Advances in Chemical Engineering and Science
Vol. 2 No. 3 (2012) , Article ID: 20817 , 9 pages DOI:10.4236/aces.2012.23037
Enhanced Electrochemical Properties of LiFePO4 as Positive Electrode of Li-Ion Batteries for HEV Application
1Physicochimie des Electrolytes, Colloïdes et Systèmes Analytiques, Université Pierre et Marie Curie, Paris, France
2Institut de Recherche d’Hydro-Québec, Varennes, Canada
3Institut de Minéralogie et Physique de la Matière Condensée, Université Pierre et Marie Curie, Paris, France
Email: *christian.julien@upmc.fr
Received August 3, 2011; revised February 29, 2012; accepted March 9, 2012
Keywords: Olivine; Nanostructured Compounds; Local Structure; Magnetometry; Lithium-Ion Batteries
ABSTRACT
LiFePO4 materials synthesized using FePO4(H2O)2 and Li2CO3 blend were optimized in view of their use as positive electrodes in Li-ion batteries for hybrid electric vehicles. A strict control of the structural properties was made by the combination of X-ray diffraction, FT-infrared spectroscopy and magnetometry. The impact of the ferromagnetic clusters (γ-Fe2O3 or Fe2P) on the electrochemical response was examined. The electrochemical performances of the optimized LiFePO4 powders investigated at 60˚C are excellent in terms of capacity retention (153 mAh·g–1 at 2C) as well as in terms of cycling life. No iron dissolution was observed after 200 charge-discharge cycles at 60˚C for cells containing Li foil, Li4Ti5O12, or graphite as negative electrodes.
1. Introduction
Since the introduction of lithium-ion batteries based on lithium cobaltate (LiCoO2) by Sony in 1991, great efforts have been addressed to find an alternative material with both sides of the battery. However, the expansion of their applications from the portable market to the electric (EVs) and hybrid vehicles (HEVs) requests lower cost and better safety characteristic electrode material. Among the well-known Li-insertion compounds, the olivine LiFePO4 (LFP) compound is being extensively investigated as a positive electrode material for Li-ion batteries because of its low cost, low toxicity, and relatively high theoretical specific capacity of 170 mAh·g–1 [1,2]. The current debate for the utilization of LiFePO4 in large-size batteries (for HEV, for instance), is mainly focused on the perceived poor rate capability because of a low electronic conductivity. Another aspect concerns the material purity and the non-migration of iron ions through the electrolyte. The high-temperature performance is also a critical issue because batteries may be operated at elevated temperatures (around 60˚C). The early drawback of highly resistive LiFePO4 has been resolved by painting the particle surface with carbon [3-6].
Recently, significant effort has been underway to improve LiFePO4 by developing a new synthesis route via carbon coating [7]. The 1D Li channels make the olivine performance sensitive not only to particle size, but also to impurities and stacking faults that block the channels. Various types of iron-based impurities have been identified in the olivine framework: for examples γ-Fe2O3, Fe3O4, Li3Fe2(PO4)3, Fe2P2O7, Fe2P, Fe3P, Fe75P15C10, etc. Critical quality control of the product is necessary to obtain a complete understanding of synthesis conditions using combination of experiments such as Raman spectroscopy and magnetic measurements [8-12].
In this paper, we report the results obtained on several samples of LiFePO4 (LFP) with special attention to the new generation of phospho-olivine materials used in lithium cells operating at 60˚C. The magnetic properties are correlated with the electrochemical performance of the positive electrode materials. Magnetization and susceptibility measurements appear to be a powerful probes for impurity detection at very low concentration of trivalent iron (<1 ppm). Electrochemical performances of Liion cells with Li4Ti5O12 (LTO) negative electrode are reported with a strict control of iron dissolution by postmortem analysis.
2. Experimental
The optimized LiFePO4 material was synthesized by solidstate reaction. Samples were prepared from FePO4(H2O)2 and Li2CO3. A stoichiometric amount of precursors was thoroughly mixed together in isopanol. After drying, the blend was heated at 500˚C - 800˚C for 8 h under reducing atmosphere. Four samples have been considered heated at carbon-coated LiFePO4 (C-LFP) was prepared with sucrose and cellulose acetate as the carbon precursors in acetone solution according to the following procedure. The carbon-free powder was mixed with the carbon precursors. The dry additive corresponded to 5 wt% carbon in LiFePO4. After drying, the blend was heated at 700˚C for 4 h under argon atmosphere. The quantity of carbon coat represents about 1 wt% of the material (C-detector, LECO Co., CS 444). It should be noted that the choice of this moderate sintering temperature minimizes the amount of Fe3+ ions present in the powder since the presence of Fe3+ has been detected by Mössbauer experiments at sintering temperatures below 500˚C, and both trivalent Fe2O3 and Li3Fe2(PO4)3 are formed in such large quantities that they are detected by X-rays by sintering above 800˚C [13]. Nevertheless, we know from our prior work [10,11] that LiFePO4, even with an intermediate sintering temperature in the range 500˚C - 800˚C, does contain Fe2O3 nanoparticles, although in such small quantities that they can be detected only by investigation of magnetic properties.
