Advances in Enzyme Research
Vol.2 No.1(2014), Article ID:43901,13 pages DOI:10.4236/aer.2014.21001
Characterization of a Thermostable, Recombinant Carboxylesterase from the Hyperthermophilic Archaeon Metallosphaera sedula DSM5348
Rushyannah Killens-Cade1, Rachel Turner2, Christine MacInnes2, Amy Grunden1*
1Department of Plant and Microbial Biology, North Carolina State University, Raleigh, USA
2Department of Biology, North Carolina State University, Raleigh, USA
Email: *amgrunde@ncsu.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 24 October 2013; revised 5 December 2013; accepted 23 December 2013
ABSTRACT
Lipid-producing microalgae are emerging as the leading platform for producing alternative biofuels in response to diminishing petroleum reserves. Optimization of fatty acid production is required for efficient conversion of microalgal fatty acids into usable transportation fuels. Microbial lipases/esterases can be used to enhance fatty acid production because of their efficacy in catalyzing hydrolysis of esters into alcohols and fatty acids while minimizing the potential poisoning of catalysts needed in the biofuel production process. Although studies have extensively focused on lipases/esterases produced by mesophilic organisms, an understanding of lipases/esterases produced by thermophilic, acidic tolerant microbes, such as Metallosphaera sedula, is limited. In this work, the carboxylesterase from Metallosphaera sedula DSM5348 encoded by Msed_1072 was recombinantly expressed in Escherichia coli strain BL21 (λDE3). The purified enzyme either with a hexahistidine (His6)-tag (Msed_1072Nt and Msed_1072Ct) or without the hexahistidine (His6)-tag (Msed_1072) was biochemically characterized using a variety of substrates over a range of temperatures and pH and in the presence of metal ions, organic solvents, and detergents. In this study, the fusion of the protein with a hexahistidine (His6)-tag did not result in a change in substrate specificity, but the findings provide information on which enzyme variant can hydrolyze fatty acid esters in the presence of various chemicals, and this has important implication for their use in industrial processes. It also demonstrates that Metallosphaera sedula Msed_1072 can have application in microalgae-based biofuel production systems.
Keywords:Carboxylesterase; Metallosphaera sedula; Hyperthermoacidophile; Biofuels; Algae; Lipids

1. Introduction
Lipases (EC 3.1.1.3) and esterases (EC 3.1.1.1) are enzymes that catalyze the synthesis of ester compounds and hydrolyze triglycerides [1] . As hydrolases, these lipolytic enzymes act on the ester bonds of the triglycerides to liberate fatty acids and glycerols. Although both enzyme types share structural characteristics, the fundamental difference between esterases and (true) lipases is that the former acts on shorter chain triglycerides (shorter than 10 carbon atoms) which are water-soluble, whereas the latter hydrolyzes longer chain triglycerides [2] [3] . Based on their regioand stereo-specific properties, cofactor-independent activity, broad substrate specificity, and stability in the presence of organic solvents, ester-hydrolyzing enzymes are attractive biocatalysts [3] . As such, they are used widely in a number of industrial applications, including medical biotechnology, resin removal from pulp, detergent production, organic synthesis, flavor and aroma synthesis, food related processes, and biofuel production [4] [5] .
The increasing consumption of energy, population growth, and environmental concerns have resulted in an urgent need to explore more sustainable energy sources. Lipid-producing microalgae are emerging as a leading platform for producing alternative sustainable energy, mainly because of desirable characteristics such as rapid growth rate, production of valuable co-products, and cultivation in various marine and freshwater environments that do not compete with arable crop lands [6] -[8] . Their adaptability to a range of growth environments may result in the ability to produce lipids of diverse compositions [8] . These include membrane lipids as well as lipids involved in carbon and energy storage such as triacylglycerides (TAG), which are neutral lipids that consist of three fatty acids esterified to glycerol. In addition to having a diversified lipid composition that may aid survival in harsh environments some algae may also alter lipid biosynthetic pathways leading to the accumulation of TAG during unfavorable environmental or stress conditions [9] .
Recent genetic and metabolic engineering research has focused on increasing lipid yields in microalgae by targeting the carbon flux through the fatty acid synthesis pathway. However, for microalgae to be a cost effective and reliable alternative energy source, the process for the direct conversion of lipidic biomass feedstock to combustible fuel must become more efficient. Specifically, there is a need to save energy during the conversion process and avoid extensive degumming. A synthetic biology approach which targets increasing the yield and quality of algal feedstock is one way of overcoming these difficulties. By transforming algae to express thermostable lipases/esterases, fatty acid release from TAGs that are stored in lipid bodies can be achieved during the high temperature conversion of lipids to fuel. Having enzymes that are active during this process can reduce the time required for lipid to fuel conversion, thereby saving energy and improving cost effectiveness.
For our microalgae based biofuel system, we selected a homolog of an Escherichia coli acyl-ACP thioesterase, TesA. TesA, expressed in an E. coli expression system, has been shown to hydrolyze long chain fatty acyl-ACPs (products of the fatty acid synthesis pathway), resulting in relief of feedback inhibition and increased production of total free fatty acids (FFAs) [10] [11] . It also has been shown that TesA has a wide substrate specificity and is able to hydrolyze long-chain fatty acyl-ACPs to produce FFAs containing 12 to 18 carbons [12] . This hydrolysis profile works to the advantage of biofuel systems because aromatic hydrocarbons with 12 to 14 carbon atoms are ideal for production of petroleum-derived jet fuels.
To evaluate its application for algae transformation and subsequent biofuel production, we have recombinantly expressed and characterized variants of a putative alpha/beta hydrolase (Msed_1072) from the hyperthermoacidophilic archaeon Metallosphaera sedula DSM5348 (optimal growth at 75˚C and a pH range of 1.0 and 4.5) which was isolated from an acidic drain of a hot water pond at Pisciarelli Solfatara, near Naples, Italy [13] . M. sedula Msed_1072, originally targeted for analysis based on its similarity to E. coli TesA (19% similarity; conserved catalytic triad amino acids Ser-Asp-His) was fused with a hexahistidine (His6)-tag and produced recombinantly in E. coli. It has been shown that fusing a thioesterase with a C-terminal hexahistidine (His6)-tag can result in a shift in substrate specificity [14] ; therefore, N-terminal histidine-tagged, C-terminal histidinetagged, and untagged variants of Msed_1072 were evaluated in order to satisfy the overall goal of this study which was to identify a thermophilic carboxylesterase with an affinity towards long carbon chain substrates.
