have shown that the D1 agonist, SKF-82958 acts on D1 receptors in the SNr to increase extracellular levels of GABA [55]. This in turn causes an increase in motor activity which is dependent on the stimulation of GABA-A receptors. Stimulation of GABA-A receptors increased the phosphorylation of DARPP-32 in the striatum and substantia nigra (Snyder et al., 1994). DARPP-32, a DA and cyclic AMP regulated phosphoprotein, is highly enriched in the striatum and substantia nigra [56]. DARPP-32 has been shown to induce the phosphorylation of ERK1/2 in the dopamine-depleted striatum after L-DOPA administration (Santini et al., 2007). In dyskinetic mice, sensitized cAMP/cAMP-dependent protein kinase/DARPP- 32 signaling leads to activation of ERK1/2 following DA depletion (Santini et al., 2007). In the present study it is possible that D1 agonist SKF-38393 increases GABA release in SN that in turn can trigger the phosphorylation of DARPP-32 which is linked to induction of ERK1/2.
The phosphorylation of ERK1/2 may rely on the activation of glial cells in SN, which have been implicated to have a crucial role in the initiation and progression of PD [57]. In situ hybridization analyses have indicated that striatal neurons, which include GABAergic neurons, may indeed express protective neurotrophic factors such as GDNF [52]. In one study where GDNF was delivered via encapsulated cells to a 6-OHDA lesioned rat striatum, GDNF was found to be present in the striatal GABAergic neurons and was subject to be transported along the striatofugal pathway to the SNr [58]. Studies have shown that the SNr contains GABAergic neurons which express the GDNF family receptor α1 (GFRα1) and the transmembrane tyrosine kinase Ret [59-61]. When GDNF binds to the GFRα1 receptor, it induces tyrosine phosphorylation which in turn triggers various intracellular pathways of which include the Ras/ERK/MAPK pathways [60]. In addition, glial cells that form as a result of reactive gliosis following dopaminergic lesion may also be a potential source of GDNF [62].
GDNF has been shown to be neuroprotective in animal models of the Parkinson’s disease [63]. DA agonists increase the synthesis of GDNF in striatal and mesencephalic neuronal cultures [64] and in astroglial cultures [65]. Exposure to DA agonists including SKF-38393 increases GDNF production in a fetal human astrocyte cell line [66]. The selective lesion of nigrostriatal dopaminergic neurons in animal models of Parkinson’s disease alters mRNA and/or GDNF protein levels both in the striatum and in the SN [49]. In the intact adult brain, GDNF expression is largely neuronal; upon injury, glial cells appear to become the predominant source of trophic factors [49]. The effects of DA agonists both in cell cultures and in animal models of Parkinson’s disease indicate that DA receptors present in neurons and astrocytes might control GDNF expression [49]. It has been shown that LDOPA induces GDNF upregulation at the mRNA and protein levels in SN neuron-glial cell cultures [61].
Based on the foregoing discussion it appears that nigral GDNF release may be a potential mechanism for the nigral induction of ERK1/2 following SKF-38393 administration in dopaminergic denervated rat. GDNF may bind to the GFRα1/RET receptor of GABAergic neurons, inducing tyrosine phosphorylation which in turn triggers the MAPK pathway and ultimately the phosphorylation of ERK1/2.
In summary, the present study confirms and extends previous observations that D1 receptor stimulation in dopaminergic denervated animals induces robust ERK1/2 phosphorylation in the striatum in a time dependent fashion. Our study also reveals for the first time that parallel changes occur in the SN, suggesting an involvement of the entire striatonigral pathway following DA agonist administration. These results are relevant to our further understanding of the plasticity of transduction mechanisms involved in the pathophysiology, treatment and/or side effects of dopaminergic drugs used in Parkinson’s disease.
5. Acknowledgements
This work was in part supported by a Research Enhancement Award from IUPUI. Thanks are due to Dr. J.-S. Hong, NIEHS, Research Triangle Park, for the generous supply of SP antiserum used in this study.
REFERENCES
- Y. Smith and J. Z. Kieval, “Anatomy of the Dopamine System In the Basal Ganglia,” Trends in Neuroscience, Vol. 23, Supplement 1, 2000, pp. S28-S33. doi:10.1016/S1471-1931(00)00023-9
- S. C. Sealfon and C. W. Olanow, “Dopamine Receptors: From Structure to Behavior,” Trends in Neuroscience, Vol. 23, Supplement, 2000 pp. S34-S40. doi:10.1016/S1471-1931(00)00025-2
- M. Lebel, P. Robinson and M. Cyr, “Canadian Association of Neurosciences Review: The Role of Dopamine Receptor Function in Neurodegenerative Diseases,” Canadian Journal of Neurological Sciences, Vol. 34, No. 1, 2007, pp. 18-29.
