Advances in Enzyme Research
Vol. 1 No. 2 (2013) , Article ID: 33419 , 13 pages DOI:10.4236/aer.2013.12002
Cooperative binding of calcium ions modulates the tertiary structure and catalytic activity of Matrix-Metalloproteinase-9
![]()
Department of Chemistry and Biochemistry, North Dakota State University, Fargo, USA; *Corresponding Authors: tobwalas@mst.edu, Dk.srivastava@ndsu.edu
Copyright © 2013 Shakila Tobwala, D. K. Srivastava. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 3 April 2013; revised 10 May 2013; accepted 25 May 2013
Keywords: Matrix Metalloproteinase-9; Gelatinase-B; Fluorescence Lifetime; Quenching; Acrylamide; Stern-Volmer Constant
ABSTRACT
To ascertain the molecular basis of Ca2+-mediated activation of matrix metalloproteinase-9 (MMP-9), we determined the accessibility of tryptophan residues to externally added acrylamide as quencher in the absence and presence of the metal ion. The steady-state and time resolved fluorescence data revealed that MMP-9 possesses two classes of tryptophan residues, “exposed” and “buried” which are quenched by the collisional rate constants (kq) of 3.2 ( 109 M−1∙s−1and 7.5 ( 108 M−1∙s−1, respectively. These values are impaired by approximately two and three-fold, respectively, in the presence of 10 mM Ca2+. The Stern-Volmer constants (Ksv values) predicted from the time resolved fluorescence data (in the absence of Ca2+) satisfied the dynamic quenching model of the enzyme’s tryptophan residues. This was not the case in the presence of Ca2+; the steady-state acrylamide quenching data could only be explained by a combination of “dynamic” and “static” quenching models. A cumulative account of these data led to the suggestion that the binding of Ca2+ modulated the tertiary structure of the protein by decreasing the dynamic flexibility of the enzyme, which is manifested in further structuring of the enzyme’s active site pocket toward facilitating catalysis. Arguments are presented that the binding of Ca2+ at distal sites “dynamically” communicates with the active site residues of MMP-9 during catalysis.
1. INTRODUCTION
Matrix metalloproteinases (MMPs) are involved in dynamically controlling the synthesis and degradation of extracellular matrix assembly (ECM), which is primarily constituted of different types of collagen fibers, in the physiological milieu. Under normal condition, these “coordinated” processes are responsible for embryonic development, reproduction, wound healing, and angiogenesis. Any factor (genetic and/or environmental), which obviates the above dynamic control, either results in an excessive accumulation of the ECM assembly or its degradation, causing a variety of human diseases. For example, depending on the tissue type, excessive ECM degradation leads to the onset of a variety of pathological conditions such as, chronic wounds, dermal photo-ageing, bullous skin diseases, atherosclerosis, rheumatoid arthritis, and cancer and metastasis [1-6].
Of 26 different types of MMPs present in human tissues, MMP-9 (also known as gelatinase-B) is involved in the degradation of collagen type IV and gelatins, and it has been widely recognized to be involved in the progression and metastasis of a variety of diseases [7-23]. In view of an extensive pathophysiological role of MMP-9, this enzyme has been considered as being the high priority target for drug design. However, earlier efforts in designing MMP inhibitors as potential drugs have not been successful, and they suffered partly due to the lack of detailed structural-functional and mechanistic studies on MMPs including those on MMP-9 [24].
From the structural point of view, MMP-9 is one of most complex MMPs, as it is comprised of a “Pre” domain (an ~20 amino acid residue N-terminal signal peptide sequence), a “Pro” domain (an ~80 amino acid residue inhibitory peptide fragment), a catalytic domains, three fibronectin (type-II ) domain, and a “hemopexin” like domains [25]. Of these, the fibronectin domain bridges the two catalytic half domains of the enzyme, and the overall assembly is believed to be responsible for the recognition and unwinding of the triple helical collagen IV peptide substrates during the enzyme catalysis. The X-ray crystallographic data of MMP-9 reveals that the enzyme contains two Zn2+ and three to five Ca2+ binding sites [26-28], of which one Zn2+ (coordinated by three histidine residues of HEXXHXXGXXH motif) is unequivocally demonstrable to exhibit the catalytic function. Due to their relative locations on the protein surface (i.e., far removed from the active site), non-catalytic Zn2+ and all Ca2+ appear to exhibit structural rather than catalytic roles (Figure 1). Contrary to this expectation, several investigators noted that besides performing the structural roles [29-31], the binding of Ca2+ enhances the catalytic activities of several MMPs [32-34], including those of MMP-2 and MMP-9 [35-38]. By employing dynamic simulation studies on MMP-2, Diaz and Suarez [35] demonstrated that both non-catalytic Zn2+ and Ca2+ (seemingly the “structural metal ions”) influence the position of several solvent-exposed loop regions that are placed near the active site cleft, and the position of these loops modulates the accessibility of the enzyme’s substrate.
Our interest towards investigating the influence of Ca2+ emerged from our preliminary comparative data on the MMP-9 and MMP-10 catalyzed reactions. To our
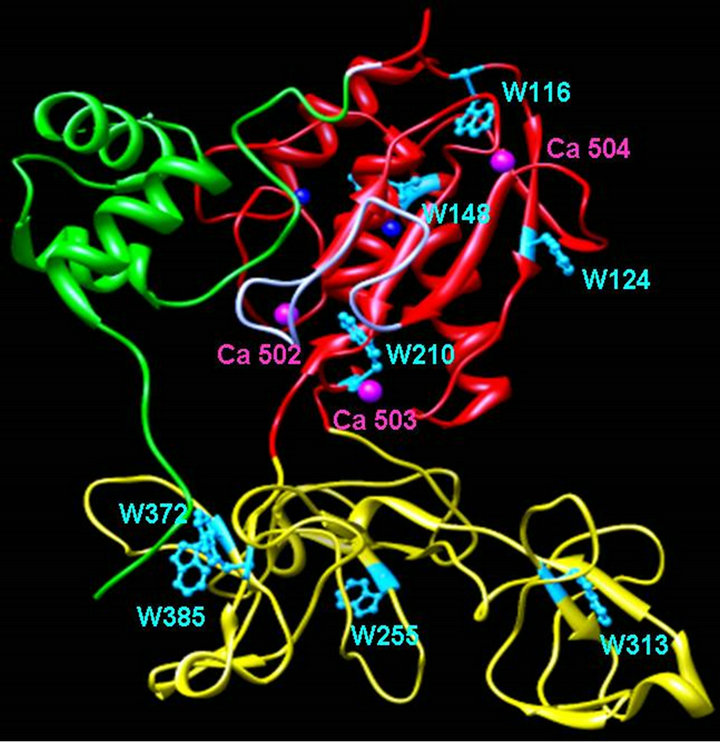
Figure 1. Ribbon diagram of MMP-9 structure. The X-ray crystallographic structure of proMMP-9 (PDB file: 1l6j.pdb) showing the tryptophan residues and bound Ca2+. The catalytic and fibronectin domains are shown in red and gold color, respectively. The green ribbon represents the pro-domain of the enzyme.
interest, we observed that the apo form of MMP-10 acquires nearly full activity upon addition of Zn2+ (and it is not further activated upon addition of Ca2+), whereas the activation of the apo form of MMP-9 requires the addition of both Zn2+ and Ca2+ ions (to be published subsequently). This functional difference was particularly noteworthy in the light of the fact that Ca2+ ions are far removed from the catalytic sites of both enzymes. Evidently, the binding of Ca2+ ions to the distal site on MMP-9 structure “allosterically” activates its substrate binding and/or the chemical transformation step. While we are currently investigating the structural-functional features of the above noted differential effects of Ca2+ ions on the activation of MMP-9 versus MMP-10, we surmised that the Ca2+- dependent activation of MMP-9 is mediated via the dynamic fluctuation in the protein structure. This has been in accord with the widely conceived notion that conformational dynamics, in conjunction with the motion of amino acid residues, in protein structures are intimately involved in promoting enzyme catalysis [39].
One of the approaches of experimentally evaluating the contributions of conformational dynamics in proteins has been to monitor the signals associated with the diffusion of different solutes within their interior cores [40,41]. In this paradigm, quenching of intrinsic fluorescence of proteins by acrylamide has been frequently utilized to probe the differential accessibility of tryptophan residues in relation to the space and time [42-44]. Although such studies have unraveled several molecular details in several proteins, they have not been extensively implemented with MMPs. In limited studies, Nagase and his collaborators [45] employed acrylamide and potassium iodide quenching of tryptophan residues of MMP- 2 and MMP-9 to probe the accessibility of those residues during the course of activation and binding with their natural inhibitor, TIMP (tissue inhibitor of metalloproteinases). With these studies in mind, we proceeded to investigate the differential accessibility of tryptophan residues in MMP-9 via monitoring the steady-state and lifetime fluorescence in the presence of acrylamide as a neutral quencher. We also studied the effects of calcium ion on protein structure using tryptophan’s intrinsic steady state fluorescence and circular dichroism and probed its relationship to changes in enzyme’s catalytic activity. Our studies have led to the elucidation of the dynamic role of Ca2+ in decelerating the collisional rate constant of the quenchers, altering the mechanistic pathway of protein’s quenching, and modulating the tertiary structure of MMP-9 resulting in overall tightening of the MMP-9 structure.