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, Enzymes, and Reagents
The genomic DNA of Metallosphaera sedula DSM5348 was kindly provided by Dr. Robert Kelly of North Carolina State University. The Escherichia coli XL1-Blue strain (Novagen, EMD Biosciences) was used for cloning and maintaining the recombinant plasmid. BL21(DE3)LysS (Novagen, EMD Biosciences) cells were used for over-expression of the proteins. The expression plasmids pET21b and pET28a were obtained from Novagen, EMD Biosciences. Restriction endonucleases, T4 DNA ligase, and DNA polymerase were purchased from New England Biolabs. Synthetic oligonucleotides were synthesized by Eurofins MWG Operon (Huntsville, AL). The PCR products were purified using a QIAquick PCR purification kit (Qiagen). The plasmids were purified using a QIAprep spin miniprep kit (Qiagen). All assay substrates were purchased from Sigma-Aldrich. The following chromatography columns were utilized for the enzyme purification step: 5 ml HisTrap Nickel Sepharose™, HiTrap Q Sepharose™, and HiTrap SP Sepharose™ from GE Healthcare Life Sciences.
2.2. Cloning of a Thermoactive Carboxylesterase from Metallosphaera sedula
The Msed_1072Nt and Msed_1072 genes were amplified using the forward primer: 5’-TTAATACGACCATAT GCCCCTACATCCAGAGGTAGAAATTAC-3’, with an NdeI site (bold), and the reverse primer: 5’-GATACA TTCGGATCCGTGGATAGGTTCATCTCGG-3’, with a BamHI site (bold). The Msed_1072Ct gene was amplified using the forward primer: 5’-ATACGACCATATGCCCCTACATCCAGAGGTAAAGAAATTACTT TCCCAGCAGAGGTAAGAAATTAC-3’, with an NdeI site (bold), and the reverse primer: 5’-TACGGATCC AGGACAGATCTCAGAACCCCA-GCAATGTG 3’, with a BamHI site (bold) and Phusion® high fidelity DNA polymerase in a 50 µl PCR reaction solution containing 10 µl 5× Phusion® HF buffer, 1 µl dNTPs (10 mM), 0.5 µl forward primer (40 uM), 0.63 µl reverse primer (40 uM), 0.5 µl Phusion® DNA polymerase, and 2 µl M. sedula genomic DNA (50 ng/µl). The PCR reactions were carried out using a thermal cycler (Bio-Rad) under the following conditions: an initial denaturing step at 98˚C for 30 sec; 30 cycles at 98˚C for 10 sec (denaturing), annealing at 64.4˚C [Msed_1072 and Msed_1072 Nt]/67.9˚C [Msed_1072Ct] for 30 sec, and extension at 72˚C for 30 sec; a final extension cycle of 72˚C for 7 min, and preservation at 4˚C. The amplicons were purified and visualized using a 1% (w/v) agarose gel. The purified DNA products were subjected to a double digestion, overnight, with NdeI and BamHI to render sticky ends needed for cloning. For construction of pET21b-Msed_1072, pET28a-Msed_1072Nt, and pET21b-Msed_1072Ct the plasmids were digested with NdeI and BamHI, gel purified, and ligated to the compatible sticky end DNA molecule of Msed_1072, Msed_1072Nt and Msed_1072Ct, respectively. CaCl2-competent cells of E. coli XL1-Blue were transformed with the ligated reaction and plated on solid LB agar media supplemented with ampicillin (100 µg/ml) or kanamycin (50 µg/ml) for selection of pET21b-Msed_1072, pET21b-Msed_1072Ct and pET28a-Msed_1072Nt, respectively. A positive clone of each construct was sequenced by Eurofins MWG Operon (Huntsville, AL) to ensure that no mutations were generated during the amplification process. The sequence information was analyzed by using MacVector (Accelrys) computer software.
2.3. Over-Expression of Recombinant Thermoactive Carboxylesterase Msed_1072
For the over-expression of M. sedula carboxylesterase, each construct and the rare arginine, leucine, and isoleucine tRNA encoding plasmid pRIL were used to transform E. coli BL21(DE3)pLysS cells. For the control expression samples, the plasmids without the Msed_1072 gene were also used to transform E. coli BL21(DE3) pLysS. The transformants were plated and selected on LB agar plates containing the appropriate antibiotics after incubation overnight at 37˚C. The E. coli BL21(DE3)pLysS cells harboring the recombinant plasmids were used to inoculate a 2 L culture of auto-induction media supplemented with the appropriate antibiotics for large scale expression of the protein [15] . The cells were incubated with shaking at 200 rpm for 16 h at 37˚C. Cells were harvested by centrifugation (9500 rpm, 50 min, 4˚C) and stored at −20˚C prior to preparing cell lysates. The expression of the recombinant protein was evaluated by SDS-PAGE using a 12.5% polyacrylamide gel with the Bio-Rad Mini-PROTEAN® apparatus and visualized using Coomassie brilliant blue R250. The molecular mass of the recombinant carboxylesterase was determined to be approximately 33.2 kDa by SDS-PAGE. The molecular weight of the poly-histidine tagged proteins was determined to be approximately 36.10 kDa by SDSPAGE. Protein concentrations were determined by the Bradford protein assay (Bio-Rad).
2.4. Recombinant Thermoactive Carboxylesterase Purification
The E. coli BL21(DE3)pLysS cell pellets containing the Msed_1072, Msed_1072Nt, and Msed_1072Ct, proteins were resuspended and lysed for 10 min with 10 mL B-PER® bacterial protein extraction reagent (Pierce, Rockford, IL) containing 1 mM benezamidine-HCl. The use of the nonionic detergent in the initial lysing step was critical for the solubility of the protein. The cells were further lysed by three passages of the lysate through a French pressure cell (20,000 lb/in2). Cellular debris was removed by centrifugation (15,000 rpm, 45 min, 4˚C). The supernatant was collected and heated at 70˚C for 15 min. The heat-treated supernatant was centrifuged (15,000 rpm, 15 min, 4˚C) to remove denatured proteins.