- S. P. Onn, A. R. West and A. A. Grace, “Dopamine-Mediated Regulation of Striatal Neuronal and Network Interactions,” Trends in Neuroscience, Vol. 23, Suppl. 1, 2000, pp. S48-S56.
- A. M. Graybiel, C. W. Ragsdale Jr. and P. C. Emson, “Biochemical Anatomy of the Striatum,” Raven Press, New York, 1983, pp. 427-504.
- J. D. Berke, R. F. Paletzki, G. J. Aronson, S. E. Hyman and C. R. Gerfen, “A Complex Program of Striatal Gene Expression Induced by Dopaminergic Stimulation,” Journal of Neuroscience, Vol. 18, No. 14, 1998, pp. 5301- 5310.
- C. R. Gerfen, “D1 Dopamine Receptor Supersensitivity in the Dopamine-Depleted Striatum: Animal Model of Parkinson’s Disease,” Neuroscientist, Vol. 9, No. 6, 2003, pp. 455-462. doi:10.1177/1073858403255839
- K. A. Neve, J. K. Seamans and H. Trantham-Davidson, “Dopamine Receptor Signaling,” Journal of Receptor and Signal Transduction Research, Vol. 24, No. 3, 2004, pp. 165-205. doi:10.1081/RRS-200029981
- J. W. Kebabian, “Brain Dopamine Receptors: 20 Years of Progress,” Neurochemical Research, Vol. 18, No. 1, 1993, pp. 101-104. doi:10.1007/BF00966928
- C. R. Gerfen, T. M. Engber, L. C. Mahan, Z. Susel, T. N. Chase, F. J. Monsma and D. R. Sibley, “D1 and D2 Dopamine Receptor-Regulated Gene Expression of Striatonigral and Striatopallidal Neurons,” Science, Vol. 250, No. 4986, 1990, pp. 1429-1432. doi:10.1126/science.2147780
- C. R. Gerfen, “Molecular Effects of Dopamine on Striatal Projection Pathways,” Trends in Neuroscience, Vol. 23, Suppl. 1, 2000, pp. S64-S70.
- A. Nadjar, C. R. Gerfen and E. Bezard, “Priming for LDopa-Induced Dyskinesia in Parkinson’s Disease: A Feature Inherent to the Treatment or the Disease?” Progress in Neurobiology, Vol. 87, No. 1, 2009, pp. 1-9. doi:10.1016/j.pneurobio.2008.09.013
- G. S. Robertson, S. R. Vincent and H. C. Fibiger, “Striatonigral Projection Neurons Contain D1 Dopamine Receptor-Activated c-fos,” Brain Research, Vol. 523, No. 2, 1990, pp. 288-290. doi:10.1016/0006-8993(90)91498-6
- H. Steiner and C. R. Gerfen, “Dynorphin Opioid Inhibition of Cocaine-Induced, D1 Dopamine Receptor-Mediated immediate-Early Gene Expression in the Striatum,” Journal of Comparative Neurology, Vol. 353, No. 2, 1995, pp. 200-212. doi:10.1002/cne.903530204
- H. Steiner and C. R. Gerfen, “Dynorphin Regulates D1 Dopamine Receptor-Mediated Responses in the Striatum: Relative Contributions of Preand Postsynaptic Mechanisms in Dorsal and Ventral Striatum Demonstrated by Altered Immediate-Early Gene Induction,” Journal of Comparative Neurology, Vol. 376, No. 4, 1996, pp. 530-541. doi:10.1002/(SICI)1096-9861(19961223)376:4<530::AID-CNE3>3.0.CO;2-2
- J. D. Sweatt, “The Neuronal MAP Kinase Cascade: A Biochemical Signal Integration System Subserving Synaptic Plasticity and Memory,” Journal of Neurochemistry, Vol. 76, No. 1, 2001, pp. 1-10. doi:10.1046/j.1471-4159.2001.00054.x
- G. M. Thomas and R. L. Huganir, “MAPK Cascade Signaling and Synaptic Plasticity,” Nature Reviews Neuroscience, Vol. 5. No. 3, 2004, pp.173-183. doi:10.1038/nrn1346
- D. Chuderland and R. Seger, “Protein-Protein Interactions in the Regulation of the Extracellular Signal-Regulated Kinase,” Molecular Biotechnology, Vol. 29, No. 1, 2005, pp. 57-74. doi:10.1385/MB:29:1:57
- G. Pearson, F. Robinson, T. Gibson, B. E. Xu, M. Karandikar, K. Berman and M. H. Cobb, “Mitogen-Activated Protein (MAP) Kinase Pathways: Regulation and Physiological Functions,” Endocrinology Reviews, Vol. 22, No. 2, 2001, pp. 153-183. doi:10.1210/er.22.2.153
- G. R. Post and J. H. Brown, “G Protein-Coupled Receptors and Signaling Pathways Regulating Growth Responses,” FASEB Journal, Vol. 10, No. 7, 1996, pp. 741- 749.