2. MATERIALS AND METHODS
2.1. Materials
Zinc chloride, calcium chloride, ampicillin, chloram phenicol, Tris, and IPTG were purchased from Life Science Resources, Milwaukee, WI; yeast extracts, and tryptone were purchased from Becton Dickinson, Sparks, MD; PMSF, acrylamide, and gelatin agarose column material was obtained from Sigma. The expression cells, BL21codon plus DE3 (RIL) were from Stratagene, La Jolla, CA. All other chemicals were of reagent grade, and were used without further purification.
2.2. Cloning of Human MMP-9
The plasmid pOTB7 containing the full length cDNA clone of human matrix metalloproteinase 9 (Gelatinase B) was obtained from ATCC (MGC-12688). The nucleotide fragment from the residue Phe-106—Leu-442 was amplified by the hot start PCR method [46] using the above plasmid as the DNA template, and following synthetic primers. The sense and anti-sense primers were 5’-GGA ATT C-CATATGTTC CAA ACC TTT GAG GGC GAC -3’ and 5’-CCG-CTCGAG-TTA ATA GAG GTG CCG GAT GC-3’, respectively. These primers contained the NdeI restriction enzyme sequence in the sense primer and XhoI restriction enzyme sequence in the antisense primer (underlined). The bold sequences in both sense and anti-sense primers are the additional sequences for recognition by their respective enzymes. The PCR reaction mixture contained the template DNA (~50 ng), primers (4 ml of 25 mM), dNTPs (1 ml of 10 mM), MgCl2 (10 ml of 25 mM), and pfu DNA polymerase (5 units) in a total volume of 100 ml. The PCR reaction conditions were set as follows: 30 seconds at 94˚C denaturation, 1 min and 30 seconds at 55˚C for annealing, 15 min at 72˚C for extension, for a total number of 25 reaction cycles. The PCR reaction product and the expression vector pET 20b (+) were digested with Nde I and XhoI and purified from an agarose gel using the QIAquick gel extraction kit. The purified products were ligated with T4 DNA ligase (forming the MMP-9-pET plasmid) and transformed into DH5α cells for plasmid propagation [46]. The plasmid was transformed into E. coli BL21 (DE3) for protein expression. The cloning of the coding regions of MMP-9 in the pET 20b (+) vector was confirmed by sequencing the plasmid at the University of Chicago Cancer Research Center.
2.3. Expression and Purification of Recombinant Human MMP-9
The MMP-9-pET expression system was grown in LB medium, supplemented with 100 mg of ampicillin/ml, 50 mg/ml of chloramphenicol at 37˚C until A600 was 0.6. The expression of MMP-9 was induced by addition of 400 mM IPTG. The cells were incubated further at 37˚C for 5 hours. The cell suspension was centrifuged at 8000 g for 20 min. The pellet was washed in 50 mM Tris-HCl (buffer A) pH 7.5, and resuspended in the same buffer followed by the addition of 1mM PMSF (in 2-propanol) prior to sonication. The cells were sonicated for a total time of 10 min in a Branson sonifier utilizing 40% duty cycle in an ice cold bath. The sonicated extract was centrifuged at 15,000 rpm for 30 min, and the pellet was collected. The pellet containing inclusion bodies was washed thrice with buffer A, and then once with 50 mM Tris-HCl buffer containing 2 M urea, pH 7.5, and then dissolved in the above buffer containing 6 M urea by stirring overnight at 4˚C. The 6 M urea extract was centrifuged at 15,000 rpm for 30 min and its supernatant was collected for further purification. To generate the active form of recombinant human MMP-9, the enzyme suspension (in urea solution) was folded by 10-fold (vol/vol) by snap-dilution in 50 mM Tris-HCl buffer, pH 7.5, containing 10 mM CaCl2 and 10 mM ZnCl2. The solution was further stirred for 30 min, and left undisturbed for 12 hours at 4˚C. The refolded protein was concentrated, and loaded onto a gelatin-agarose column (1 ´ 10 cm), previously equilibrated with 50 mM Tris-Cl, 10 mM CaCl2, 10 mM ZnCl2, pH 7.5. The column was washed with buffer containing 50 mM Tris-Cl, 10 mM CaCl2, 10 mM ZnCl2, 0.5 M NaCl, pH 7.5, and the enzyme was eluted by supplementing the above buffer with 10% DMSO. The active enzyme fractions were pooled, concentrated, and stored at −20˚C freezers. At times the protein after snap dilution was ³90% pure, and the column purification step was eliminated. The activity of the recombinant MMP-9 was measured in 50 mM Tris-HCl buffer, pH 7.5, containing 10 mM CaCl2 at 25˚C, utilizing thiopeptolide TPL (Ac-Pro-Leu-Gly-(2-mercapto-4-methylpentanoyl)- Leu-Gly-OE) as substrate [38]. The protein concentration was determined by BCA assay, utilizing BSA as the standard protein. The purified enzyme was judged to be homogeneous by the SDS-PAGE analysis. The purified MMP-9 showed single band on SDS gel electrophoresis. The yield from 1 liter of bacterial culture was in the range of 20 - 30 mg. To obtain Ca2+ -free MMP-9, the urea extract of inclusion bodies was folded in the absence of calcium ions. To ensure that no Ca2+ tagged along with MMP-9 during the course of purification, we dialyzed the purified enzyme by four changes (for 8 hours at 4˚C) of 50 mM Tris-HCl buffer, pH 7.5, prepared by using Chelex-100-treated water. The enzyme thus prepared did not exhibit any activity, but it restored full activity upon addition of 10 mM Ca2+ (aside from 10 mM Zn2+). All steady state and time resolved fluorescence decay experiments in the absence and presence of CaCl2 were performed in 50 mM Tris-HCl buffer, pH 7.5. Steady State Fluorescence Measurements Steady state spectrofluorometric studies were performed on a Perkin Elmer lambda 50-B spectrofluorometer, equipped with a magnetic stirrer and thermostated water bath. The emission spectra of MMP-9 in the absence and presence of calcium ions were acquired by fixing the excitation wavelength at 295 nm while maintaining both excitation and emission slits at 5 mm without any cut-off filter. All measurements were performed in a standard quartz cuvette at room temperature. For quenching experiments, aliquots of acrylamide were added directly to the sample in small steps. Fluorescence spectra were corrected for the dilution of the titrant. The fluorescence intensities were corrected for the acrylamide absorbance using e295 = 0.25 M−1∙cm−1.
2.4. Time-Resolved Fluorescence Studies
Fluorescence lifetime measurements were performed on a custom designed Photon Technology International (PTI) Fluorescence-Lifetime instrument. The excitation source for measuring the time resolved fluorescence decay was the Light Emitting diode (LED) with maximum power output at 280 nm. For all experiments, the excitation and emission wavelengths were set to 280 and 335 nm, respectively. The emission slits were 6 mm. The emitted light was detected (at right angle of the excitation source) by means of a stroboscopic emission monochromator configured at appropriate wavelengths. The instrument response function was recorded by collecting scattered light from a starch suspension. The data were collected in 200 channels with an integration time of 1 sec, and six data sets were averaged to obtain better resolution of the fluorescence traces. The time resolved fluorescence decay curves were analyzed to obtain the lifetimes of the protein resident tryptophan residues under different experimental conditions by the aid of the PTI’s software, Felix 32. The kinetic traces were fitted by the single and double exponential rate equations of the following format (Eq. 1).
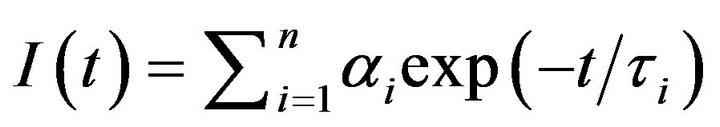 (1)
(1)
Where 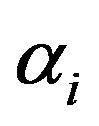 and τi are amplitude and fluorescence lifetime for the ith component, respectively. The relative contribution of component i (fi) was calculated from the following relationship (Eq. 2).
and τi are amplitude and fluorescence lifetime for the ith component, respectively. The relative contribution of component i (fi) was calculated from the following relationship (Eq. 2).