The supernatant containing the solubilized his-tagged recombinant proteins (Msed_1072Nt, Msed_1072Ct) was added to a 5 ml HisTrap Ni Sepharose™ column for purification. Prior to the addition of the protein, the Ni2+-charged column was equilibrated with 50 mM sodium phosphate buffer (pH 8.0), 300 mM NaCl, and 10 mM imidazole (buffer A). After binding the protein to the metal charged column, the column was washed with the equilibration buffer to remove all unbound protein. The thermostable esterase bound to the affinity column was then eluted with a linear 10 mM to 250 mM imidazole gradient. The peak fractions were visualized using 12.5% SDS-PAGE gels. To remove the imidazole, fractions containing the recombinant proteins were pooled and dialyzed against the storage buffer, 50 mM Tris-HCl buffer (pH 7.0), for 16 h at 4˚C.
The supernatant containing solubilized untagged Msed_1072 was loaded onto a 5 mL HiTrapTM Q anion exchange column equilibrated with 20 mM Tris-HCl buffer (pH 8.0), 0.025% Tween-20, and 0.1% CHAPS (buffer B) and eluted with a linear 0-to-1 M NaCl gradient. The fractions containing carboxylesterase activity were collected and dialyzed against 50 mM MES (pH 6.0), 0.025% Tween-20, and 0.1% CHAPS (buffer C) overnight for purification by a 1 mL HiTrapTM SP cation exchange column. The column was equilibrated with the same buffer (buffer C). Bound protein was eluted by linearly increasing NaCl concentration from 0 to 1.0 M. The fractions were collected and analyzed for carboxylesterase activity. Active fractions containing carboxylesterase activity were pooled and dialyzed against the storage buffer, 50 mM Tris-HCl buffer (pH 8.0) and 0.025% Tween-20, for 16 h at 4˚C.
2.5. Measurement of Carboxylesterase Activity
Enzyme activity was determined by a spectrophotometric assay using 4-nitrophenyl-octanoate as substrate, which was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 0.1 M. The reaction mixture (1 ml) consisted of 50 mM MOPS buffer, pH 7.0, 0.025% Tween-20, and 1 mM substrate. The reaction mixture was pre-heated with enzyme at 65˚C for 5 min to thermo-activate the enzyme. The reaction was initiated by the addition of substrate and allowed to proceed for 5 min at determined optimal temperatures. The enzyme reaction was stopped by adding 97.5 µl of chilled 0.25 M sodium carbonate. Controls were carried out as above, minus the addition of enzyme, to monitor the background hydrolysis of the substrate. The amount of liberated p-nitrophenol (pNP) was determined by reading absorbance of the sample at 410 nm. Esterase samples were assayed in 12 technical replicates. One unit (U) of enzyme activity was defined as the amount of enzyme required to hydrolyze 1 µmol of substrate per min per mg of protein; thus, activities were expressed in units per milligram of protein.
For assays conducted at different pH values, the following buffers were used at a final concentration of 50 mM: pH 4.0 and 5.0, sodium acetate; pH 6.0, MES; pH 7.0 MOPS; pH 8.0 - 10.0 CAPS. For assays that determined the optimal temperature, the following temperatures were used: 37˚C, 60˚C, 65˚C, 70˚C, 75˚C, 80˚C, 85˚C, 90˚C, and 95˚C. When evaluating substrate preference, the following pNP esters were used at a final concentration of 1 mM: pNP-butyrate (C4), pNP-octanoate (C8), pNP-decanoate (C10), pNP-dodecanoate (C12), pNP-myristate (C14), pNP-palmitate (C16), and pNP-stearate (C18). To measure the effect of metal ions and a chelating agent on the catalytic activity, the following were used at a final concentration of 1 mM: CaCl2, CuSO4, EDTA, KCl, MgCl2, and ZnCl2. For assays that examined the effect of detergents and an oxidizing agent on the enzyme activity, detergents Triton, Tween-20, Tween-80, CHAPS, and the oxidizing agent hydrogen peroxide were used at final concentrations of 0.1% (w/v) and 1% (w/v). The effect of SDS on the enzyme activity was also evaluated at final concentrations of 1 mM and 5 mM. To investigate the effect of organic solvents on the enzyme activity, the following organic solvents were used at a final concentration of 10% (v/v): acetone, ethanol, isopropanol, methanol, and n-butanol. Thermostability was measured after incubation of Msed_1072Nt and Msed_1072Ct (0.02 mg/ml) at 70˚C and 90˚C, respectively, at various intervals of time.
2.6. Analysis of Fatty Acid Methyl Esters
A reaction mixture (500 uL) consisting of 50 mM MOPS buffer (pH 7.0), 0.025% Tween-20, 10 g tri-palmitate, and 78 ug histidine-tagged enzyme (Msed_1072Nt and Msed_1072Ct) was incubated at 65˚C for 3 h. After incubation, the reaction products were converted to fatty acid methyl esters as described by Bannon et al. [16] . The hexane layer was directly analyzed with a GC HP5890 series II equipped with a FID (Hewlett Packard, refurbished by Primera Scientific LLC, Princeton, New Jersey) and a 7673 A autosampler (Alpha Omega Technologies, Inc., refurbished by Primera Scientific LLC, Princeton, New Jersey). Separation was achieved in an Rtx-2330 capillary column (Hewlett Packard). The temperature was programmed to include an initial 3 min at 60˚C, and then was increased to 230˚C at a rate of 4˚C/min. Both injector and detector temperatures were set at 265˚C. Injections were performed under the splitless mode [17] . Data acquisition and analysis were completed using the GC Chemstation Rev. A.08.03 (847) software. Calculations for total FAs were completed using the relative response factor (RRF) method [17] and area ratios were compared to the internal standard (KEL-FIMFAME-5 Mixture, Matreya, LLC, Pennsylvania, USA).
3. Results and Discussion
3.1. Amino Acid Sequence Comparison
The BLASTP tool was used to find identities of Msed_1072 among the non-redundant protein sequence data deposited at the National Center for Biotechnology Information database. The deduced amino acid sequence for the M. sedula carboxylesterase gene (Msed_1072) showed 46%, 47%, and 47% identity with those from a metagenomic DNA library, Sulfolobus tokodaii strain 7, and Archaeoglobus fulgidus DSM 4304, respectively (Figure 1). The amino acid sequence of the M. sedula carboxylesterase was the most similar (65% identity) to that of a putative alpha/beta hydrolase from Metallosphaera cuprina Ar-4 [18] . The Msed_1072 sequence has 53% identity to a putative 308 amino acid alpha/beta hydrolase from Sulfolobus islandicus M.14.25 [19] and 52% identity to a 311 amino acid lipase from Sulfolobus solfataricus P2 [20] . In addition, sequence alignment revealed that the M. sedula carboxylesterase contains the typical catalytic triad composed of Ser-Asp-His and the consensus sequence (Gly-X-Ser-X-Gly) around the active-site serine (Figure 1) [21] . Sequence alignment also showed that the amino acid residues Gly-78 and Gly-79 (within the conserved sequence motif HGGG, that is observed in the hormone-sensitive lipase (HSL) family [22] ) and Ala-151 form the oxyanion hole in the Msed_1072 sequence.