X-ray diffractometry (XRD) was carried out with a Philips X’Pert apparatus equipped with a CuKα X-ray source (λ = 1.5406 Å). Slice views were examined with a scanning electron microscope (SEM, Philips XL30). Fourier transform infrared (FTIR) absorption spectra were recorded with a Fourier transform interferometer (model Bruker IFS113v) in the wavenumber range 150 - 1400 cm–1 at a spectral resolution of 2 cm–1. Magnetic measurements (susceptibility and magnetization) were carried out with a fully automated magnetometer (MPMS-5S from Quantum Design) using an ultra-sensitive Superconducting Quantum Interference Device (SQUID) in the temperature range 4 - 300 K. The experimental details are given elsewhere [11]. The electrochemical properties of LiFePO4 were measured at 60˚C in cells with metallic lithium as the negative electrode. The electrolyte was 1 M LiPF6 in EC/DEC (1/1) solvent. The measurements were carried out following the experimental procedure previously described [14] using the coffee-bag technology developed at Hydro-Québec. Coffee-bag or laminated battery technology was described by Zaghib and Armand [15].
3. Results and Discussion
3.1. Structure and Morphology of LiFePO4
Figure 1 shows the typical XRD patterns of the LiFePO4 electrode material. The XRD pattern of sample synthesized from the mixture FePO4(H2O)2 + Li2CO3 agrees very well with that of phospho-olivine LiFePO4 [16] and no impurity was detected. The XRD diagram of the new
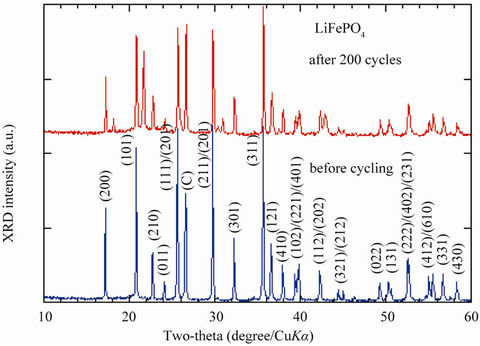
Figure 1. XRD pattern of as-prepared LiFePO4 material (lower curve) and positive electrode after 200 cycles (lower curves). Bragg lines are indexed in the Pmna space group. Notice that the olivine framework remains intact after cycling at 60˚C.
generation of LFP after 200 cycles (47 days) at 60˚C are also shown in Figure 1. There is no change in the olivine structure after cycling at 60˚C. We observed Bragg lines with the same intensity as that for the pristine material. The capacity loss was below 3% in 100 cycles for this optimised electrode material, which also displays excellent capacity retention. First, we present an overview of the high-temperature performance for an optimised LFP sample. The coffee-bag cell was charged and discharged at C/8 for the first cycle followed by 12 cycles at C/4 with 1 h rest before each charge and discharge. This high temperature test was made at 60˚C, which is the appropriate condition to investigate possible iron dissolution in non-aqueous electrolytes.
The local structure of LFP materials was studied by FTIR in the spectral range 150 - 1400 cm–1). Figure 2 presents the FTIR spectra of several LiFePO4 samples in the low-wavenumber region (300 - 600 cm–1) involving bending modes and in the high-wavenumber domain (600 - 1300 cm–1) involving stretching vibrations. A comparison can be established between impurity-containing samples (A-type) and the optimised phospho-olivine material (B-type). In the low-wavenumber region of the active symmetric and asymmetric (ν2 - ν4) bending modes of P-O bonds, we observe two well-resolved doublets at 349 - 377 and 468 - 500 cm–1 for the optimised C-LiFePO4. When an impurity such as Li3PO4 (sample A) is present, the FTIR spectrum displays some modifications in the shape of the doublets and an additional IR band grows at 424 cm–1. The presence of an impurity was also observed in the high-wave number region of the symmetric and asymmetric (ν1 - ν3) modes of PO4 groups. Introduction of LiFeP2O7 is detected by the appearance of two sets of IR bands at 762 and 1180 cm–1. The band at 762 cm–1 is
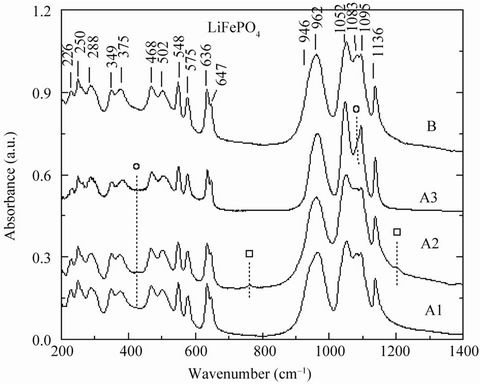
Figure 2. FTIR spectra of LiFePO4 samples. Patterns show bands due to Li3PO4 (o) and LiFeP2O7 (□) impurity. The best LiFePO4 material is the B-10 sample (upper curve).