 (2)
(2)
The parameters describing the true decay function were extracted from the experimental data by a nonlinear least squares convolution process. The goodness of the exponential fit of fluorescence curves was determined by the reduced χ2, the Durbin Watson, and Z values.
3. RESULTS
The experimental data presented herein were prompted by our preliminary observation that unlike MMP-10 (which does not require Ca2+ for the catalytic activity), MMP-9 catalyzed reaction is absolutely contingent on the presence of both Ca2+ and Zn2+ (to be published subsequently). To probe the dynamic contribution of Ca2+ in activating MMP-9, we decided to investigate the accessibility of the enzyme’s tryptophan residues by monitoring their fluorescence properties in the presence of acrylamide. Although in a cursory manner, it has been demonstrated [45] that both acrylamide and iodide serve as quenchers of the tryptophan fluorescence in MMP-9, no detailed studies on the molecular mechanism of such quenching (to the best of our knowledge) have been performed.
Given that the steady-state fluorescence measurements are often unreliable due to several uncontrollable variables, such as “scattering”, “inner-filter” effects etc., we decided to first determine the lifetime profiles of the enzyme’s tryptophan residues in the presence of increasing concentrations of acrylamide. Figure 2(a) shows the excited state fluorescence decay profiles of MMP-9 (lex = 280 nm, lem = 335 nm) in the absence and presence of 122 mM acrylamide. The experimental data were best fitted (solid smooth lines) by the biphasic rate equation for the short (ts) and long (tl) lifetimes of 1.7 ns and 5.9 ns in the absence of acrylamide, and 1.0 ns and 3.4 ns in the presence of 122 mM acrylamide, respectively. Our attempts to fit these data by the single exponential rate equation yielded systematic deviation of their residuals. We ascribe the observed lifetimes, ts and tl, to the exposed and buried tryptophan residues of MMP-9, although we cannot rule out the possibility of alternate rotamer distribution of the above residues [47].
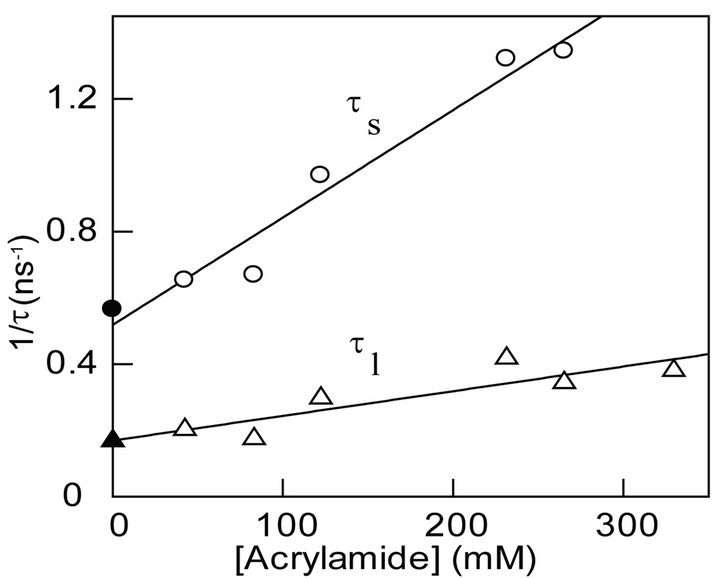
We investigated the influence of acrylamide concentration on ts and tl. Figure 2(b) shows that as the concentration of acrylamide increases, the magnitudes of both ts and tl decrease linearly, and the overall profile adheres to the Stern-Volmer relationship in the following format (Eq. 3) [40,41,48].
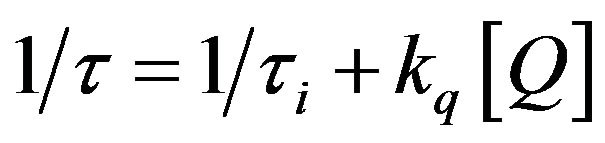 (3)
(3)
where t0 and t are the lifetimes of the tryptophan fluorescence in the absence and presence of acrylamide, respectively, and kq is the bimolecular rate constant of quenching.
The solid lines of Figure 2(b) are the best fit of the acrylamide quenching data for t0 and kq values, derived from shorter lifetime (ts) component, of 1.9 ns and 3.2 ´ 109 M−1∙s−1, and the corresponding values derived from the longer lifetime component (tl) of 5.9 ns and 7.5 ´ 108 M−1∙s−1, respectively. Note that the intercept values (obtained from the extrapolations of 1/t versus [Q] plots) match fairly closely to the independently determined lifetimes (t0) of the enzyme’s tryptophan in the absence of acrylamide. It is further noteworthy that the kq value derived from the shorter lifetime component is about 4-fold higher than that derived from the longer lifetime component. Since acrylamide is known to quench the surface exposed tryptophan residues more efficiently than those buried in the interior core of the enzyme [40,41,48], we assign high and low kq values to the surface exposed and buried tryptophan residues of MMP-9, respectively.
It has been known that the fluorescence emission band of tryptophan residues are sensitive to the polarity of the medium; lower polarity shifts the emission band to blue, higher polarity shifts it to the red [48]. If MMP-9 harbors two classes of tryptophan residues (i.e., exposed and buried), the preferential quenching of the exposed residues would shift the fluorescence emission spectrum to blue. This feature is evident from the data of Figure 2(c). Note that upon addition of 0.5 M acrylamide, the fluorescence emission band of MMP-9 shifts from 343.5 nm to 337 nm, suggesting that the preferential quenching of the surface exposed tryptophan residues augments the fluorescence (with concomitant blue shift) of the buried tryptophan residues [48]. Although it would be tempting to quantitate the relative distribution of the surface ex posed versus buried tryptophan residues from the data of Figure 2(c), such deduction would unlikely to be accuched (at least partially) by acrylamide.
 (b)
(b) (a)
(a)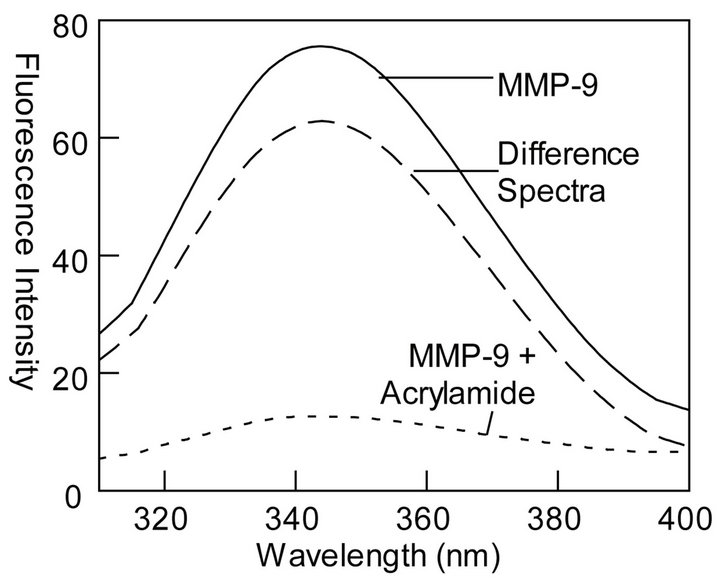 (c)
(c)
Figure 2. Fluorescence profiles for quenching of tryptophan residues of MMP-9 by acrylamide. The experiments were performed in 50 mM Tris-HCl buffer at pH 7.5. (a): The time resolved decay of tryptophan fluorescence of MMP-9 ([MMP-9] = 28 mM) (lex = 280 nm, and lem = 335 nm) in the absence and presence of acrylamide ([Acrylamide] = 122 mM). The solid smooth lines are the best fit of the data according to Eq. 1 with ts = 1.7 ns ± 0.07 and αs (amplitude) = 0.38 ± 0.01; tl = 5.9 ns ± 0.43 and αl = 0.08 ± 0.01 for MMP-9 in the absence of metal ions and ts = 1.0 ns ± 0.04 and αs = 0.27 ± 0.01; tl = 3.4 ns ± 0.16 and αl = 0.06 ± 0.01 in the presence of acrylamide, respectively. The bottom panels show the residuals of the fit of the individual lifetime traces; (b): Acrylamide concentration dependence of τs and τl. The solid lines are the linear regression analyses of the data according to Eq. 3 for kq and ts values of 3.2 ´ 109 M−1∙s −1 and 1.9 ns, respectively for the shorter lifetime component, and kq and tl values of 7.5 ´ 108 M−1∙s −1 and 5.9 ns for the longer lifetime component, respectively; (c): Effect of acrylamide on the steady-state fluorescence spectrum of MMP-9. Note a blue shift in the fluorescence spectrum of MMP-9 (lex = 295) from 343.5 nm to 337 nm upon addition of acrylamide. The dashed line represents the difference spectrum, i.e., the spectrum of MMP-9 minus that in the presence of acrylamide.