3.2. Expression and Purification of Recombinant Carboxylesterase from M. sedula
The carboxylesterase gene variants were heterogeneously expressed in E. coli BL-21 (DE3)pLysS cells under the control of the T7 promoter to allow for a high level of expression. The expression of the targeted protein was induced by α-lactose monohydrate while culturing in auto-induction media [17] . Cells from a two liter culture were incubated for 16 h at 37˚C, harvested, and lysed by the addition B-PER® bacterial protein extraction reagent followed by passage through a French pressure cell. The cell-free supernatant was heat-treated (to remove native proteins), centrifuged, and filtered through a 0.45 uM membrane. A histidine-tag on the targeted proteins (Msed_1072Nt and Msed_1072Ct) allowed for a single-step purification using nickel affinity chromatography. Msed_1072 (untagged enzyme) was purified by a two-step procedure utilizing anion exchange chromatography followed by cation exchange chromatography. Initially, between the purification steps for Msed_1072, the targeted protein was dialyzed in the appropriate start buffer with a pH 4.6. This pH was selected for dialysis based upon the isoelectric point of Msed_1072; however, at this low pH for the start buffer, the partially purified Msed_1072 formed aggregates. Thus, the purification buffer pH levels were increased to 6.0 with the expectation that Msed_1072 would be collected in the flow-through while the E. coli native proteins would bind the column. Detergents, 0.025% Tween-20 and 0.1% CHAPS, were also added to the purification buffers to avoid aggregation of the carboxylesterase. After column purification, enzyme purity was confirmed with a 12.5% SDS-PAGE gel (Figure 2). The purified carboxylesterase variants were stored at 4˚C and were used to characterize the enzyme.

Figure 1. Alignment of amino acid sequences of Msed_1072 (this study), a hyperthermophilic carboxylesterase (EstE1) from a metagenomic DNA library [23] , a thermostable carboxylesterase (Sto-Est) from Sulfolobus tokodaii strain 7 [24] , and a hyperthermophilic carboxylesterase (AFEST) from Archaeoglobus fulgidus DSM 4304 [22] . The putative catalytic triad (Ser150, Asp244, His274) on Msed_1072 deduced from the alignment is indicated by an arrow.
3.3. Substrate Specificity
The substrate specificity of each purified carboxylesterase variant was investigated using several pNP-esters as substrates for the enzyme assays (Figure 3). The highest specific activity was observed with pNP-octanoate for all three enzyme variants (Msed_1072Nt, Msed_1072Ct, and Msed_1072). This result suggests that the presence of the histidine-tag does not change the substrate preference. It is also similar to that of a well-studied thermostable esterase Thermotoga maritima EstA [25] . EstA demonstrated an optimal activity towards pNP-acetate and pNP-octanoate. When EstA hydrolyzed pNP-octanoate, the catalytic activity (kcat) was 37 and the catalytic efficiency (kcat/Km) was 1370. Until recently, most hyperthermophilic carboxylic ester hydrolases were character-
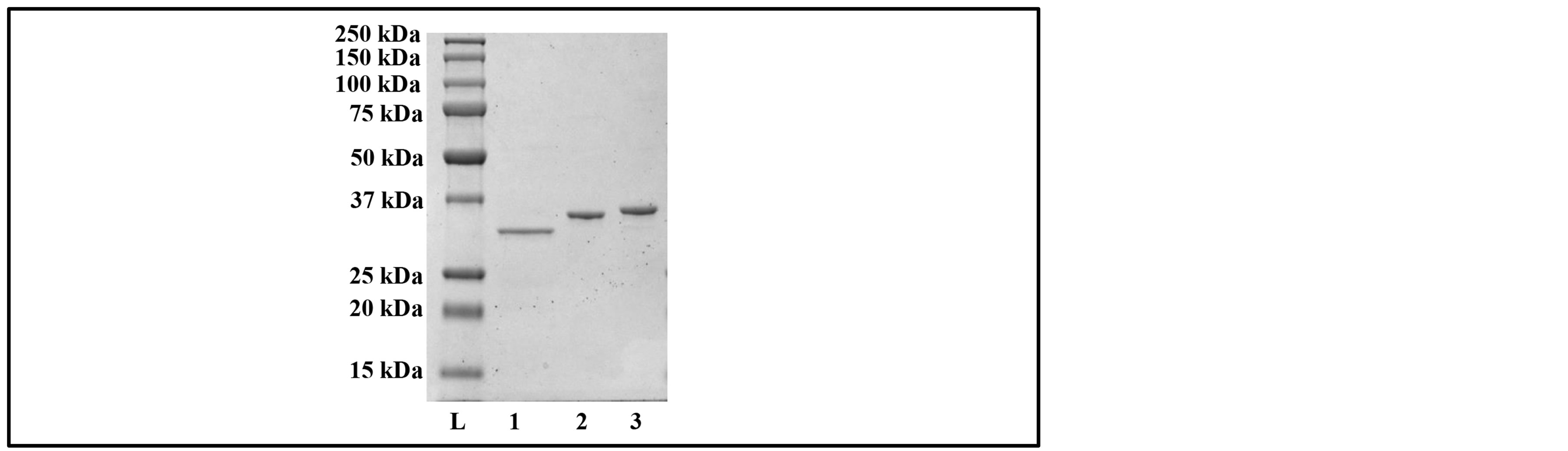
Figure 2. SDS-PAGE of purified M. sedula carboxylesterase variants. L: Molecular Weight Marker (in kDa), Lane 1: Purified Msed_1072 (1 ug), Lane 2: Purified Msed_1072Nt (1 ug), Lane 3: Purified Msed_1072Ct (1 ug).

