Advances in Enzyme Research
Vol.1 No.1(2013), Article ID:29397,15 pages DOI:10.4236/aer.2013.11001
Dexamethasone treatment alters kinetics properties of liver mitochondrial F0.F1-ATPase and membrane lipid profiles in developing and adult rats
![]()
1Department of Biochemistry, Faculty of Science, the Maharaja Sayajirao University of Baroda, Vadodara, India; *Corresponding Author: Jignesh.Pandya@uky.edu 2Spinal Cord and Brain Injury Research Center (SCoBIRC), University of Kentucky, Lexington, USA
Received 27 January 2013; revised 1 March 2013; accepted 9 March 2013
Keywords: Dexamethasone; Development; Glucocorticoids; Liver Mitochondria; F0.F1-ATPase; Substrate and Temperature Kinetics; Lipid and Phospholipid Profiles
ABSTRACT
Dexamethasone—a potent synthetic glucocorticoid—has multiple diagnostic and therapeutic applications in wide range of age groups. However, the side-effects of dexamethasone (Dex) treatment including those on development are becoming increasingly apparent. Since the developmental processes are energy-dependent, we examined the effects of chronic Dex treatment on kinetics properties of liver mitochondrial F0.F1-ATPase and mitochondrial membrane lipid profiles in rats belonging to different developmental age groups (2, 3, 4 and 5 weeks) and in adults (~8 weeks). The animals were treated with a subcutaneous dose of 2 mg of Dex/kg body weight (or saline as vehicle) for three alternative days (at around 7.00 A.M.) prior to the day of sacrifice. Dex treatment resulted in significant reduction in F0.F1-ATPase activity in developmental age groups and in adults as compared to their age-matched vehicle-treated control group. The substrate kinetics analysis of F0.F1-ATPase resolved Km and Vmax values in 3 components in all the control age groups; whereas Dex treatment significantly altered the Km and Vmax values or abolished the entire components in age-specific manner. Dex treatment significantly lowered the energy of activation and altered phase transition temperature (Tt˚C) in all the developmental age groups and in adults. Dex treatment significantly increased the contents of total phospholipid (TPL), individual phospholipids classes and cholesterol (CHL) in all the developmental age groups whereas opposite pattern was observed in adults. The mitochondrial membrane became more fluidized in the developing age groups (2, 4 and 5 weeks); whereas no change was observed in 3-week and adult groups following Dex treatment. In present study, our data demonstrate comprehensive deleterious effects of chronic Dex treatment on liver mitochondrial membrane structure and F0.F1-ATPase functional properties with respect to energy metabolism. At the same time, our data also warns against excessive repeated use of antenatal DEX in treatments in growing and adult human patients.
1. INTRODUCTION
Dexamethasone (Dex) is considered as a drug of choice for diagnosis and treatment of a wide range of pathological conditions in all the age groups in humans [1,2]. It is a potent synthetic anti-inflammatory agent with higher glucocorticoid activity and negligible mineralocorticoid activity compared to the natural glucocorticoid cortisol. Due to its distinct anti-inflammatory properties, it is being used for the treatment of wide range of diseases. These include skin diseases; bronchial asthma; chronic lung disease (CLD) in extremely low birth weight (ELBW) and very low weight (VLBW) infants; bronchopulmonary dysplasia (BPD); metastasis of brain, spine and meninges, respiratory distress syndrome (RDS) in fetuses and rheumatoid arthritis [1,3-8]. Glucocorticoids like dexamethasone and betamethasone are choice drugs of clinicians to prevent lung diseases in immature newborns. Multiple Dex courses are prescribed in 98% of cases of pregnant women who are thought to be at the risk of pre-term delivery [2,9,10]. However, more clinical trials are required to decide right formulation and dosage [4].
The synthetic glucocorticoids are believed to have lesser or no side-effects or toxicity [1]. It also remains a critical issue if the glucocorticoids were not being used in proper dose, routes and formulations [10,11]. The use of Dex as potent glucocorticoids as well as an anti-inflammatory agent is recommended by American National Institutes of Health [2]. However, when it is used in pharmacological doses, adverse side-effects are seen in as high as 50% of the cases [12]. The major risks of serious adverse neuro-developmental effects remain after antenatal, pre-natal and post-natal glucocorticoid administration [12-15].