3.1. Effect of Ca2+ on Acrylamide Dependent Changes in the Fluorescence Profile of MMP-9
To ascertain the contribution of Ca2+ on the acrylamide dependent changes in the fluorescence profile of MMP-9, we repeated the experiment of Figure 2(a) in the presence of 10 mM Ca2+ (Figure 3(a)). As observed with apo (Ca2+-free) MMP-9, the excited state fluorescence decay profile of the enzyme in the presence of Ca2+ once again conformed to the biphasic rate equation (Eq. 1) with ts and tl values of 1.7 ns and 5.9 ns, respectively. Note that these values are comparable to those obtained in the absence of Ca2+. However, the presence of Ca2+
altered the amplitude of the two phases. Whereas the binding of Ca2+ decreased the amplitude of the shorter lifetime component from 83 to 76%, it increased the amplitude of the longer lifetime component from 17 to 24%. When we performed these experiments in the presence of 122 mM acrylamide, no significant effect of Ca2+ on the short and long life time components of MMP-9 was observed. However, in all these conditions, the excited state decay profiles invariably conformed to the biphasic decay profile as shown by the residuals at the bottom panel of the figure. To further ascertain whether Ca2+ influenced the magnitudes of kq and t0 values of the shorter and longer components, we determined the tryptophan
 (a)
(a) 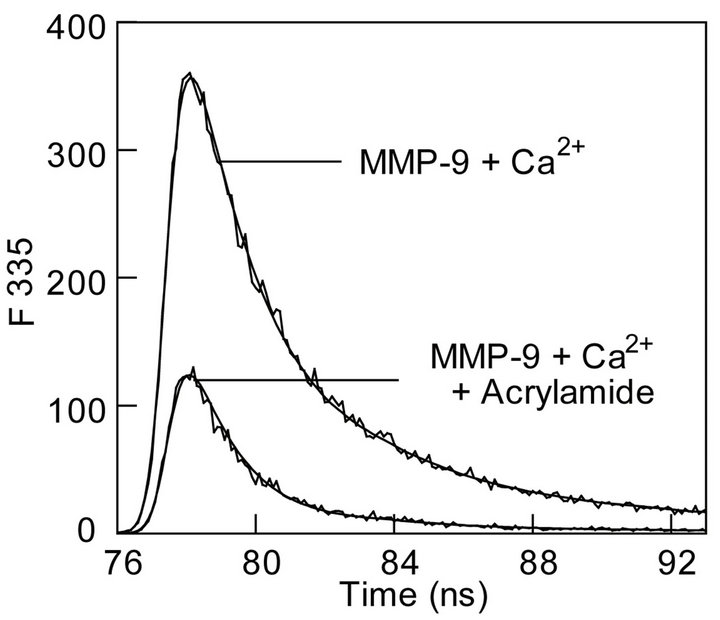
 (b)
(b) 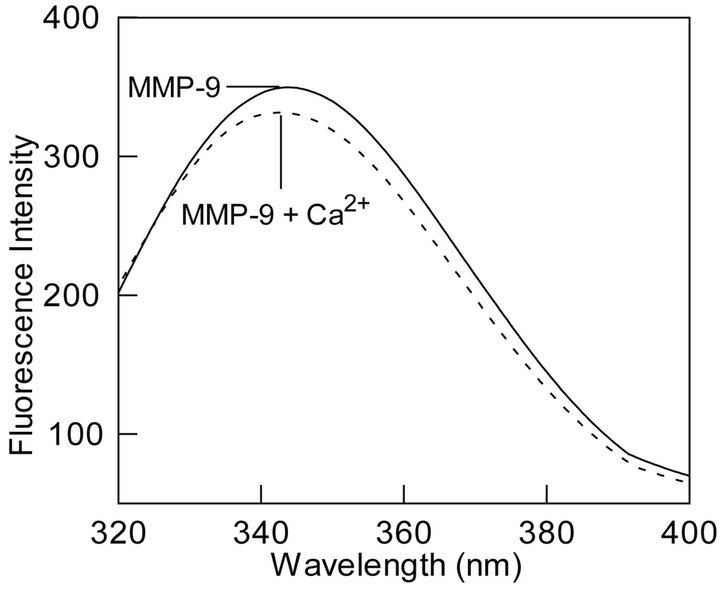 (c)
(c)
Figure 3. Effect of calcium ions on the fluorescence profiles of the tryptophan residues of MMP-9. (a): The time resolved fluorescence decay of the tryptophan residues of MMP-9 + Ca2+ (10 mM) in the absence and presence of acrylamide. [MMP-9] = 28 mM, [acrylamide] = 122 mM, lex = 280 nm, and lem = 335 nm. Other conditions were same as in Figure 2. The solid smooth lines are the best fit of the data according to the Equation (1) with ts = 1.7 ns ± 0.05 and αs = 0.53 ± 0.01; tl = 5.9 ns ± 0.20 and αl = 0.170 ± 0.001 for MMP-9 in the presence of 10 mM Ca2+ ions, and ts = 1.3 ns ± 0.04 and αs = 0.34 ± 0.01; tl = 4.3 ns ± 0.15 and αl = 0.09 ± 0.01 in the presence of acrylamide. The bottom panels show the residuals of the fitted lines; (b): Acrylamide concentration dependence of the shorter (τs) and longer (τl) lifetime components. The solid lines are the least squares fits the data according to Eq. 3 with kq and ts values of 1.4 ´ 109 M−1∙s−1 and 1.9 ns, respectively for the shorter component, and the corresponding parameters for the longer components being equal to 2.2 ´ 108 M−1∙s−1 and 5.9 ns, respectively; (c): Calcium-induced changes in the steady-state fluorescence spectrum of MMP-9. Note a calcium ion induced shift in emission peak from 343.5 nm to 340.5 nm in the presence of 10 mM CaCl2.
lifetimes of MMP-9 in the presence of increasing concentrations of acrylamide (Figure 3(b)). The linear regression analyses of the data yielded the kq and t0 values of 1.4 × 109 M−1∙s−1 and 1.9 ns from the shorter lifetime component, and 2.2 × 108 M−1∙s−1 and 5.9 ns from the longer lifetime component, respectively. A casual comparison of the data of Figure 2(b) and Figure 3(b) reveal that the presence of Ca2+ slightly impairs the bimolecular rate of quenching (kq) of the enzyme’s tryptophan residues of MMP-9 by acrylamide. This coupled with the fact that the presence of Ca2+ alters the relative amplitudes of the short and long lifetime phases of the enzyme’s tryptophan residues (Figure 3(a)) lead to the suggestion that binding of metal ion to the enzyme buries some of the surface exposed tryptophan residues to the interior.
3.2. Effect of Ca2+ on the Secondary and Tertiary Structure and Activation Profile of MMP-9
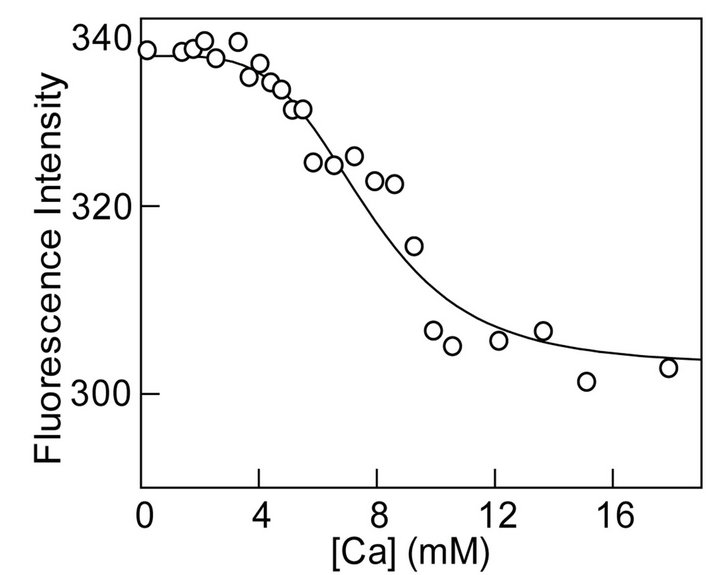
To further attest our hypothesis of burial of surface exposed or partially exposed tryptophan residues we studied the effect of Ca2+ on the secondary and tertiary structure of MMP-9. CD spectrum of MMP-9 in the presence and absence of calcium ions were indistinguishable indicating no change in the secondary structure of the protein upon binding calcium ions. However, binding of Ca2+ to MMP-9 resulted in about 3 nm blue shift in the steady-state fluorescence emission band of the enzyme’s tryptophan residues indicating changes in the tertiary structure MMP-9 (Figure 3(c)). In addition, decrease in emission intensity upon binding Ca2+ indicates movement of surface exposed or partially exposed tryptophan residues to the interior of the protein. Given the changes in steady-state fluorescence upon binding of Ca2+ to MMP-9, we proceeded to determine the binding profile of the enzyme-metal complex. Figure 4(a) shows the titration of a fixed concentration of MMP-9 (16 mM) with increasing concentration of Ca2+ while monitoring the decrease in the fluorescence emission intensity at 343.5 nm (lex = 295 nm). Note that as the concentration of Ca2+ increases, the fluorescence intensity decreases in a sigmoidal fashion indicating local structural changes caused by the binding of calcium ions. However, given that there are multiple binding sites of Ca2+ on the protein surface [26-28], we believe the origin of the sigmoidal profile of Fig. 4A lies in the cooperative interaction of Ca2+ to MMP-9, resulting in the quenching of the enzyme’s tryptophan fluorescence. Microscopically, the above feature can originate due to the Ca2+ dependent changes in the conformational states of MMP-9.