Figure 3. The specific activities of purified Msed_1072 (gray bar), Msed_1072Nt (white bars), and Msed_1072Ct (black bars) toward pNP-esters with different acyl chain lengths were determined under standard conditions at optimal temperatures of 70˚C (Msed_1072Nt), 95˚C (Msed_1072), and 95˚C (Msed_1072Ct). The highest specific activities were observed using pNP-octanoate as the substrate, and they were as follows: Msed_1072Nt (4079 U/mg), Msed_1072Ct (19089 U/mg), and Msed_1072 (19961 U/mg). Error bars represent standard error.
rized as esterases, preferring medium chain (acyl chain length of 6) p-nitrophenyl substrates [26] [27] . The location of the histidine-tag, however, does influence the specific activity. The C-terminal histidine-tagged protein had activity towards medium-chain substrates (C8-C12) comparable to that of the untagged protein, whereas, the N-terminal histidine-tagged protein had less comparable activity for this range of substrates. All enzyme variants had higher activity toward pNP-esters of carboxylic acids with medium chain length (C8-C12) when compared to those of longer chain length (C14-C18). In addition, the apparent rate of hydrolysis decreased as the carbon chain length increased. Interestingly, the carboxylesterase variant with a polyhistidine-tag fused to the N-terminal region demonstrated hydrolytic activity within a broader specific specificity range using pNP-esters as a substrate (C4-C18), albeit at a lower rate with long chain esters. It is important to note that at high temperatures, such as 95˚C, there is an increase in breakdown of pNP-esters with acyl chains of short length (C4) and long length (C16 and C18). For this reason, 4-methylumbelliferone was used to measure Msed_1072 and Msed_1072Ct ability to hydrolyze carbon chains of these lengths [28] . The activity of Msed_1072 and Msed_1072Ct towards these substrates was undetectable (data not shown). Based on the conserved domain program on NCBI, the substrate binding pocket is located in the N-terminus of the protein. An explanation could be the presence of the poly-histidine affinity tag on the N-terminus influenced the conformation of the substrate binding pocket. Utilizing another substrate (triacylglycerol), which was tested by GC-FID, the tagged esterase variants were able to hydrolyze a long-chain substrate (C16). Msed_1072Nt had a total fat of 2.86 mg/g and Msed_1072Ct had a total fat of 45.78 mg/g (data not shown). Generally, the algal fatty acid synthesis pathway produces a carbon chain length of 14 to 18, saturated or mono-unsaturated fatty acid [29] . For algae-based biofuel production, these results suggest that recombinant Msed_1072 is a carboxylesterase that will enhance the free fatty acid pool by releasing the fatty acids previously sequestered during cell growth as part of algal lipid bodies.
3.4. Influence of Temperature and pH on Enzyme Activity
The activity response of each esterase variant was determined by monitoring the hydrolysis of pNP-octanoate over a temperature range of 37˚C to 95˚C using the standard spectrophotometric assay as described in Methods and Materials. The results (Figure 4) indicate that all enzyme variants were active over a broad temperature range, with the highest hydrolytic activities measured at 70˚C (Msed_1072Nt), 95˚C (Msed_1072Ct), and 95˚C (Msed_1072). For the temperature (75˚C) consistent with what is typically found in the habitat of M. sedula, all variants of the enzyme exhibited catalytic activity. Thermostability for Msed_1072Nt and Msed_1072Ct was also examined as described in Methods and Materials. At 70˚C, Msed_1072Nt was not thermostable (data not shown), but the half-life of Msed_1072Ct at 90˚C was ~28 min (data not shown). The unstable characteristics of the N-terminal tagged protein may be due to the position of the poly-histidine tag. Mandrich and colleagues reported that the NH2-terminal region contributes to enzyme activity and stability in esterases of the HSL family [30] .
The effect of pH on the activity of each Msed_1072 variant was investigated using pNP-octanoate (Figure 5).

Figure 4. Temperature profile of purified Msed_1072 (gray bar), Msed_1072Nt (white bars), and Msed_1072Ct (black bars). Enzyme activity of each esterase variant was determined over a temperature range from 37˚C to 95˚C, using pNP-octanoate as the substrate. The reaction mixture consisted of 50 mM MOPS buffer, pH 7.0, 0.025% Tween-20, and 1 mM pNP-octanoate. Error bars represent standard error.

Figure 5. Effect of pH on the activity of purified Msed_1072 (gray bar), Msed_1072Nt (white bars), and Msed_1072Ct (black bars) was determined over a pH range from 4 to 10, at the optimal temperature for each esterase variant. Assays contained 1 mM pNP-octanoate, 0.025% Tween-20, and 50 mM of the following buffers: pH 4.0 - 5.0, sodium acetate; pH 6.0, MES; pH 7.0, MOPS; pH 8.0 - 10.0, CAPS. Error bars represent standard error.
Activity was measured within a pH range of 4 to 10. A broad pH range for activity was observed for all variants of the enzyme. Nevertheless, Msed_1072Nt, Msed_1072Ct, and Msed_1072 each displayed the highest activity at neutral pH, with an optimum pH of 7. This result suggests that Msed_1072 is likely an intracellular protein. Although M. sedula thrives in an acidic environment (pH 1 - 4), the organism is able to maintain a neutral internal environment with membrane localized proton pumps, a feature found in many microorganisms living in extreme pH conditions.
3.5. Effects of Detergents/Oxidizing Agent, Metal Ions/Chelating Agent, and Organic Solvents
The stability of enzyme activity in the presence of detergents and oxidizing agents is generally considered a desired feature of industrial enzymes. Our results demonstrated that the position of the poly-histidine tag likely determines the stability of the Msed_1072 variants when they are incubated with a surfactant or an oxidizing agent. Enzyme activity toward pNP-octanoate was evaluated for Msed_1072Nt, Msed_1072Ct, and Msed_1072 in the presence of nonionic (Tween-20, Tween-80, and Triton-X100), zwitterionic (CHAPS), and ionic (SDS) detergents as well as H2O2 (oxidizing agent). These results are summarized in Table 1. In the presence of nonionic detergents, activity for both tagged enzyme variants was inhibited as compared to the no-detergent control. In general, relative enzyme activity decreased as the concentration of these detergents increased, which was also observed with the ionic detergent. The exception was Msed_1072Nt activity, which was not affected by the presence of 1.0% Tween-20. Msed_1072Nt and Msed_1072Ct retained 32% and 100% activity, respectively, in the presence of 1 mM sodium dodecyl sulfate (SDS); however, there was no activity observed for either Msed_1072 variants in the presence 5 mM SDS (data not shown). The addition of 0.1% Zwitterionic 3-[(3-cholamidopropyl) dimethyl-ammonio]-1-propanesulfonate (CHAPS) had a slight activating effect on both variants of the enzyme. With regards to the enzyme assay, the addition of a detergent (0.025% Tween-20) to the buffer was critical. When a small amount of this non-ionic surfactant was added, it likely decreased formation of substrate micelles as described for Bacillus stearothermophilus NCA2184 carboxylesterase [31] . Reduced micelle formation then increased enzyme accessibility to the substrate and observed enzyme activity improved. Supplementation with 0.1% H2O2 resulted in a significant reduction of activity for Msed_1072Ct; however, Msed_1072 activity was not affected significantly. It should be noted that the activity of the enzymes (Msed_1072Ct and Msed_1072) were measured in 1.0% H2O2; however, due to breakdown of the substrate (pNP-octanoate) at that concentration, an accurate determination of the enzyme activity could not be made.