Studies with Dex in isolated mitochondria from brain cortex and liver indicate that under in Vitro conditions Dex induces membrane permeability transition (MPT) pore formation and disrupts calcium homeostasis thereby triggers apoptosis form of cell death [16,17]. Although, the literature survey majorly focuses on adverse effect of Dex on development of brain [18], there are only a few reports which indicate its hepatotoxicity [16]. Observations from our present studies together with finding from a few other laboratories indicate that Dex may have adverse effects on liver function [16]. Our laboratory has demonstrated earlier that repeated exposure of Dex alters oxidative energy metabolism of mitochondria isolated from liver [19].
As it is well recognized the developmental processes are energy-dependent and the mitochondria play major role in ATP synthesis [20]. It is also well recognized that the processes of energy transduction are localized in the inner mitochondrial membrane and its functions are dependent on specific phospholipid classes [21,22]. The effects of treatment with repeated doses of Dex during developmental and adult period on mitochondrial ATP synthesizing ability by F0.F1-ATPase and on the mitochondrial lipid profiles is not well understood. In the present study, we have analyzed the structure-function relations of mitochondria by checking kinetics properties of F0.F1-ATPase and by performing lipid/phospholipid analysis of mitochondrial membrane. It is suggested that the results of our present study may provide possible explanation for energy functions/dysfunctions involved during developmental period and in adult life following chronic exposure to Dex.
2. MATERIALS AND METHODS
2.1. Chemicals
Silica Gel G was purchased from E. Merck, Germany. Dexamethasone phosphate and 1,6 diphenyl-1,3,5 hexatriene (DPH) was purchased from Sigma Chemical Company, St. Louis, MO, USA. ADP was purchased from Fluka, Basel, Switzerland. Bovine serum albumin (BSA) fraction V was purchased from SRL, Mumbai, India. All other chemicals were of analytical-reagent grade and were purchased locally.
2.2. Animals and Treatment with Dex
Male albino rats of Charles-Foster strain belonging to different developmental age groups i.e. 2, 3, 4 and 5 weeks and adults (8 - 10 weeks) were used for this study. The animals were injected with Dex solution subcutaneously (s.c.) at a dose of 2 mg Dex/Kg body weight for three alternative days prior to the day of sacrifice [8, 18-20]. Thus in the 2-week group, the animals received Dex treatment on day 8, 10 and 12; and the animals were sacrificed on day 14. Dex solution were prepared fresh daily in saline and the animals were injected between 6:00 A.M. to 7:00 A.M. when the plasma corticosterone levels in the rat are the lowest and thereby the influence of circadian rhythm for all the age groups was similar [23]. The control groups received equivalent amount of saline vehicle. The experimental design and all the procedural protocols were approved by the Animal Use and Care Committee of the Department of Biochemistry, Faculty of Science, The Maharaja Sayajirao University of Baroda, India.
2.3. Isolation of Mitochondria
The animals were sacrificed by decapitation and their livers were quickly removed and placed in beakers containing chilled (0˚C - 4˚C) isolation medium. The isolation medium contained 0.25 M sucrose, 10 mM Tris-HCl buffer, pH 7.4, 1 mM EDTA and 250 mg BSA/ml. The tissue was minced and was repeatedly washed with the isolation medium to remove adhering blood and 10% (w/v) homogenate was prepared using a Potter Elvehjem type glass-Teflon homogenizer. The nuclei and cell debris were sedimented by centrifugation at 650 × g for 10 min and discarded. The supernatant was subjected to a further centrifugation at 7500 × g for 10 min. The resulting mitochondrial pellet was washed by re-suspending gently in the isolation medium and by resedimenting at 7500 × g for 10 min. Finally the mitochondria were suspended in the isolation medium to give a protein concentration of about 30 - 50 mg/ml [19,24].