- P. Rogue and A. N. Malviya, “Regulation of Signaling Pathways to the Nucleus by Dopaminergic Receptors,” Cell Signal, Vol. 6, No. 7, 1994, pp. 725-733. doi:10.1016/0898-6568(94)00044-1
- S. Maudsley, B. Martin and L. M. Luttrell, “The Origins of Diversity and Specificity in G Protein-Coupled Receptor Signaling,” Journal of Pharmacology and Experimental Therapeutics, Vol. 314, No. 2, 2005, pp. 485-494. doi:10.1124/jpet.105.083121
- R. J. Kelleher III, A. Govindarajan, H. Y. Jung, H. Kang and S. Tonegawa, “Translational Control by MAPK signaling in Long-Term Synaptic Plasticity and Memory,” Cell, Vol. 116, No. 3, 2004, pp. 467-479. doi:10.1016/S0092-8674(04)00115-1
- L. Pozzi, K. Hakansson, A. Usiello, A. Borgkvist, M. Lindskog, P. Greengard and G. Fisone, “Opposite Regulation by Typical and Atypical Anti-Psychotics of ERK1/2, CREB and Elk-1 Phosphorylation in Mouse Dorsal Striatum,” Journal of Neurochemistry, Vol. 86, No. 2, 2003, pp. 451-459. doi:10.1046/j.1471-4159.2003.01851.x
- J. Chen, M. Rusnak, R. R. Luedtke and A. Sidhu, “D1 Dopamine Receptor Mediates Dopamine-Induced Cytotoxicity via the ERK Signal Cascade,” Journal of Biological Chemistry, Vol. 279, No. 38, 2004, pp. 39317- 39330. doi:10.1074/jbc.M403891200
- J. McDaid, M. P. Graham and T. C. Napier, “Methamphetamine-Induced Sensitization Differentially Alters pCREB and Delta FosB throughout the Limbic Circuit of the Mammalian Brain,” Molecular Pharmacology, Vol. 70, No. 6, 2006, pp. 2064-2074. doi:10.1124/mol.106.023051
- L. Zhang, D. Lou, H. Jiao, D. Zhang, X. Wang, Y. Xia, J. Zhang and M. Xu, “Cocaine-Induced Intracellular Signaling and Gene Expression Are Oppositely Regulated by the Dopamine D1 and D3 Receptors,” Journal of Neuroscience, Vol. 24, No. 13, 2004, pp. 3344-3354.
- D. Tardito, J. Perez, E. Tiraboschi, L. Musazzi, G. Racagni and M. Popoli, “ Signaling Pathways Regulating Gene Expression, Neuroplasticity, and Neurotrophic Mechanisms in the Action of Antidepressants: A Critical Overview,” Pharmacological Reviews, Vol. 58, No.1, 2006, pp. 115- 134.
- E. Valjent, V. Pascoli, P. Svenningsson, S. Paul, H. Enslen, J. C. Corvol, A. Stipanovich, J. Caboche, P. J. Lombroso, A. C. Nairn, P. Greengard, D. Herve and J. A. Girault, “Regulation of a Protein Phosphatase Cascade Allows Convergent Dopamine and Glutamate Signals to Activate ERK in the Striatum,” Proceedings of National Academy of Sciences USA, Vol. 102, No. 2, 2005, pp. 491-496. doi:10.1073/pnas.0408305102
- N. Pavon, A. B. Martin, A. Mendialdua and R. Moratalla, “ERK Phosphorylation and FosB Expression Are Associated with L-DOPA-Induced Dyskinesia in Hemiparkinsonian Mice,” Biological Psychiatry, Vol. 59, No. 1, 2006, pp. 64-74. doi:10.1016/j.biopsych.2005.05.044
- C. R. Gerfen, S. Miyachi, R. Paletzki and P. Brown, “D1 Dopamine Receptor Supersensitivity in the DopamineDepleted Striatum Results from a Switch in the Regulation of ERK1/2/MAP Kinase,” Journal of Neuroscience, Vol. 22, No. 12, 2002, pp. 5042-5054.