Table 1. Enzyme activity in the presence of various detergents.
Prior to the standard activity assay, the enzyme variants were pre-incubated for 5 min at 65˚C in the reaction mixture, containing 50 mM MOPS buffer, pH 7.0, 0.025% Tween-20, and one detergent. The reaction was initiated by the addition of 1 mM pNP-octanoate. Hydrolysis of pNP-octanoate was carried out at 70˚C and 95˚C for Msed_1072Nt and Msed_1072Ct, respectively. The reaction was terminated by the addition of 97.5 µl of chilled 0.25 M sodium carbonate. Enzyme activity determined in the absence of a detergent was defined as 100% activity.
Enzyme activity for each Msed_1072 variant was also evaluated when the enzymes were pre-incubated with and tested for activity in the presence of either a metal ion or a chelating agent (Figure 6). As compared to the control reactions with no added metal ion, the presence of either Cu2+, Mn2+, Zn2+, Ni2+ reduced enzyme activity for each enzyme variant. Reduced activity was most significant for Cu2+ and Zn2+, suggesting that these ions are potent enzyme inhibitors. Ca2+ had no significant impact on the enzyme activity of Msed_1072 and Msed_1072Ct, but reduced the activity of Msed_1072Nt. Interestingly, magnesium ion (Mg2+) stimulated the activity of Msed_1072Ct, reduced the activity of Msed_1072Nt, and had no significant impact on Msed_1072. Potassium ion (K+) significantly reduced the activity of Msed_1072Nt and Msed_1072Ct, while having no significant impact on the activity of Msed_1072. There was also no effect on enzyme activity upon incubation with the metal chelator EDTA for either variant of the enzyme (Msed_1072Nt and Msed_1072Ct), but the activity of Msed_1072 was slightly stimulated in the presence of the chelator. Our findings suggest that Msed_1072 is not a metalloenzyme, in agreement with the fact that lipases and esterases generally do not require cofactors for catalysis [32] .
Enzyme activity toward pNP-octanoate was evaluated for Msed_1072Nt and Msed_1072Ct in the presence of various organic solvents, and the results are summarized in Table 2. While esterase activity of Msed_1072Nt
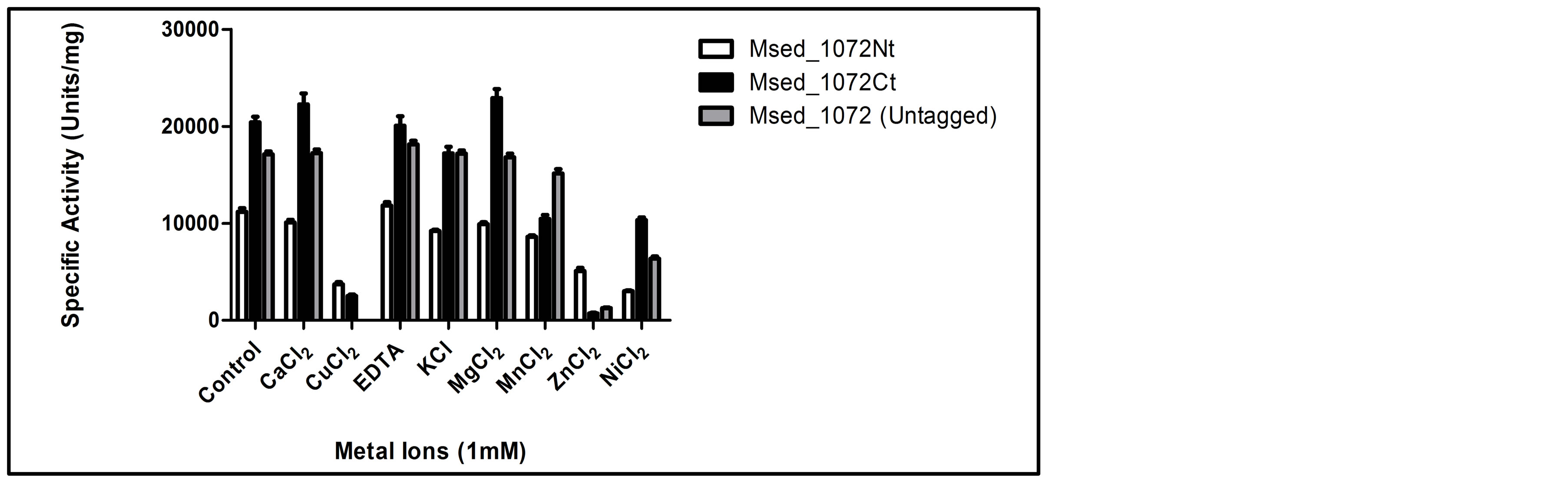
Figure 6. Enzyme activity after pre-incubation with and in the presence of either metal ion or chelating agent supplementation. Prior to the standard activity assay, each enzyme variant was pre-incubated for 5 min at 65˚C in the reaction mixture, containing 50 mM MOPS buffer, pH 7.0, 0.025% Tween-20, and either a metal ion or chelating agent. The reaction was initiated by the addition of 1 mM pNP-octanoate. Hydrolysis of pNP-octanoate was carried out at 70˚C (Msed_1072Nt) and 95˚C (Msed_1072Ct and Msed_1072). The reaction was terminated by the addition of 97.5 µl of chilled 0.25 M sodium carbonate. Enzyme activity determined in the absence of metals or chelator was defined as 100% activity.
Table 2. Enzyme activity in the presence of selected organic solvents.