due to the symmetric stretching mode of P2O7 pyrophosphate groups, while the high-frequency band at 1180 cm–1 is assigned to the vibration of the PO3 terminals [17-19]. These two spectral features are fingerprints of the diphosphate impurity. The inclusion of Li3PO4 is observed by additional spectral features such as enhancement of the asymmetric stretching (ν3) vibration of (PO4)3– oxo-anions. It is worth noting that FTIR measurements (like X-ray diffractometry) are not sensitive to detect of low concentrations of impurities (at the ppm level). The FTIR features show that the carbon does not penetrate significantly inside the LiFePO4 particles, although it is very efficient in reducing Fe3+, which prevents γ-Fe2O3 clustering, thus pointing to a gas-phase reduction process.
Figure 3(a) presents the typical SEM image of the prepared LiFePO4 material. The powders are composed of agglomerated crystallites of size 150 nm in average. The existence of the carbonaceous film is also indicated by the bright-field TEM image presented in Figure 3(b). This image displays representative primary particles with a network of carbon in the interstitial grain-boundary region. In the micrograph, the LFP crystallites appear as the darker regions while the carbon coating is surrounding the primary particle as the grayish region.
3.2. Iron(III) Nanoclusters in LiFePO4
LiFePO4 undergoes a transition to antiferromagnetic (AFM) order at a Néel temperature TN = 52 K [20]. Figure 4 shows the temperature dependence of the reciprocal magnetic susceptibility for three samples. While nano-sized ferromagnetic particles were evidenced in previously prepared LiFePO4, such clusters do not exist in the optimized LiFePO4 [10,11]. Magnetic measurements illus-

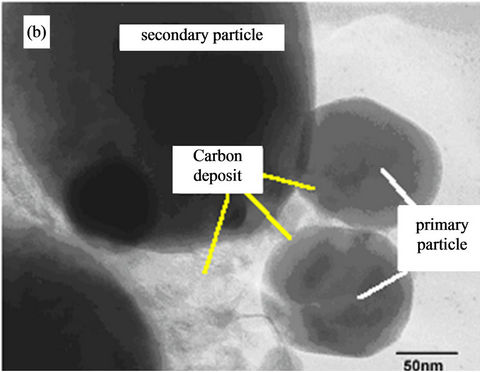
Figure 3. SEM image (a) of the carbon-coated sample showing the shape of the secondary particles. HRTEM image (b) showing the amorphous carbon layer deposited onto the LiFePO4 crystallite. Particle size is expressed in nm.
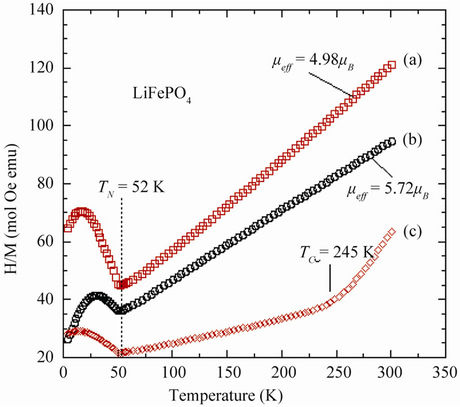
Figure 4. Temperature dependence of the reciprocal magnetic susceptibility of different LiFePO4 samples. (a) Optimized pure LiFePO4; (b) Fe2O3-containing sample; and (c) x-1(T) Fe2P-containing sample.
trate that the magnetization M(H) is the superposition of two contributions M(H) = xmH + Mextrin. The intrinsic part, xmH, is linear in the applied magnetic H and an extrinsic component, Mextrin = Nnµ£(ξ), is easily saturated by the application of H due to ferromagnetic impurities. Here, £(ξ) is the Langevin function, N is the number of magnetic clusters made of n magnetic moments µ.
Analysis of the magnetic properties gives an average separation of the magnetic clusters that is too large for interaction between particles (superparamagnetic model). This hypothesis must be released where the number n of magnetic clusters is so large that magnetic interactions between the ferrimagnetic particles become important [9]. At high fields, Mextrin saturates to Nnµ so that this quantity is readily determined as the ordinate at H = 0 of the intersection of the tangent to the magnetization curves at large fields. As a result, we find that Nnµ does not depend significantly on temperature below 300 K. We are in the situation where the cluster magnetization is temperature independent, which amounts to say that the Curie temperature TC inside the clusters is much larger than 300 K. This is important information on the nature of the ferromagnetic clusters. In particular, this feature precludes the existence of Fe2P clusters in some LFP samples prepared according to a different procedure [10], since the Curie temperature of these clusters is only 220 K. The nature of the strongly ferromagnetic clusters in the present case is most likely maghemite (γ-Fe2O3).