- D. S. Kim, R. D. Palmiter, A. Cummins and C. R. Gerfen, “Reversal of Supersensitive Striatal Dopamine D1 Receptor Signaling and Extracellular Signal-Regulated Kinase Activity in Dopamine-Deficient Mice,” Neuroscience, Vol. 137, No. 4, 2006, pp. 1381-1388. doi:10.1016/j.neuroscience.2005.10.054
- S. T. Papadeas, B. L. Blake, D. J. Knapp and G. R. Breese, “Sustained Extracellular Signal-Regulated Kinase 1/2 Phosphorylation in Neonate 6-Hydroxydopamine-Lesioned Rats after Repeated D1-Dopamine Receptor Agonist Administration: Implications for NMDA Receptor Involvement,” Journal of Neuroscience, Vol. 24, No. 26, 2004, pp. 5863-5876. doi:10.1523/JNEUROSCI.0528-04.2004
- S. P. Sivam and C. Moreno, “Dopamine Agonist Mediated Striatonigral ERK1/2 Signaling in a Rat Model of Parkinson’s Disease,” Society of Neuroscience Abstracts, Washington DC, 2008.
- G. Paxinos and C. Watson, “The Rat Brain in Stereotaxic Coordinates,” Elsevier Academic Press, New York, 2005.
- G. R. Breese, A. A. Baumeister, T. J. McCown, S. G. Emerick, G. D. Frye, K Crotty and R. A. Mueller, “Behavioral Differences between Neonatal and Adult 6-Hydrocydopamine-Treated Rats to Dopamine Agonists: Relevance to Neurological Symptoms in Clinical Syndromes with Reduced Brain Dopamine,” Journal of Pharmacology and Experimental Therapeutics, Vol. 231, No. 2, 1984, pp. 343-354.
- S. J. Li, S. P. Sivam, J. F. McGinty, Y. S. Huang and J. S. Hong, “Dopaminergic Regulation of Tachykinin Metabolism in the Striatonigral Pathway,” Journal of Pharmacology and Experimental Therapeutics, Vol. 243, No. 2, 1987, pp. 792-798.
- L. K. Nisenbaum, W. R. Crowley and S. T. Kitai, “Partial Striatal Dopamine Depletion Differentially Affects Striatal Substance P and Enkephalin Messenger RNA Expression,” Molecular Brain Research, Vol. 37, No. 1-2, 1996, pp. 209-216. doi:10.1016/0169-328X(95)00317-L
- S. P. Sivam, G. R. Breese, J. E. Krause, T. C. Napier, M. A. Mueller and J. S. Hong, “Neonatal and Adult 6-Hydroxydopamine-Induced Lesions Differentially Alter Tachykinin and Enkephalin Gene Expression,” Journal of Neurochemistry, Vol. 49, No.5, 1987, pp. 1623-1633. doi:10.1111/j.1471-4159.1987.tb01036.x
- J. E. Westin, L. Vercammen, E. M. Strome, C. Konradi and M. A. Cenci, “Spatiotemporal Pattern of Striatal ERK1/2 Phosphorylation in a Rat Model of L-DOPA-induced Dyskinesia and the Role of Dopamine D1 Receptors,” Biological Psychiatry, Vol. 62, No. 7, 2007, pp. 800-810. doi:10.1016/j.biopsych.2006.11.032
- E. Valjent, J. C. Corvol, J. M. Trzaskos, J. A. Girault and D. Herve, “Role of the ERK Pathway in Psychostimulant-Induced Locomotor Sensitization,” BMC Neuroscience, Vol. 7, No. 1, 2006, p. 20.