Prior to the standard activity assay, the enzyme variants were pre-incubated for 5 min at 65˚C in the reaction mixture, containing 50 mM MOPS buffer, pH 7.0, 0.025% Tween-20, and an organic solvent. The reaction was initiated by the addition of 1 mM pNP-octanoate. Hydrolysis of pNP-octanoate was carried out at 70˚C and 95˚C for Msed_1072Nt and Msed_1072Ct, respectively. The reaction was terminated by the addition of 97.5 µl of chilled 0.25 M sodium carbonate. Enzyme activity determined in the absence of an organic solvent was defined as 100% activity.
was increased with the addition of ethanol, this polar solvent had an inhibitory effect on Msed_1072Ct. The presence of n-butanol had a significant inhibitory effect on both enzyme variants, suggesting that this solvent may be toxic to the enzyme. Supplementation with acetone and ispropanol each resulted in reduced activity for Msed_1072Ct; however, Msed_1072Nt activity was not affected significantly. Again, our research findings revealed that the recombinant carboxylesterase can tolerate exposure to organic solvents, and this tolerance depends on the position of the poly-histidine tag. For example, although only Msed_1072Nt demonstrated stability towards ethanol and 2-propanol, the activities of both tagged variants were significantly enhanced by methanol. There are only a few reports showing stability of native lipases, isolated from non-solvent-tolerant microorganisms, in the presence of an organic solvent. For example, a novel enantioselective lipase produced by Acinetobacter species SY-01 had a relative activity value of 100% after 1 hour at 50˚C in methanol [33] . In the case of increased activity of Msed_1072Nt and Msed_1072Ct in the presence of methanol, a likely explanation is that methanol provides enhanced access of the enzyme to the substrate. The ability of Msed_1072 variants to hydrolyze fatty acid esters in the presence of organic solvents does provide strong evidence that this carboxylesterase could be used for high temperature organic syntheses (60˚C - 95˚C).
4. Conclusion
This is the first report of the cloning, functional, and biochemical characterization of a thermostable carboxylesterase enzyme from Metallosphaera sedula DSM5348 for the purpose of microalgae based biofuel production. Although the fusion of the protein with a poly-histidine tag did not shift substrate specificity to carboxylic acids of longer chain length (C12-C18), it did provide insight on which carboxylesterase variation can hydrolyze fatty acid esters in the presence of various chemicals that can be beneficial to industrial processes. To achieve the goal of increasing the enzyme’s affinity towards longer acyl chains, future experiments should focus on altering the acyl binding pocket via site-directed and saturation mutagenesis experiments [34] .
Acknowledgements
The authors thank Drs. Lisa Dean and Stephanie Mixson for their help with the GC-FID protocol and analysis. This work was supported by the National Science Foundation EFRI program under Grants EFRI 093772 and 1332341.
References
- Jaeger, K.E. and Eggert, T. (2002) Lipases for Biotechnology. Current Opinion in Biotechnology, 13, 390-397. http://dx.doi.org/10.1016/S0958-1669(02)00341-5
- Gilham, D. and Lehner, R. (2005) Techniques to Measure Lipase and Esterase Activity in Vitro. Methods, 36, 139-147. http://dx.doi.org/10.1016/j.ymeth.2004.11.003
- Jaeger, K.E., Dijkstra, B.W. and Reetz, M.T. (1999) Bacterial Biocatalysts: Molecular Biology, Three-Dimensional Structures, and Biotechnological Applications of Lipases. Annual Review of Microbiology, 53, 315-351. http://dx.doi.org/10.1146/annurev.micro.53.1.315
- Panda, T. and Gowrishankar, B.S. (2005) Production and Applications of Esterases. Applied Microbiology and Biotechnology, 67, 160-169. http://dx.doi.org/10.1007/s00253-004-1840-y
- Salameh, M.A. and Wiegel, J. (2007) Purification and Characterization of Two Highly Thermophilic Alkaline Lipases from Thermosyntropha lipolytica. Applied and Environmental Microbiology, 73, 7725-7731. http://dx.doi.org/10.1128/AEM.01509-07
- Menetrez, M.Y. (2012) An Overview of Algae Biofuel Production and Potential Environmental Impact. Environmental Science and Technology, 46, 7073-7085. http://dx.doi.org/10.1021/es300917r
- Pienkos, P.T. and Darzins, A. (2009) The Promise and Challenges of Microalgal-Derived Biofuels. Biofuels, Bioproducts, & Biorefining, 3, 431-440. http://dx.doi.org/10.1002/bbb.159
- Guschina, I.A. and Harwood, J.L. (2006) Lipids and Lipid Metabolism in Eukaryotic Algae. Progress in Lipid Research, 45, 160-186. http://dx.doi.org/10.1016/j.plipres.2006.01.001
- Hu, Q., Sommerfeld, M., Jarvis, E., Ghirardi, M., Posewitz, M., Seibert, M. and Darzins, A. (2008) Microalgal Triacylglycerols as Feedstocks for Biofuel Production: Perspectives and Advances. Plant Journal, 54, 621-639. http://dx.doi.org/10.1111/j.1365-313X.2008.03492.x
- Liu, X., Sheng, J. and Curtiss, R. (2010) Fatty Acid Production in Genetically Modified Cyanobacteria. Proceedings of the National Academy of Sciences, 108, 6899-6904. http://dx.doi.org/10.1073/pnas.1103014108
- Voelker, T.A. and Davies, H. (1994) Alteration of the Specificity and Regulation of Fatty Acid Synthesis of Escherichia coli by Expression of a Plant Medium-Chain Acyl-Acyl Carrier Protein Thioesterase. Journal of Bacteriology, 176, 7320-7327.
- Barnes, E.M. and Wakil, S.J. (1968) Studies on the Mechanism of Fatty Acid Synthesis. XIX. Preparation and General Properties of Palmityl Thioesterase. Journal of Biological Chemistry, 243, 2955-2962.
- Huber, G., Spinnler, C., Gambacorta, A. and Stetter, K.O. (1989) Metallosphaera sedula gen. and sp. nov. Represents a New Genus of Aerobic, Metal-Mobilizing, Thermoacidophilic Archaebacteria. Systematic and Applied Microbiology, 12, 38-47. http://dx.doi.org/10.1016/S0723-2020(89)80038-4
- Lee, Y.L., Su, M.S., Huang, T.H. and Shaw, J.F. (1999) C-Terminal His-Tagging Results in Substrate Specificity Changes of the Thioesterase I from Escherichia coli. Journal of the American Oil Chemists’ Society, 76, 1113-1118. http://dx.doi.org/10.1007/s11746-999-0082-7
- Studier, F. (2005) Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expression and Purification, 41, 207-234. http://dx.doi.org/10.1016/j.pep.2005.01.016
- Bannon, C.D., Craske, J.D., Hai, N.T., Hai, N.L., Happer, N.L. and O’Rourke, K.L. (1982) Analysis of Fatty Acid Methyl esters with High Accuracy and Reliability : II. Methylation of Fats and Oils with Boron Trifluoride-Methanol. Journal of Chromatography A, 247, 63-69. http://dx.doi.org/10.1016/S0021-9673(00)84856-6
- Sparkman, O.D., Penton, Z.E. and Kitson, F.G. (2011) Gas Chromatography and Mass Spectrometry: A Practical Guide. 2nd Edition, Elsevier, Inc., Burlington.