It is remarkable from Figure 4 that the A-type sample displays different magnetic features i.e. with a magnetic moment µeff = 5.72µB, due to the existence of Fe(III) containing impurities. The first consequence is an ambiguity in what is called the magnetic susceptibility xm since M/H is distinct from dM/dH. The magnetic susceptibility measured with a SQUID at H = 10 kOe shows the non-linearity of the magnetic moments attributed to the presence of γ-SFe2O3. The best material shows the lowest Curie constant 3.09 emu·K/mol. The effective magnetic moment µeff = 4.98µB is close to theoretical value 4.90µB calculated from the spin-only value of Fe2+ in its highspin configuration. Departure from the spin-only value may reflect the presence of Fe3+ ions and/or an orbitalmomentum contribution from the Fe2+ ions [12].
3.3. Influence of the Fe2P Nanoclusters
The electrochemical properties of LFP are known to be sensitive to the mode of preparation and the structural properties [21]. This can be an advantage for potential applications since it allows for an optimization of the material if we can correlate the mode of preparation with the structural and the physical properties. To address this issue, we investigated this relationship in LiFePO4 sample that were grown at different conditions. Undesirable impurities in the lattice can be introduced during the growth process. For instance, the presence of Fe2P can increase the electronic conductivity, but on the other hand it also decreases the ionic conductivity so that both the capacity and cycling rates are degraded with respect to C-LFP. The presence of a small concentration (>0.5%) of Fe2P is evidenced in Figure 4 by the appearance of an abnormal x(T) behaviour with the occurrence of a shoulder near TC = 265 K, the Curie temperature of the ferromagnet Fe2P. Figure 5 shows the Arrhenius plot of the electronic conductivity, σelec, of three LiFePO4 samples: a pure material, a Fe2P-containing sample, and a C-LFP. It is obvious that addition of either iron phosphide or carbon enhances greatly σelec but to the detriment of the capacity for the former compound, as it will be discussed next.
Furthermore, we know that hydrogen, carbon monoxide, or carbon can reduce Fe2O3 through different reduction steps that depend on temperature and other physical parameter such as particle size. Although we anticipate that over 1000˚C carbon might reduce Fe3+ ions or through the formation of CO gas to prevent the formation of γ-Fe2O3, other factors may be involved. We believe that the carbon deposition process, which was organic precursors to make C-coated samples, generates a reductive gas such as hydrogen that is more kinetically active and reduces Fe3+ impurities in the 400˚C - 700˚C temperature range used in our studies. This model is also favoured by the fact that the organic precursor is usually mixed with the LFP material or with the LiFePO4 chemical precursors by solution processes at a molecular-size level. However, as presented in Figure 5, the carbon coating greatly enhances the electronic conductivity of the LFP
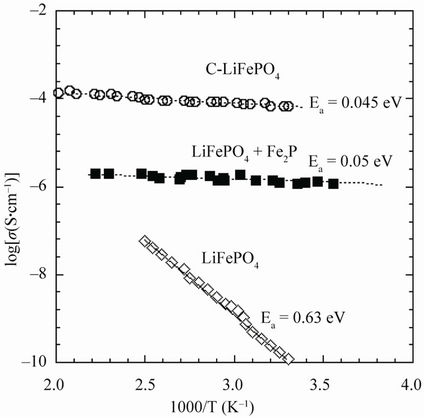
Figure 5. Electronic conductivity of LiFePO4 samples. (a) Pure material; (b) Fe2P-containing sample; and (c) Carboncoated LiFePO4.
particles that allows high rate for the charge discharge process.
Figure 6 displays the electrochemical charge-discharge profiles of Li//LFP cells cycled at room temperature with pure LiFePO4 and with Fe2P-containing electrode material. It is obvious that at the rate 2C, the capacity retention decreases significantly for the material containing few% of Fe2P. A close examination was made for the detection of any iron dissolution that could occur after long-term cycling. The analysis of iron species was investigated at the separator/lithium (SL) interface by SEM cross-section (slice view) as shown in Figures 7(a) and (b). The micrograph (Figure 7(a)) obtained from evaluation of the earlier generation material shows the presence of iron islands at the SL interface. Obviously, some iron particles (or ions) migrate through the electrolyte from the LiFePO4 positive electrode to the lithium negative. The net effect of this migration is a large decrease in capacity retention of the Li//LFP cell. Figure 7(b) shows the post-mortem micrograph obtained from tests with an optimised electrode in a Li cell with a lithium foil negative. In this case, there is no iron detected at the SL interface, which remained intact after 100 cycles. In fact, this high performance was possible not only because the optimised synthesis of the LFP powders, but also because of strict control of the structural quality of the materials. Several physical methods were utilized to analyse the local structure and the electronic properties of the phospho-olivine framework.