To probe the functional consequences of the observed cooperativity in binding of Ca2+ to MMP-9, we measured the enzyme activity as a function of increasing concentration of Ca2+. As shown in Figure 4(b), the rate of the MMP-9 catalyzed reaction increases with the increase in Ca2+ concentration. Note that although the overall activation profile of Figure 4(b) appears to be hyperbolic in nature (as expected for the non-cooperative binding model), the initial region of such profile shows sigmoidal behavior (inset of Figure 4(b)). Due to hyperbolic nature, the entire range of data of Figure 4 could be analyzed according to the Michaelis-Menten Equation with Vmax
 (a)
(a)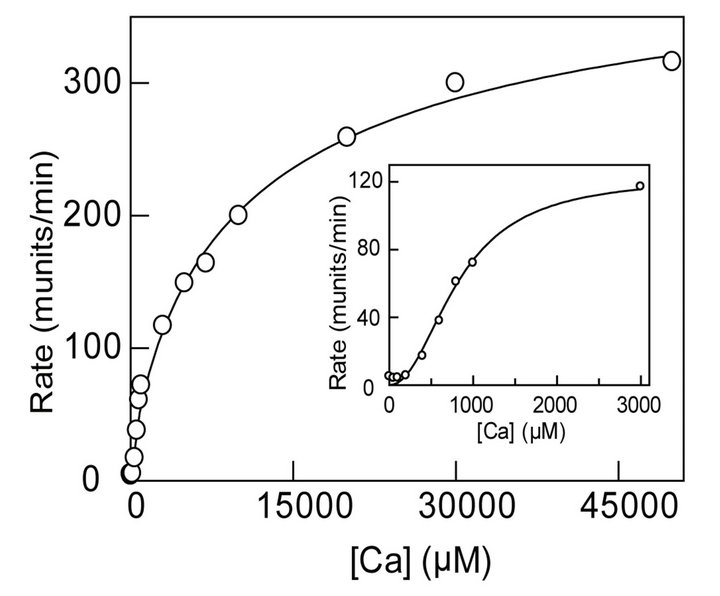 (b)
(b)
Figure 4. Calcium-induced changes in the steady-state fluorescence and activity of MMP- 9. (a): Binding profile for the interaction of calcium ions with MMP-9. The decrease in the fluorescence emission intensity of protein at 343.5 nm (lex = 295 nm) upon titration of a fixed concentration of the MMP-9 (16 µM) as a function of the total concentration of calcium ions is shown. The solid smooth line is the best fit of the data according to Hill equation with K0.5 and nH (Hill Coefficient) values as being equal to 6.7 mM and 5.7, respectively; (b): Effect of calcium ions on MMP-9 catalyzed reaction. An increase in rate of 0.5 µM MMP-9 reaction is shown. The experiments were performed in 50 mM Tris-HCl, 100 mM NaCl buffer at pH 7.5. The solid smooth line is the best fit of the data according to MichaelisMenten Equation with Vmax and Km values of 352 munits/min and 6.6 mM, respectively. The inset shows the sigmoidal profile of Ca2+ - dependent activation of the enzyme, at lower calcium ion concentrations.
and Km values of 352 munits/min and 6.6 mM, respectively. Although we could not reliably analyze the trun cated (sigmoidal) region of the Ca2+ dependent activation data (inset of Figure 4(b)) due to uncertainty in the extrapolated Vmax value, a rough estimate of the Hill Coefficient (nH) value emerged out to be in the range of 1.4 - 1.8. The number of Ca2+ moieties responsible for cooperatively producing fluorescence changes (cooperatively) in the enzyme’s tryptophan residues appears to be different than those involved in the (cooperative) activation of the enzyme. We believe the origin of such discrepancy lies in the differential avidity of Ca2+ in influencing the fluorescence versus activity changes in MMP-9. Irrespectively, both processes being cooperative are suggestive of their common molecular origin.
3.3. Steady-State Fluorescence Quenching and Stern-Volmer Relationship
Given the magnitudes of t0 and kq values, derived from the acrylamide concentration dependent fluorescence lifetime data (Figure 2(b) and Figure 3(b)), we could predict the intrinsic Stern-Volmer constants (Ksv = t0 × kq) for the two classes (viz., exposed and buried) of tryptophan residues of MMP-9 in the absence and presence of Ca2+. To ascertain whether the predicted Ksv values can reliably explain the steady-state fluorescence quenching data, we performed the latter studies in the presence of increasing concentrations of acrylamide in the absence and presence of Ca2+. Towards this goal, we measured the steady-state fluorescence of MMP-9 (lex =295 nm, lem = 335 nm) in the presence of increasing concentrations of acrylamide. Figure 5 shows the SternVolmer plot (F0/F versus [Acrylamide]) for the quenching of the enzyme’s tryptophan fluorescence (F0 and F are representative of fluorescence intensity in the absence and presence of acrylamide, respectively). Since our lifetime data conformed to the two classes of the tryptophan residues in MMP-9, the data of Figure 5 were analyzed by the modified form of the Stern-Volmer relationship ((Eq. 4); [40-42]).
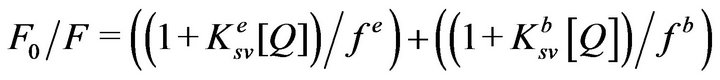 (4)
(4)
where fe and fb refer to the fractions of exposed and buried tryptophan residues, and Kesv and Kbsv are the corresponding Stern-Volmer constants, respectively. We analyzed the data of Figure 5 by Eq. 4 using the Ksv values derived from the fluorescence lifetime data of Figure 2. The solid smooth line is the best fit of the experimental data for the magnitudes of fe and fb values as being equal to 3.9 and 1.1, respectively. In accord with our working model, the above analysis led to the suggestion that about 78% of the MMP-9 resident tryptophan residues are exposed and the remaining 22% are buried within the interior core of MMP-9. These values attest to our initial
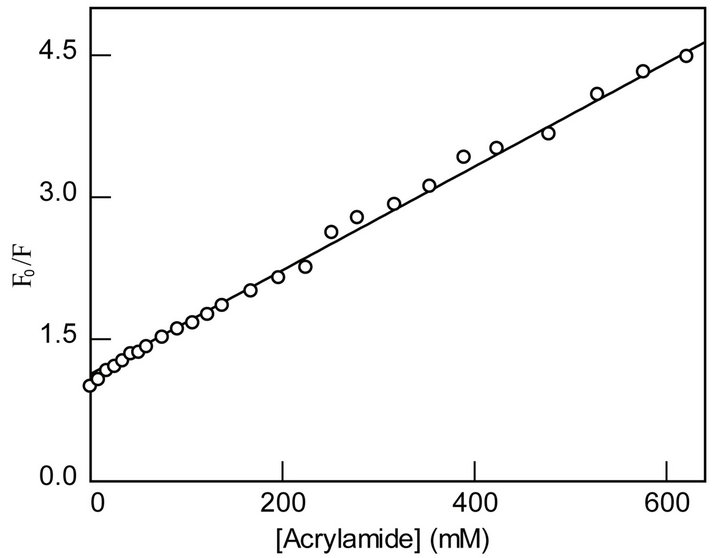
Figure 5. Stern-Volmer plot for the acrylamide quenching of the tryptophan residues of MMP-9. The relative quenching (F0/F) of the steady-fluorescence of the enzyme’s tryptophan residues (lex = 295 nm, lem = 335 nm) as a function of the acrylamide concentration (open circle) is shown. The solid line is the best fit of the data according to Eq. 4, by fixing the magnitudes of Ksv’s (derived from the data of Figure 2(b)) and allowing to vary the amplitude parameters, fe and fb. The latter parameters were discerned to be 3.9 ± 0.20 and 1.1 ± 0.02, respectively.
assignment (based on the amplitudes of the lifetime traces of Figure 2(a)) that the relative distribution of the surface exposed and buried tryptophan residues in MMP- 9 is 83% and 17%, respectively. We must emphasize that our approach of deriving the Ksv values from the lifetime data, and fixing them while analyzing the steady-state data of Figure 5 by Eq. 4 reliably yielded the relative distribution of the surface exposed and buried tryptophan residues in MMP-9. However, an interesting aspect of the above analytical outcome has been the apparent lack of the “static” contribution during the acrylamide quenching of MMP-9 in the absence of Ca2+. This is clearly not the case in the presence of Ca2+.