- Liu, L.J., You, X.Y., Zheng, H., Wang, S., Jiang, C.Y. and Liu, S.J. (2011) Complete Genome Sequence of Metallosphaera cuprina, a Metal Sulfide-Oxidizing Archaeon from a Hot Spring. Journal of Bacteriology, 193, 3387-3388. http://dx.doi.org/10.1128/JB.05038-11
- Reno, M.L., Held, N.L., Fields, C.J., Burke, P.V. and Whitaker, R.J. (2009) Biogeography of the Sulfolobus islandicus Pan-Genome. Proceedings of the National Academy of Sciences, 106, 8605-8610. http://dx.doi.org/10.1073/pnas.0808945106
- She, Q., Singh, R.K., Confalonieri, F., Zivanovic, Y., Allard, G., Awayez, M.J., Chan-Weiher, C.C., Clausen, I.G., Curtis, B.A., De Moors, A., Erauso, G., Fletcher, C., Gordon, P.M., Heikamp, D.E., Jong, I., Jeffries, A.C., Kozera, C.J., Medina, N., Peng, X., Thi-Ngoc, H.P., Redder, P., Schenk, M.E., Theriault, C., Tolstrup, N., Charlebois, R.L., Doolittle, W.F., Duguet, M., Gaasterland, T., Garrett, R.A., Ragan, M.A., Sensen, C.W. and van der Oost, J. (2001) The Complete Genome of the Crenarchaeon Sulfolobus solfataricus P2. Proceedings of the National Academy of Sciences, 98, 7835-7840. http://dx.doi.org/10.1073/pnas.141222098
- Jaeger, K.E., Ransac, S., Dijkstra, B.W., Colson, C., van Heuvel, M. and Misset, O. (1994) Bacterial Lipases. FEMS Microbiology Reviews, 15, 29-63. http://dx.doi.org/10.1016/0168-6445(94)90025-6
- De Simone, G., Menchise, V., Manco, G., Mandrich, L., Sorrentino, N., Lang, D., Rossi, M. and Pedone, C. (2001) The Crystal Structure of a Hyper-Thermophilic Carboxylesterase from the Archaeon Archaeoglobus fulgidus. Journal of Molecular Biology, 314, 507-518. http://dx.doi.org/10.1006/jmbi.2001.5152
- Byun, J.S., Rhee, J.K., Kim, D.U., Oh, J.W. and Cho, H.S. (2006) Crystallization and Preliminary X-Ray Crystallographic Analysis of EstE1, a New and Thermostable Esterase Cloned from a Metagenomic Library. Acta Crystallographica Section F: Structural Biology and Crystallization Communications, 62, 145-147. http://dx.doi.org/10.1107/S1744309106000832
- Angkawidjaja, C., Koga, Y., Takano, K., and Kanaya, S. (2012) Structure and Stability of a Thermostable Carboxylesterase from the Thermoacidophilic Archaeon Sulfolobus tokodaii. FEMS Microbiology Letters, 276, 3071-3084. http://dx.doi.org/ 10.1111/j.1742-4658.2012.08687.x
- Levisson, M., Sun, L., Hendriks, S., Swinkels, P., Akveld, T., Bultema, J., Barendregt, A., van den Heuvel, R., Dijkstra, B., ver der Oost, J. and Kengen, S. (2009) Crystal Structure and Biochemical Properties of a Novel Thermostable Esterase Containing an Immunoglobulin-Like Domain. Journal of Molecular Biology, 385, 949-962. http://dx.doi.org.prox.lib.ncsu.edu/10.1016/j.jmb.2008.10.075
- Atomi, H., Fukui, T., Kanai, T., Morikawa, M. and Imanaka, T. (2004) Description of Thermococcus kodakaraensis sp. nov., a Well Studied Hyperthermophilic Archaeon Previously Reported as Pyrococcus sp. KOD1. Archaea, 1, 263-267. http://dx.doi.org/10.1155/2004/204953
- Levisson, M., van der Oost, J. and Kengen, S.W. (2009) Carboxylic Ester Hydrolases from Hyperthermophiles. Extremophiles, 13, 567-581. http://dx.doi.org/10.1007/s00792-009-0260-4
- Sehgal, A.C., Callen, W., Mathur, E.J., Short, J.M. and Kelly, R.M. (2001) Carboxylesterase from Sulfolobus solfataricus P1. Methods in Enzymology, 330, 461-471. http://dx.doi.org/10.1016/S0076-6879(01)30398-1
- Harwood, J. (1998). Membrane lipids in algae. In Lipids in Photosynthesis: Structure, Function, and Genetics. Kluwer Academic Publishers, Dordrecht, 53-64.
- Mandrich, L., Merone, L., Pezzullo, M., Cipolla, L., Nicotra, F., Rossi, M. and Manco, G. (2005) Role of the N Terminus in Enzyme Activity, Stability and Specificity in Thermophilic Esterases Belonging to the HSL Family. Journal of Molecular Biology, 345, 501-512. http://dx.doi.org/10.1016/j.jmb.2004.10.035
- Matsunaga, A., Koyama, N. and Noso, Y. (1974) Purification and Properties of Esterase from Bacillus stearothermophilus. Archives of Biochemistry and Biophysics, 160, 504-513. http://dx.doi.org/10.1016/0003-9861(74)90427-5
- Jaeger, K.E. and Reetz, M.T. (1998) Microbial Lipases form Versatile Tools for Biotechnology. Trends in Biotechnology, 16, 396-403. http://dx.doi.org/10.1016/S0167-7799(98)01195-0
- Han, S.J., Back, J.H., Yoon, M.Y., Shin, P.K., Cheong, C.S., Sung, M.H., Hong, S.P., Chung, I.Y. and Han, Y.S. (2003) Expression and Characterization of a Novel Enantioselective Lipase from Acinetobacter Species SY-01. Biochimie, 85, 501-510. http://dx.doi.org/10.1016/S0300-9084(03)00057-9
NOTES

*Corresponding author.