- C. J. Marshall, “Specificity of Receptor Tyrosine Kinase Signaling: Transient versus Sustained Extracellular Signal-Regulated Kinase Activation,” Cell, Vol. 80, No. 2, 1995, pp. 179-185. doi:10.1016/0092-8674(95)90401-8
- J. Chen, M. Rusnak, P. J. Lombroso and A. Sidhu, “Dopamine Promotes Striatal Neuronal Apoptotic Death via ERK Signaling Cascades,” European Journal of Neuroscience, Vol. 29, No. 2, 2009, pp. 287-306. doi:10.1111/j.1460-9568.2008.06590.x
- L. Colucci-D'Amato, C. Perrone-Capano and U. di Porzio, “Chronic Activation of ERK and Neurodegenerative Diseases,” Bioessays, Vol. 25, No. 11, 2003, pp. 1085-1095. doi:10.1002/bies.10355
- C. R. Gerfen and W. S. Young III, “Distribution of Striatonigral and Striatopallidal Peptidergic Neurons in Both Path and Matrix Compartments: An in Situ Hybridization Histochemistry and Fluorescent Retrograde Tracing Study,” Brain Research, Vol. 460, No. 1, 1988, pp. 161-167. doi:10.1016/0006-8993(88)91217-6
- S. M. Hersch, B. J. Ciliax, C. A. Gutekunst, H. D. Rees, C. J. Heilman, K. K. Yung, J. P. Bolam, E. Ince, H. Yi and A. I. Levey, “Electron Microscopic Analysis of D1 and D2 Dopamine Receptor Proteins in the Dorsal Striatum and their Synaptic Relationships with Motor Corticostriatal Afferents,” Journal of Neuroscience, Vol. 15, No. 7, 1995, pp. 5222-5237.
- L, Pei, F. J. Lee, A. Moszczynska, B. Vukusic and F. Liu, “Regulation of Dopamine D1 Receptor Function by Physical Interaction with the NMDA Receptors,” Journal of Neuroscience, Vol. 24, No. 5, 2004, pp. 1149-1158. doi:10.1523/JNEUROSCI.3922-03.2004
- G. L. Snyder, G. Fisone and P. Greengard, “Phosphorylation of DARPP-32 Is Regulated by GABA in Rat Striatum and Substantia Nigra,” Journal of Neurochemistry, Vol. 63, No. 5, 1994, pp. 1766-1771. doi:10.1046/j.1471-4159.1994.63051766.x
- A. Saavedra, G. Baltazar and E. P. Duarte, “Driving GDNF Expression: The Green and the Red Traffic Lights,” Vol. 86, No. 3, 2008, pp. 186-215.
- T. Gonzalez-Hernandez and M. Rodriguez, “Compartmental Organization and Chemical Profile of Dopaminergic and GABAergic Neurons in the Substantia Nigra of the Rat,” Journal of Comparative Neurology, Vol. 421, No. 1, 2000, pp. 107-135. doi:10.1002/(SICI)1096-9861(20000522)421:1<107::AID-CNE7>3.0.CO;2-F
- K. K. Yung, J. P. Bolam, A. D. Smith, S. M. Hersch, B. J. Ciliax and A. I. Levey, “Immunocytochemical localization of D1 and D2 Dopamine Receptors in the Basal Ganglia of the Rat: Light and Electron Microscopy,” Neuroscience, Vol. 65, No. 3, 2008, pp. 709-730. doi:10.1016/0306-4522(94)00536-E
- J. L. Bizon, J. C. Lauterborn and C. M. Gall, “Subpopulations of Striatal Interneurons Can Be Distinguished on the Basis of Neurotrophic Factor Expression,” Journal of Comparative Neurology, Vol. 408, No. 2, 1999, pp. 283-298. doi:10.1002/(SICI)1096-9861(19990531)408:2<283::AID-CNE9>3.0.CO;2-2
- M. G. Rosales, D. Martinez-Fong, R. Morales, A. Nunez, G. Flores, J. L. Gongora-Alfaro, B. Floran and J. Aceves, “Reciprocal Interaction between Glutamate and Dopamine in the Pars Reticulata of the Rat Substantia Nigra: A Microdialysis Study,” Neuroscience, Vol. 80, No. 3, 1997, pp. 803-810. doi:10.1016/S0306-4522(97)00160-7
- F. Windels and E. A. Kiyatkin, “GABAergic Mechanisms in Regulating the Activity State of Substantia Nigra Pars Reticulata Neurons,” Neuroscience, Vol. 140, No. 4, 2006, pp. 1289-1299. doi:10.1016/j.neuroscience.2006.03.064
- T. Trevitt, B. Carlson, M. Correa, A. Keene, M. Morales and J. D. Salamone, “Interactions between Dopamine D1 Receptors and Gamma-Aminobutyric Acid Mechanisms in Substantia Nigra Pars Reticulata of the Rat: Neurochemical and Behavioral Studies,” Psychopharmacology, Vol. 159, No. 3, 2002, pp. 229-237. doi:10.1007/s002130100908
- C. C. Ouimet, P. E. Miller, H. C. Hemmings Jr., S. I. Walaas and P. Greengard, “ DARPP-32, a Dopamine and Adenosine 3':5'-Monophosphate-Regulated Phosphoprotein Enriched in Dopamine-Innervated Brain Regions. III. Immunocytochemical Iocalization,” Journal of Neuroscience, Vol. 4, No. 1, 1984, pp. 111-124.