3.4. Electrochemical Properties of Optimized LiFePO4
To study the capacity fade of LiFePO4 at 60˚C, we used three different negative electrodes, namely lithium metalgraphite, and Li4Ti5O12. A lithium metal anode gave exact capacity during charge-discharge process with Li metal excess 2.5 times to LiFePO4 cathode material. Due to the large excess of lithium metal, it was difficult to observe the capacity fade with this anode at 60˚C. Graphite anode was 5% of excess to LiFePO4 cathode. This type of anode can detect easily the dissolution of iron because the passivation layer of graphite anode is ionic conductor and electronically insulator; so the dissolution of iron from the cathode side to the anode increases the electronic conductivity of passivation layer of graphite that results on the capacity fade of the cell. LTO has been used because is passivation layer free and zero strain material. The anode has 0% excess to the cathode that gives the high stability of the cycling and also prevents side reactions or reduction of electrolyte to the potential of LTO (1.5 V vs Li+/Li0).
Figures 8(a)-(c) present the electrochemical performance of cell in several configurations using LFP as positive electrode. The typical electrochemical profile of the C-LFP/1 mol·L–1 LiPF6-EC-DEC/Li cell at 60˚C is shown in Figure 8(a). The charge-discharge curves were obtained by cycling at C/4 rate (about 35 mA/g) in the voltage range 2.2 - 4.0 V vs Li0/Li+. The optimized LFP exhibits a reversible capacity that is maintained over many charge-discharge cycles. The 10th and 120th cycle shows a similar specific capacity of 160 mAh·g–1. These results illustrate the excellent electrochemical performance of the carbon-coated olivine material. The electrode can be fully charged up to 4 V, which is its most reactive state. This remarkable performance is attributed to the optimized carbon-coated particles and their structural integrity under a large current in the electrode. Even at such a high cycling rate, C-LiFePO4 exhibits rapid kinet-
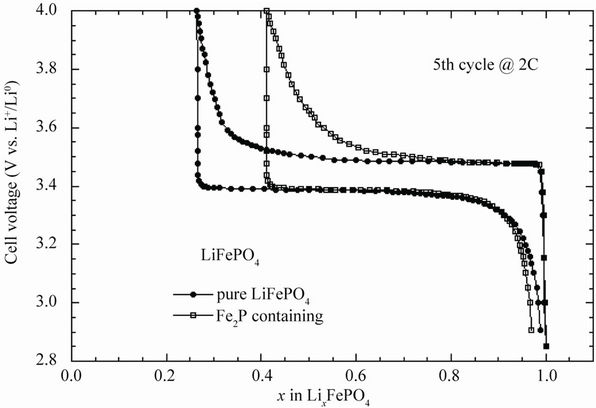
Figure 6. Electrochemical charge-discharge profiles of Li//LiFePO4 cells cycled at room temperature. (a) With pure LiFePO4; and (b) With Fe2P-containing electrode material.


Figure 7. Post-mortem SEM images of the detection of iron species at the separator/lithium interface. (a) Formation of iron islands at the interface with an earlier generation of LiFePO4; (b) No iron was detected at the surface of Li foil with the optimized LiFePO4.
ics of lithium extraction, and realizes most of its theoretical capacity (170 mAh·g–1). The discharge profile appears with the typical voltage plateau (at ca. 3.45 V vs Li0/Li+) attributed to the two-phase reaction of the (1 – x) FePO4 + xLiFePO4 system.
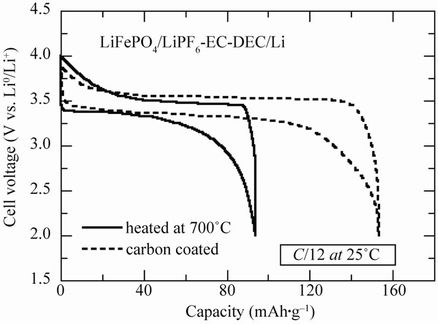
To investigate the impact of the crystallization of the surface layer of LiFePO4 particles on the electrochemical properties, we have reported the first charge/discharge of a coin cell in Figure 8(b), in which the active cathode element was non-coated LFP heated at 700˚C during 4 h and C-LFP for comparison. The same powder (particles of average thickness 40 nm) was used to avoid any size effect of the particles on the electrochemical particles. The experiments were performed at rate C/12 at room temperature. The results show that the capacity of the cell prepared with LFP heat treated at 700˚C is very small despite the crystallization of the surface layer of the LiFePO4 particles. However, the capacity only reaches ca. 55% of its theoretical value, while, after carbon coating, the capacity of these particles is close to the theoretical value. This is indeed the evidence that, even in the case of nano-scaled particles, the carbon coating alone is far from sufficient to recover the full capacity of C-LFP, for the reasons we have recalled in the introduction. In our previous exploration of the surface properties of the LFP particle, we have shown by Raman spectroscopy that the deposit is a disordered graphite-type carbon [22]. The small amount of carbon (<2 wt%) can be viewed as a film of irregular thickness, 3 nm thick on average, with gaps. The above experimental condition (ca. 60˚C) has a severe impact on the kinetics of the Fe2+/Fe3+ redox reaction, but the recent report from Hydro-Québec Research Labs showed that this type of C-LFP electrode can be cycled at 60˚C without significant capacity loss for over 200 cycles [7]. Optimized particle size in the range 200 - 300 nm agrees well with the average diameter of grains L that validates the characteristic diffusion time τ = L2/4π2D* [23], where D* is the chemical diffusion coefficient of Li+ ions in the LiFePO4 matrix (typically 10–14 cm2·s–1) when compared with the experimental discharge rate up to 5C.