To ascertain the contribution of Ca2+ on the acrylamide dependent quenching of the steady-state fluorescence of MMP-9, we performed a similar experiment as described above. As shown in Figure 6, the acrylamide dependent quenching of the MMP-9 fluorescence in the presence of 10 mM Ca2+ shows an upward curvature in the SternVolmer plot. To our surprise, such curvature emerged only when we used Ca2+ in the enzyme preparation, and there was no sign of such curvature in the absence of Ca2+. Clearly, Ca2+ modulates the overall microscopic pathway for the acrylamide assisted quenching of MMP-9. When we attempted to analyze the data of Figure 6 by Eq. 4 (by fixing the magnitudes of Ksv’s, derived from the lifetime data of Figure 3(b)), there was a marked viation between the experimental data and the fitted line(see dotted line of Figure 6). The upward curvature in Stern-Volmer plot has been conceived to originate due to involvement of the “static” (i.e., aside from the dy-

Figure 6. Influence of Ca2+ on the Stern-Volmer plot for the quenching of the tryptophan residues of MMP-9 by acrylamide. The experimental conditions were the same as in Figure 5, except for the presence of 10 mM Ca2+ in the enzyme preparation. The dotted line is least squares fit of the data according to Eq. 4 using the fixed Ksv values (obtained from the data of Figure 3(b)). Note a marked deviation from the experimental and fitted line. The data was best fitted (by Eq. 5) on the basis that aside from “dynamic” quenching, the presence of Ca2+ induces “static” quenching of the enzyme’s tryptophan residues by acrylamide. The magnitudes of fe, fb and Vb were derived (from the best of fit of the data) as being equal to 3.0 ± 0.73, 1.1 ± 0.09, and 1.2 M−1, respectively.
namic) component in overall quenching pathway [40,41]. Hence, the recourse was made to analyze the SternVolmer plot of Figure 6 by Eq. 5 [40-42].
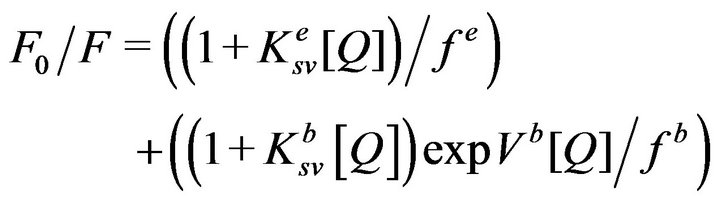 (5)
(5)
where Vb is the static quenching constant of the buried residues. The solid line of Figure 6 is the best fit of the data (while fixing the magnitudes Ksv’s) by Eq. 5 for the magnitudes of fe, fb, and Vb values as being equal to 3.0, 1.1, and 1.2 M−1 respectively. Note a marked correspondence between the experimental data and the fitted (solid) line. These parameters along with those derived from the data of Figures 2, 3, and 5 are summarized in Table 1. A cumulative account of the data of Table 1 lead to the suggestion that aside from promoting the “static” quenching of the enzyme’s tryptophan residues, Ca2+ also alters the fractional accessibility of both exposed versus buried tryptophan residues to acrylamide. We believe the Ca2+- mediated “static” quenching arises from a weaker inter action of acrylamide with MMP-9. The latter feature is evident from the fact that acrylamide serves as a competitive inhibitor of the enzyme with a ki value of 589 mM (data not shown).

Table 1. Acrylamide-dependent quenching of tryptophan fluorescence of MMP-9.
4. DISCUSSION
This paper provides a detailed account of the acrylamide assisted quenching of tryptophan fluorescence in MMP-9 in the absence and presence of Ca2+. Both excited state fluorescence decay profile of the enzyme’s tryptophan residues and the acrylamide quenching data are consistent with the fact that MMP-9 harbors two classes of tryptophan residues, which can be formalistically assigned as the “surface exposed” and “buried” residues. Of eight tryptophan residues in the coding sequence of our construct [27], four of them are present in the catalytic domain and the remaining four are present in the fibronectin domain. The structural data of catalytic domain reveal that whereas three tryptophan residues (W116, W124, and W148) are partially to fully surface exposed, one tryptophan residue (W210) is buried to the interior core of the MMP-9 structure. Of four tryptophan residues present in the fibronectin domain (see Figure 1), two (W255 and W313) are exposed and the other two residues (W372 and W385) appear to be buried in the protein structure [27].
The fact that the short (ts) and long (tl) tryptophan lifetimes of MMP-9 decrease upon increasing concentration of acrylamide suggests that both surface exposed and buried tryptophan residues are quenched. However, the effectiveness of quenching (as judged from the derived bimolecular quenching constant; kq) is about 4-fold more pronounced for the surface exposed tryptophan residues than those buried in the protein mass. This is not surprising on consideration that although buried tryptophan residues are also accessible to acrylamide, the overall process has somewhat higher energy barrier than those for the exposed residues. This is presumably due to the impaired flexibility (on a dynamic scale) of the buried amino acid (e.g., tryptophan) residues at the interior of the protein core.
The fact that the Ksv values derived from the lifetime data (Figure 2(b)) could be used to reliably simulate the steady-state fluorescence data of the Stern-Volmer plot of Figure 5 attests to the internal consistency of our quenching results in the absence of Ca2+. Our strategy also allowed us to determine the fractional contribution of the surface exposed versus buried tryptophan residues in MMP-9 from the Stern-Volmer plot, and the derived parameters (fe versus fb) are similar to the relative amplitudes of the lifetime traces. It should be emphasized that the fractional accessibility of two classes of tryptophan residues to acrylamide quenching would have remained obscured if we had solely relied on the steady-state fluorescence data in analyzing the Stern-Volmer plot.
One of interesting aspect of our observation is the marked difference in the Stern-Volmer plots in the absence (Figure 5) and presence (Figure 6) of Ca2+. Since the Stern-Volmer plot in the absence of Ca2+ is nearly linear and it is devoid of any upward curvature (contrary to that observed in the presence of Ca2+), it follows that the overall acrylamide assisted tryptophan quenching of MMP-9 is “dynamically” controlled. In other words, acrylamide depopulates the excited states of the tryptophan species primarily via collisional encounters that via a diffusion limited pathway [40,41] and less due to binding in the vicinity of the tryptophan residues if it does or exhibiting apparent saturation due to inherent kinetic complexity as described by Eftink and Ghiron [40,41]. Aside from these, the differential quenching of the surface exposed versus buried tryptophan residues of MMP- 9 by acrylamide is further evident from 6 nm blue shift in the fluorescence emission spectrum of the enzyme in the presence of 0.5 M acrylamide (Figure 2(c)). As found with other proteins [48], the acrylamide-induced blue shift in the fluorescence emission spectrum of MMP-9 is likely to be caused by the preferential quenching of the surface exposed tryptophan residues such that the overall emission spectrum is dominated by the buried (blue shifted) tryptophan residues.
Our experimental data reveal that Ca2+ ions exhibit different levels of effect on the tryptophan fluorescence properties of MMP-9. 1). The binding of Ca2+ to MMP-9 changes the equilibrium distribution between the surface exposed and buried tryptophan residues to the latter. Both fluorescence lifetime and Stern-Volmer analysis reveal that an additional 7% - 8% of the surface exposed tryptophan residues, presumably, W116, W124, and W148 from the catalytic domain and W255 and W313 from the fibronectin domain partially move to the interior of the protein core (as buried residues) upon binding of Ca2+ to the enzyme. 2) The binding of Ca2+ impairs the bimolecular quenching rate constants of the surface exposed and buried tryptophan residues by 2- and 3-fold, respectively. 3) Unlike apo (Ca2+-free) MMP-9, the SternVolmer plot shows an upward curvature in the presence of Ca2+, suggesting that aside from the dynamic quenching, the binding of Ca2+ to MMP-9 induces the “static” quenching of the enzyme’s tryptophan residues by acrylamide. Finally, the binding of Ca2+ induces about 3 nm blue shift, with concomitant hypochromicity, in the fluorescence emission spectrum of the tryptophan residues of MMP-9. A cumulative account of all these data lead to the suggestion that Ca2+ modulates the structural as well as dynamic feature of MMP-9, and by doing so, it “structures” the active site cavity of the enzyme and thus facilitates catalysis.