- P. Teismann, K. Tieu, O. Cohen, D. K. Choi, D. C. Wu, D. Marks, M. Vila, V. Jackson-Lewis and S. Przedborski, “Pathogenic Role of Glial Cells in Parkinson’s Disease,” Movement Disorders, Vol. 18, No. 2, 2003, pp. 121-129. doi:10.1002/mds.10332
- A. Sajadi, J. C. Bensadoun, B. L. Schneider, C. Lo Bianco and P. Aebischer, “Transient Striatal Delivery of GDNF via Encapsulated Cells Leads to Sustained Behavioral Improvement in a Bilateral Model of Parkinson Disease,” Neurobiology of Disease, Vol. 22, No. 1, 2006, pp. 119- 129. doi:10.1016/j.nbd.2005.10.006
- P. Barroso-Chinea, I. Cruz-Muros, M. S. Aymerich, M. Rodriguez-Diaz, D. Afonso-Oramas, J. L. Lanciego and T. Gonzalez-Hernandez, “Striatal Expression of GDNF and Differential Vulnerability of Midbrain Dopaminergic Cells,” European Journal of Neuroscience, Vol. 21, No. 7, 2005, pp. 1815-1827. doi:10.1111/j.1460-9568.2005.04024.x
- A. Sarabi, B. J. Hoffer, L. Olson and M. Morales, “GFRalpha-1 mRNA in Dopaminergic and Nondopaminergic Neurons in the Substantia Nigra and Ventral Tegmental Area,” Journal of Comparative Neurology, Vol. 441, No. 2, 2001, pp. 106-117. doi:10.1002/cne.1400
- A. Saavedra, G. Baltazar, P. Santos, C. M. Carvalho and E. P. Duarte, “Selective Injury to Dopaminergic Neurons Up-Regulates GDNF in Substantia Nigra Postnatal Cell Cultures: Role of Neuron-Glia Crosstalk,” Neurobiology of Disease, Vol. 23, No. 3, 2006, pp. 533-542. doi:10.1016/j.nbd.2006.04.008
- R. L. Miller, G. Y. Sun and A. Y. Sun, “Cytotoxicity of Paraquat in Microglial Cells: Involvement of PKC Deltaand ERK1/2-Dependent NADPH Oxidase,” Brain Research, Vol. 1167, 2007, pp. 129-139.
- N. Lindgren, R. K. Leak, K. M. Carlson, A. D. Smith and M. J. Zigmond, “Activation of the Extracellular SignalRegulated Kinases 1 and 2 by Glial Cell Line-Derived Neurotrophic Factor and Its Relation to Neuroprotection in a Mouse Model of Parkinson’s Disease,” Journal of Neuroscience Research, Vol. 86, No. 9, 2008, pp. 2039- 2049. doi:10.1002/jnr.21641
- H. Guo, Z. Tang, Y. Yu, L. Xu, G. Jin and J. Zhou, “Apomorphine Induces Trophic Factors that Support Fetal Rat Mesencephalic Dopaminergic Neurons in Cultures,” European Journal of Neuroscience, Vol. 16, No. 10, 2002, pp. 1861-1870. doi:10.1046/j.1460-9568.2002.02256.x
- K. Ohta, S. Kuno, I. Mizuta, A. Fujinami, H. Matsui and M. Ohta, “Effects of Dopamine Agonists Bromocriptine, Pergolide, Cabergoline, and SKF-38393 on GDNF, NGF, and BDNF Synthesis in Cultured Mouse Astrocytes,” Life Sciences, Vol. 73, No. 5, 2003, pp. 617-626. doi:10.1016/S0024-3205(03)00321-7
- N. Kinor, R. Geffen, E. Golomb, T. Zinman and G. Yadid, “Dopamine Increases Glial Cell Line-Derived Neurotrophic Factor in Human Fetal Astrocytes,” Glia, Vol. 33, No. 2, 2001, pp.143-150. doi:10.1002/1098-1136(200102)33:2<143::AID-GLIA1013>3.0.CO;2-3
NOTES
*Corresponding author.