The electrochemical performance of optimized LFP and LTO electrode materials has been tested separately in half cell with respect to Li metal anode, using the same electrolyte mentioned above. The voltage vs capacity curves recorded under such conditions at 25˚C are reported in Figure 8(c) at low C-rate C/24 to approach thermodynamic equilibrium together with the potentialcapacity curve of the LTO//LFP lithium-ion battery. The voltage window is 2 - 4 V for LiFePO4, 1.2 - 2.5 V for Li4Ti5O12. Note in this figure (and the following ones), we have kept the conventional rule, i.e. the capacity is in mAh per gram of the active element of the cathode. That is the reason why the maximum capacity for the LFP//Li and LFP//LTO cells are the same. For LFP//Li, the first coulombic efficiency is 100 % and the reversible capacity is 148 mAh·g–1. For LTO, the first coulombic efficiency is 98% and the reversible capacity is 157 mAh·g–1. The well-known plateaus at 3.4 and 1.55 V are characteristics of the topotactic insertion/deinsertion of lithium in the two-phase systems LiFePO4-FePO4 and Li4Ti5O12- Li7Ti5O12, respectively.
The electrochemical performance of C-LiFePO4 was tested in various conditions of temperature. At 2C rate, the capacity retention was 153, 136 and 93 mAh·g–1 for cells discharged at 60˚C, 25˚C and –10˚C, respectively [24]. The Ragone plots of cell cycled at 25˚C and 60˚C are shown in Figure 9. The cells were cycled in the potential range 2.5 - 4.0 V. The discharge capacity and electrochemical utilisation, i.e. the ratio discharge/charge, vs cycle number are excellent for the C-LiFePO4/LiPF6- EC-DEC/Li cells. At 10C rate, these Li-ion cells provide coulombic efficiencies 85% at 60˚C.
4. Conclusion
In this work, we compared the physico-chemical and electrochemical properties of a series of carbon-coated LiFePO4 samples. A major effect of the carbon deposition process has been to reduce Fe3+, most probably through a gas-phase reduction process involving hydro-
 (a)
(a)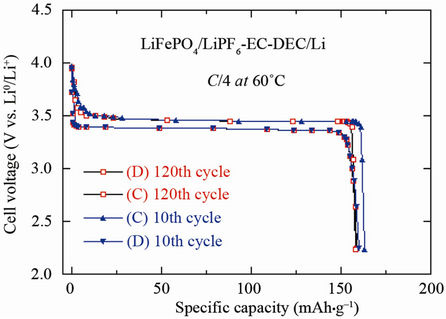 (b)
(b)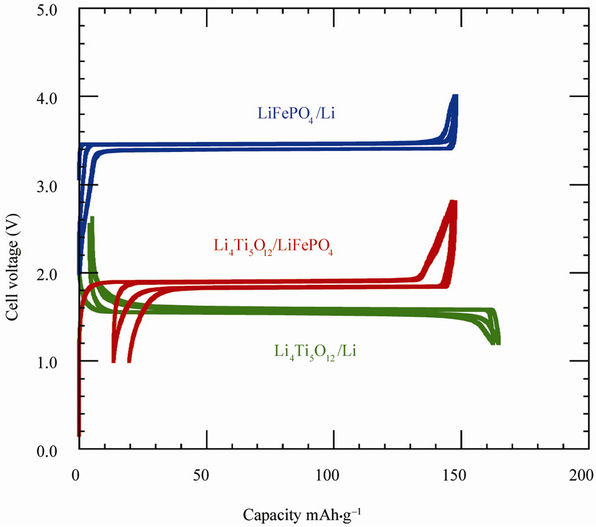 (c)
(c)
Figure 8. (a) Electrochemical performance of the C-LiFePO4//Li cell operating at 60˚C. Charge-discharge cycling was conducted at the C/4 rate; (b) Electrochemical profiles at C/12 of LiFePO4//Li cells. Positive electrodes were (i) non-coated and heated at 700˚C (c) carbon-coated; (c) Voltage-capacity cycle for LiFePO4//Li, Li4Ti5O12//Li and Li-ion cell LiFePO4//Li4Ti5O12 at C/24 rate. The capacity is in mAh per gram of the positive electrode element (LiFePO4, Li4Ti5O12 and LiFePO4, respectively). The larger hysteresis in the LiFePO4//Li4Ti5O12 cell comes from the fact that the cell in that case was a button cell instead of the more elaborate 18650-cell, but the plateau at 1.9 V is well observed. All the cells used 1 mol·L–1 LiPF6 in EC:DEC (1:1) as electrolyte.