Although acrylamide assisted “static” quenching may proceed without formation of a stable protein-quencher complex [40,41], our preliminary experimental data reveal that acrylamide, in fact, inhibits the MMP-9 catalyzed reaction with a Ki value of 589 mM. Whether or not acrylamide only binds to MMP-9 in the presence of Ca2+ (a required ingredient while measuring the enzymatic reaction) or it also binds to “apo” MMP-9 must await additional studies.
One of the interesting aspects of our experimental outcomes is the sigmoidal dependence of the binding of Ca2+ to MMP-9 and also MMP-9 activity with increasing Ca2+ concentration (Figure 4). A simple minded explanation of such sigmoidal behavior is that the initial binding of Ca2+ enhances its own binding affinity to the enzyme site, and the overall process must be “allosterically” controlled. Of the three putative Ca2+ binding sites in the catalytic domain of MMP-9 [27], presumably Ca-502 (see Figure 1), confined near the active site pocket of the enzyme, might play a major role as an activator. This Ca2+ is clamped to the S-shaped double loop connecting the b3 and b4 strands of the enzyme [27], and it is presumably responsible for “structuring” of the enzyme’s active site pocket toward enhanced catalysis.
It may appear, intuitively, that the binding of Ca2+ to MMP-9 does not markedly alter the dynamic behavior of the enzyme, as the fractional distribution between exposed and buried tryptophan residues is altered only by 7% - 8% and the bimolecular quenching rate constants are impaired only by 2 - 3 fold. In this regard, it is noteworthy that the above changes are not so small from the point of view that only 17% - 20% tryptophan residues are intrinsically buried in the MMP-9 structure. Aside from this, our deduction of the Ca2+-induced changes in MMP-9 structure exclusively relies on the signal associated with the microenvironment of the tryptophan residues, which is only representative of a small fraction of the total residues in the protein structure. In the absence of comparative structural information of MMP-9 (or any other MMPs) in the absence and presence of Ca2+, we cannot reliably predict the magnitude of the metal ion induced “static” deviation of the different residues (including tryptophan) in the protein structure. However, with precedents of the structural features of metalloenzymes involved in the hydrolytic reactions, it is apparent that the binding of metal ions does not significantly alter their cognate protein structures [49,50]. Assuming similar scenario prevails in the case of MMP-9, we are prompted to speculate that the origin of the Ca2+ induced changes in the fluorescence profiles of the enzyme’s tryptophan residues is predominantly “dynamic” in nature. These data also reveal that calcium ions are not merely a structural requirement and have a role in regulating catalytic activity of MMP-9 by structuring the enzyme’s active site. Our future studies will focus on selectively replacing the tryptophan residues with phenylalanine and assessing the influence of Ca2+ ion binding on MMP-9 structure.
5. ACKNOWLEDGEMENTS
The work was supported by the National Institutes of Health grant CA113746. The authors thank Donald Klocke for helping us clone MMP-9.
REFERENCES
- Chou, K.C., Kahari, V.M. and Saarialho-Kere, U. (1997) Matrix metalloproteinases in skin. Experimental Dermatology, 6, 199-213. doi:10.1111/j.1600-0625.1997.tb00164.x PMid:9450622
- Saarialho-Kere, U.K. (1998) Patterns of matrix metalloproteinase and TIMP expression in chronic ulcers. Archives of Dermatological Research, 290, S47-S54. doi:10.1007/PL00007453 PMid:9710383
- Nagase, H. and Woessner Jr., J.F. (1999) Matrix metalloproteinases. Journal of Biological Chemistry, 274, 21491- 21494. doi:10.1074/jbc.274.31.21491 PMid:10419448
- Kerkela, E. and Saarialho-Kere, U. (2003) Matrix metalloproteinases in tumor progression: Focus on basal and squamous cell skin cancer. Experimental Dermatology, 12, 109-125. doi:10.1034/j.1600-0625.2003.120201.x PMid:12702139
- Nelson, A.R., Fingleton, B., Rothenberg, M.L. and Matrisian, L.M. (2000) Matrix metalloproteinases: Biologic activity and clinical implications. Journal of Clinical Oncology, 18, 1135-1149.
- Liotta, L.A., Tryggvason, K., Garbisa, S., Hart, I., Foltz, C.M. and Shafie, S. (1980) Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature, 284, 67-68. doi:10.1038/284067a0 PMid:6243750
- Polette, M., Gilbert, N., Stas, I., Nawrocki, B., Noel, A., Remacle, A., Stetler-Stevenson, W.G., Birembaut, P. and Foidart, M. (1994) Gelatinase A expression and localization in human breast cancers. An in situ hybridization study and immunohistochemical detection using confocal microscopy. Virchows Archives, 424, 641-645. doi:10.1007/BF00195779
- Dalberg, K., Eriksson, E., Enberg, U., Kjellman, M. and Backdahl, M. (2004) Gelatinase A, membrane type 1 matrix metalloproteinase, and extracellular matrix metalloproteinase inducer mRNA expression: correlation with invasive growth of breast cancer. World Journal of Surgery, 24, 334-340. doi:10.1007/s002689910053 PMid:10658069
- Hanemaaijer, R., Verheijen, J.H., Maguire, T.M., Visser, H., Toet, K., McDermott, E., O’Higgins, N. and Duffy, M.J. (2000) Increased gelatinase-A and gelatinase-B activities in malignant vs. benign breast tumors. International Journal of Cancer, 86, 204-207. doi:10.1002/(SICI)1097-0215(20000415)86:2<204::AID-IJC9>3.0.CO;2-6
- Papadopoulou, S., Scorilas, A., Arnogianaki, N., Papapanayiotou, B., Tzimogiani, A., Agnantis, N. and Talieri, M. (2001) Expression of gelatinase-A (MMP-2) in human colon cancer and normal colon mucosa. Tumor Biology, 22, 383-389. doi:10.1159/000050641 PMid:11786732
- Segain, J.P., Harb, J., Gregoire, M., Meflah, K. and Menanteau, J. (1996) Induction of fibroblast gelatinase B expression by direct contact with cell lines derived from primary tumor but not from metastases. Cancer Research, 56, 5506-5512.
- Tokuraku, M., Sato, H., Murakami, S., Okada, Y., Watanabe, Y. and Seiki, M. (1995) Activation of the precursor of gelatinase A/72 kDa type IV collagenase/MMP-2 in lung carcinomas correlates with the expression of membrane-type matrix metalloproteinase (MT-MMP) and with lymph node metastasis. International Journal of Cancer, 64, 355-359. doi:10.1002/ijc.2910640513 PMid:7591310
- Nagawa, H. and Yagihashi, S. (1994) Expression of type IV collagen and its degrading enzymes in squamous cell carcinoma of lung. Japanese Journal of Cancer Research, 85, 934-938. doi:10.1111/j.1349-7006.1994.tb02971.x
- Sehgal, G., Hua, J., Bernhard, E.J., Sehgal, I., Thompson, T.C. and Muschel, R.J. (1998) Requirement for matrix metalloproteinase-9 (gelatinase B) expression in metastasis by murine prostate carcinoma. American Journal of Pathology, 152, 591-596.
- Koshiba, T., Hosotani, R., Wada, M., Fujimoto, K., Lee, J.U., Doi, R., Arii, S. and Imamura, M. (1997) Detection of matrix metalloproteinase activity in human pancreatic cancer. Surgery Today, 27, 302-304. doi:10.1007/BF00941802 PMid:9086544
- Gress, T.M., Muller-Pillasch, F., Lerch, M.M., Friess, H., Buchler, M. and Adler, G. (1995) Expression and in-situ localization of genes coding for extracellular matrix proteins and extracellular matrix degrading proteases in pancreatic cancer. International Journal of Cancer, 62, 407- 413. doi:10.1002/ijc.2910620409 PMid:7635566
- Young, T.N., Rodriguez, G.C., Rinehart, A.R., Bast Jr., R.C., Pizzo, S.V. and Stack, M.S. (1996) Characterization of gelatinases linked to extracellular matrix invasion in ovarian adenocarcinoma: Purification of matrix metalloproteinase 2. Gynecologic Oncology, 62, 89-99. doi:10.1006/gyno.1996.0195 PMid:8690299
- Hsieh, M.S., Ho, H.C., Chou, D.T., Pan, S., Liang, Y.C., Hsieh, T.Y., Lan, J.L. and Tsai, S.H. (2003) Expression of matrix metalloproteinase-9 (gelatinase B) in gouty arthritis and stimulation of MMP-9 by urate crystals in macrophages. Journal of Cellular Biochemistry, 89, 791-799. doi:10.1002/jcb.10530 PMid:12858344
- Pirila, E., Ramamurthy, N.S., Sorsa, T., Salo, T., Hietanen, J. and Maisi, P. (2003) Gelatinase A (MMP-2), collagenase-2 (MMP-8), and laminin-5 gamma2-chain expression in murine inflammatory bowel disease (ulcerative colitis). Digestive Diseases Sciences, 48, 93-98. doi:10.1023/A:1021790532723
- Pyo, R., Lee, J.K., Shipley, J.M., Curci, J.A., Mao, D., Ziporin, S.J., Ennis, T.L., Shapiro, S.D., Senior, R.M. and Thompson, R.W. (2000) Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. Journal of Clinical Investigation, 105, 1641-1649. doi:10.1172/JCI8931
- Kossakowska, A.E., Medlicott, S.A., Edwards, D.R., Guyn, L., Stabbler, A.L., Sutherland, L.R. and Urbanski, S.J. (1999) Elevated plasma gelatinase A (MMP-2) activity is associated with quiescent Crohn’s Disease. Annals of the New York Academy of Sciences, 878, 578-580. doi:10.1111/j.1749-6632.1999.tb07732.x PMid:10415778
- Kee, C., Son, S. and Ahn, B. (1999) The relationship between gelatinase A activity in aqueous humor and glaucoma. Journal of Glaucoma, 8, 51-55. doi:10.1097/00061198-199902000-00011
- Fisher, G.J. and Kang, S. (2002) Phototherapy for scleroderma: Biologic rationale, results, and promise. Current Opinion in Rheumatology, 14, 723-726. doi:10.1097/00002281-200211000-00016
- Overall, C.M. and Lopez-Otin, C. (2002) Strategies for MMP inhibition in cancer: Innovations for the post-trial era. Nature Reviews Cancer, 2, 657-672. doi:10.1038/nrc884
- Bode, W., Fernandez-Catalan, C., Tschesche, H., Grams, F., Nagase, H. and Maskos, K. (1999) Structural properties of matrix metalloproteinases. Cellular and Molecular Life Sciences, 55, 639-652. doi:10.1007/s000180050320
- Rowsell, S., Hawtin, P., Minshull, C.A., Jepson, H., Brockbank, S.M., Barratt, D.G., Slater, A.M., McPheat, W.L., Waterson, D., Henney, A.M. and Pauptit, R.A. (2002) Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. Journal of Molecular Biology, 319, 173-181. doi:10.1016/S0022-2836(02)00262-0
- Elkins, P.A., Ho, Y.S., Smith, W.W., Janson, C.A., D’Alessio, K.J., McQueney, M.S., Cummings, M.D. and Romanic, A.M. (2002) Structure of the C-terminally truncated human ProMMP9, a gelatin-binding matrix metalloproteinase. Acta Crystallographica Section D, 58, 1182- 1192. doi:10.1107/S0907444902007849
- Tochowicz, A., Maskos, K., Huber, R., Oltenfreiter, R., Dive, V., Yiotakis, A., Zanda, M., Pourmotabbed, T., Bode, W. and Goettig, P. (2007) Crystal structures of MMP-9 complexes with five inhibitors: Contribution of the flexible Arg424 side-chain to selectivity. Journal of Molecular Biology, 371, 989-1006. doi:10.1016/j.jmb.2007.05.068
- Zhang, Y., Dean, W.L. and Gray, R.D. (1997) Cooperative binding of Ca2+ to human interstitial collagenase assessed by circular dichroism, fluorescence, and catalytic activity. Journal of Molecular Biology, 272, 1444-1447.
- Lowry, C.L., McGeehan, G. and LeVine, H. (1992) Metal ion stabilization of the conformation of a recombinant 19-kDa catalytic fragment of human fibroblast collagenase. Proteins, 12, 42-48. doi:10.1002/prot.340120106
- Housley, T.J., Baumann, A.P., Braun, I.D., Davis, G., Seperack, P.K. and Wilhelm, S.M. (1993) Recombinant Chinese hamster ovary cell matrix metalloprotease-3 (MMP-3, stromelysin-1). Role of calcium in promatrix metalloprotease-3 (pro-MMP-3, prostromelysin-1) activation and thermostability of the low mass catalytic domain of MMP-3. Journal of Biological Chemistry, 268, 4481-4487.
- Lee, S., Park, H.I. and Sang, Q.X. (2007) Calcium regulates tertiary structure and enzymatic activity of human endometase/matrilysin-2 and its role in promoting human breast cancer cell invasion. Biochemical Journal, 403, 31-42. doi:10.1042/BJ20061390
- Gossas, T. and Danielson, U.H. (2006) Characterization of Ca2+ interactions with matrix metallopeptidase-12: Implications for matrix metallopeptidase regulation. Biochemical Journal, 398, 393-398. doi:10.1042/BJ20051933
- Wetmore, D.R. and Hardman, K.D. (1996) Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry, 35, 6549-6558. doi:10.1021/bi9530752
- Diaz, N. and Suarez, D. (2007) Molecular dynamics simulations of matrix metalloproteinase 2: Role of the structural metal ions. Biochemistry, 46, 8943-8952. doi:10.1021/bi700541p
- Munshi, H.G., Wu, Y.I., Ariztia, E.V. and Stack, M.S. (2002) Calcium regulation of matrix metalloproteinasemediated migration in oral squamous cell carcinoma cells. Journal of Biological Chemistry, 277, 41480- 41488. doi:10.1074/jbc.M207695200
- Winberg, J.O., Berg, E., Kolset, S.O. and Uhlin-Hansen, L. (2003) Calcium-induced activation and truncation of promatrix metalloproteinase-9 linked to the core protein of chondroitin sulfate proteoglycans. European Journal of Biochemistry, 270, 3996-4007. doi:10.1046/j.1432-1033.2003.03788.x
- Cheng, D., Shen, Q., Nan, F., Qian, Z. and Ye, Q.Z. (2003) Purification and characterization of catalytic domains of gelatinase A with or without fibronectin insert for highthroughput inhibitor screening. Protein Expression and Purification, 27, 63-74. doi:10.1016/S1046-5928(02)00530-2
- Hammes-Schiffer, S. (2006) Hydrogen tunneling and protein motion in enzyme reactions. Accounts of Chemical Research, 39, 93-100. doi:10.1021/ar040199a
- Eftink, M.R. and Ghiron, C.A. (1976) Exposure of tryptophanyl residues in proteins. Quantitative determination by fluorescence quenching studies. Biochemistry, 15, 672-680. doi:10.1021/bi00648a035
- Eftink, M.R. and Ghiron, C.A. (1981) Fluorescence quenching studies with proteins. Analytical Biochemistry, 114, 199-227. doi:10.1016/0003-2697(81)90474-7
- Chang, Y.C. and Ludescher, R.D. (1993) Tryptophan fluorescence quenching in rabbit skeletal myosin rod. Biophysical Chemistry, 48, 49-59. doi:10.1016/0301-4622(93)80041-G
- Chao, P.W. and Chow, C.S. (2007) Monitoring aminoglycoside-induced conformational changes in 16S rRNA through acrylamide quenching. Bioorganic & Medicinal Chemistry, 15, 3825-3831. doi:10.1016/j.bmc.2007.03.025
- Khan, K.K., Mazumdar, S., Modi, S., Sutcliffe, M., Roberts, G.C. and Mitra, S. (1997) Steady-state and picosecond-time-resolved fluorescence studies on the recombinant heme domain of Bacillus megaterium cytochrome P-450. European Journal of Biochemistry, 244, 361-370. doi:10.1111/j.1432-1033.1997.00361.x
- Stack, M.S., Itoh, Y., Young, T.N. and Nagase, H. (1996) Fluorescence quenching studies of matrix metalloproteinases (MMPs): Evidence for structural rearrangement of the proMMP-2/TIMP-2 complex upon mercurial activation. Archives of Biochemistry and Biophysics, 333, 163-169. doi:10.1006/abbi.1996.0377
- Sambrook, J., Fritsch, E.F. and Maniatis, T. (2000) Molecular cloning: A laboratory manual. Cold Spring Harbor Press, New York.
- Szabo, J. and Rayner, D. (1980) Fluorescence decay of tryptophan conformers in aqueous solution. Journal of American Chemical Society, 102, 554-563. doi:10.1021/ja00522a020
- Stobiecka, A. (2005) Acrylamide-quenching of Rhizomucor miehei lipase. Journal of Photochemistry and Photobiology B: Biology, 80, 9-18. doi:10.1016/j.jphotobiol.2005.02.002
- Becker, A., Schlichting, I., Kabsch, W., Groche, D., Schultz, S. and Wagner, A.F. (1998) Iron center, substrate recognition and mechanism of peptide deformylase. Nature Structural & Molecular Biology, 5, 1053-1058. doi:10.1038/4162
- Dardel, F., Ragusa, S., Lazennec, C., Blanquet, S. and Meinnel, T. (1998) Solution structure of nickel-peptide deformylase. Journal of Molecular Biology, 280, 501-513. doi:10.1006/jmbi.1998.1882