gen from the organic carbon precursor. The hydrogen prevents formation of γ-Fe2O3 nanoparticles in which iron is in the trivalent state. The magnetic measurements indicated the presence of nanoclusters, i.e. Fe2P or/and γ-Fe2O3 in non optimized samples. Thus, this study demonstrates that magnetic measurements (combination of M(H) and x(T) data) are beneficial for detecting ferric and/or ferrous impurities, as well as for the quality control of LiFePO4. Electrochemical tests have been conducted under various conditions to assess the influence of the electrolyte on stability and the influence of electrode processing. Post-mortem analysis showed that no iron species were detected at the separator-negative electrode interface in cells with lithium metal, graphite and CLi4Ti5O12. This result is attributed to the high quality of the “optimised” LiFePO4, impurity-free materials used as positive electrodes. The discharge capacity and electrochemical utilisation, i.e. the ratio discharge/charge, vs cycle number are excellent for the C-LiFePO4/LiPF6-ECDEC/Li cells. At 10C rate, these Li-ion cells provide
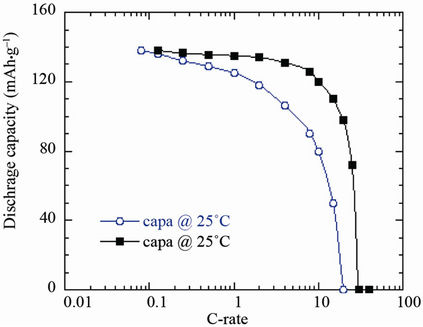
Figure 9. Ragone plots of the C-LiFePO4/LiPF6-EC-DEC/Li cells as a function of the working temperature 25˚C and 60˚C.
coulombic efficiencies 85% at 60˚C.
REFERENCES
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, “Phospho-Olivines as Positive-Electrode Materials for Rechargeable Lithium Batteries,” Journal of the Electrochemical Society, Vol. 144, No. 4, 1997, pp. 1188- 1194. doi:10.1149/1.1837571
- N. Ravet, Y. Chouinard, J. F. Magnan, S. Besner, M. Gauthier and M. Armand, “Electroactivity of Natural and Synthetic Triphylite,” Journal of Power Sources, Vol. 97- 98, 2001, pp. 503-507. doi:10.1016/S0378-7753(01)00727-3
- N. Ravet, A. Abouimrane and M. Armand, “From Our Readers: On the Electronic Conductivity of PhosphoOlivines as Lithium Storage Electrodes,” Nature Materials, Vol. 2, 2003, pp. 702-703. doi:10.1038/nmat1009b
- N. Ravet, S. Besner, M. Simoneau, A. Vallée, M. Armand and J. F. Magnan, “Electrode Materials with High Surface Conductivity,” US Patent No. 6962666, 2005.
- Y. Hu, M. M. Doeff, R. Kostecki and R. Finones, “Electrochemical Performance of Sol-Gel Synthesized LiFePO4 in Lithium Batteries,” Journal of the Electrochemical Society, Vol. 151, No. 8, 2004, pp. A1279-A1285. doi:10.1149/1.1768546
- S. L. Bewlay, K. Konstantinov, G. X. Wang, S. X. Dou and H. K. Liu, “Conductivity Improvements to SprayProduced LiFePO4 by Addition of a Carbon Source,” Materials Letters, Vol. 58, No. 11, 2004, pp. 1788-1791. doi:10.1016/j.matlet.2003.11.008
- K. Zaghib, V. Battaglia, P. Charest, V. Srinivasan, A. Guerfi and R. Kostecki, Extended Abstract of the International Battery Association & Hawaii Battery Conference, Wailoloa, 9-12 January 2006.
- A. A. Salah, P. Jozwiak, J. Garbarczyk, F. Gendron, A. Mauger and C. M. Julien, “FTIR Features of Lithium Iron Phosphates Used as Positive Electrodes in Rechargeable Lithium Batteries,” 207th ESC Meeting, Québec City, 15- 20 May 2005.
- M. M. Doeff, Y. Hu, F. McLarnon and R. Kostecki, “Effect of Surface Carbon Structure on the Electrochemical Performance of LiFePO4,” Electrochemical and SolidState Letters, Vol. 6, No. 10, 2003, pp. A207-A209. doi:10.1149/1.1601372
- A. Ait-Salah, A. Mauger, F. Gendron and C. M. Julien, “Magnetic Studies of the Carbothermal Effect on LiFePO4,” Physica Status Solidi (a), Vol. 203, No. 1, 2006, pp. R1- R3.
- A. Ait-Salah, A. Mauger, C. M. Julien and F. Gendron, “Nano-Sized Impurity Phases in Relation to the Mode of Preparation of LiFePO4,” Materials Science and Engineering: B, Vol. 129, No. 1-3, 2006, pp. 232-244. doi:10.1016/j.mseb.2006.01.022
- A. Ait-Salah, A. Mauger, K. Zaghib, J. B. Goodenough, N. Ravet, M. Gauthier, F. Gendron and C. M. Julien, “Reduction Fe3+ of Impurities in LiFePO4 from Pyrolysis of Organic Precursor Used for Carbon Deposition,” Journal of the Electrochemical Society, Vol. 153, No. 9, 2006, pp. A1692-A1701. doi:10.1149/1.2213527
- A. Yamada, S. C. Chung and K. Hinokuma, “Optimized LiFePO4 for Lithium Battery Cathodes,” Journal of the Electrochemical Society, Vol. 148, No. 3, 2001, pp. A224-A229. doi:10.1149/1.1348257
- K. Zaghib, J. Shim, A. Guerfi, P. Charest and K. A. Striebel, “Effect of Carbon Source as Additives in LiFePO4 as Positive Electrode for Lithium-Ion Batteries,” Electrochemical and Solid-State Letters, Vol. 8, No. 4, 2005, pp. A207-A210. doi:10.1149/1.1865652
- K. Zaghib and M. Armand, “Electrode Covered with a Film Obtained from an Aqueous Solution Containing a Water Soluble Inder, Manufacturing Process and Usesthereof,” Canadian Patent No. CA 2411695, 2002.
- K. Striebel, J. Shim, V. Srinivasan and J. Newman, “Comparison of LiFePO4 from Different Sources,” Journal of the Electrochemical Society, Vol. 152, No. 4, 2005, pp. A664-A670. doi:10.1149/1.1862477
- A. Ait-Salah, P. Jozwiak, J. Garbarczyk, K. Benkhouja, K. Zaghib, F. Gendron and C. M. Julien, “Local Structure and Redox Energies of Lithium Phosphates with Olivineand Nasicon-Like Structures,” Journal of Power Sources, Vol. 140, No. 2, 2005, pp. 370-375. doi:10.1016/j.jpowsour.2004.08.029
- R. Bacewicz, P. Woroniecki and J. Garbarczyk, “Raman Scattering in AgI-Ag2O-P2O5 Glasses,” Physics and Chemistry of Glasses, Vol. 40, No. 3, 1999, pp. 175-176.
- A. Adamczyk, M. Handke and W. Mozgawa, “FTIR Studies of BPO4·2SiO2, BPO4·SiO2 and 2BPO4·SiO2 Joints in Amorphous and Crystalline Forms,” Journal of Molecular Structure, Vol. 511-512, 1999, pp. 141-144. doi:10.1016/S0022-2860(99)00152-0
- R. P. Santoro and R. E. Newnham, “Antiferromagnetism in LiFePO4,” Acta Crystallographica, Vol. 22, 1967, pp. 344-347. doi:10.1107/S0365110X67000672
- G. Arnold, J. Garche, R. Hemmer, S. Ströbele, C. Vogler and M. Wohlfahrt-Mehrens, “Fine-Particle Lithium Iron Phosphate LiFePO4 Synthesized by a New Low-Cost Aqueous Precipitation Technique,” Journal of Power Sources, Vol. 119-121, 2003, pp. 247-251. doi:10.1016/S0378-7753(03)00241-6
- C. M. Julien, K. Zaghib, A. Mauger, M. Massot, A. AitSalah, M. Selmane and F. Gendron, “Characterization of the Carbon Coating onto LiFePO4 Particles Used in Lithium Batteries,” Journal of Applied Physics, Vol. 100, No. 6, 2006, Article ID: 063511. doi:10.1063/1.2337556
- S. H. Yu, C. K. Park, H. Jang, C. B. Shin and W. II Cho, “Prediction of Lithium Diffusion Coefficient and Rate Performance by Using the Discharge Curves of LiFePO4 Materials,” Bulletin of the Korean Chemical Society, Vol. 32, No. 3, 2011, pp. 852-856.
- K. Zaghib, P. Charest, M. Dontigny, A. Guerfi, M. Petitclerc and M. Duchesne, “Olivines: 10 Years R & D at Hydro-Québec in Li-Ion Batteries,” Rechargeable Lithium and Lithium Ion Batteries Battery/Energy Technology, Washington DC, 7-12 October 2007, Abstract No. 637.
NOTES
*Corresponding author.