2.4. Assay of F0.F1-ATPase Activity
The F0.F1-ATPase activity was measured in the assay medium (total volume 0.4 ml) containing 50 mM TrisHCl buffer pH 7.4, 75 mM KCl and 0.4 mM EDTA. The assays were performed in the presence of 6 mM MgCl2 and 100 mM DNP. After pre-incubating the mitochondrial protein (Ca. 100 mg) in the assay medium at 25˚C and 37˚C, the reaction was initiated by the addition of ATP at a final concentration of 5 mM [25]. The reaction was carried out for 10 min and then terminated by the addition of 0.1 ml of 5% (w/v) sodium dodecyl sulfate (SDS) solution and the amount of liberated inorganic phosphorus was estimated by the method of Fiske and Subba Row [26].
2.5. Substrate and Temperature Kinetics of F0.F1-ATPase
For substrate kinetics studies, concentration of ATP was varied in the range from 0.1 mM to 5 mM with 10 data-points. For temperature kinetics studies, experiments were carried out with fixed ATP concentration of 5 mM and the temperature was varied from 5˚C - 53˚C with an increment of 4˚C with 13 data points [25].
2.6. Data Analysis
The data for substrate kinetics were computer analyzed using three methods: Eadie-Hofstee, LineweaverBurk and Cornish-Bowden plots for determination of Km and Vmax [27,28]. The values of Km and Vmax obtained by the three methods were in close agreement and were averaged for the final presentation of the results. For graphical presentation, only the typical substrate saturation curves and corresponding Eadie-Hofstee plots have been shown in Figures 1 and 2 [25,27,28]. Hill plot analysis was carried out where it was required as shown in Figure 3 [27]. The data on temperature kinetics were analyzed for determination of energies of activation in the high and low temperature ranges (E1 and E2 respectively) and phase transition temperature (Tt˚C) according to the method described previously from the Figures 4 and 5 [25,29]. All the kinetics data were computer analyzed employing Sigma plot version 5.0, Jandel Corporation, San Rafael, California, USA [25,28].
2.7. Extraction and Separation of Phospholipids
Aliquots of mitochondrial suspension containing 4 - 8 mg protein were extracted in a freshly prepared chloroform: methanol mixture (2:1 v/v) as described by Folch et al. [30]. After extraction, suitable aliquots were taken for the estimation of cholesterol [31] and total phospholipid (TPL) [32] and separation of individual phospholipid classes by thin layer chromatography (TLC) using Silica Gel G [25,33,34]. All the spots of separated phospholipids classes were visualized by brief exposure of iodine vapor, marked immediately on the TLC plate, scraped and transferred to marked test tubes. The phosphorus content of the individual phospholipids released by digestion with H2SO4 and was quantified according to the previously published procedure of Bartlett as described earlier [32,33].
2.8. Determination of Mitochondrial Membrane Fluidity
Membrane fluidity determination was carried out at 25˚C spectrophotoflourimetrically using DPH as the probe. Stock DPH solution (2 mM) was prepared in tetrahydrofuran and stored at 0˚C - 4˚C in an amber colored bottle. For measurement of fluorescence polarization, samples were taken in 3 ml of buffered sucrose solution (0.25 M sucrose containing 10 mM Tris-HCl, pH 7.4) at a final protein concentration of 0.2 mg/ml, and an aliquot of stock DPH solution was added so that the molar ratio of probe to lipid was between 1:200 to 1:300 [35,36]. The mixture was vortexed vigorously and left in dark for 30 min to permit equilibration of probe into the membranes. Fluorescence polarization was measured in a Shimadzu RF 5000 spectrophotofluorimeter with a polarizer attachment. Excitation and emission wavelengths were 360 nm and 430 nm; bandwidths were 5 nm and 10 nm respectively. Data were accumulated for 5 sec for each polarization setting: vertical (parallel) and horizontal (perpendicular) [36]. The instrument has program for calculation of fluorescence polarization (p). The details of the methods have been described previously [33,37].
Protein estimation was by the method of Lowry et al. with bovine serum albumin used as the standard [38]. Results are given as mean ± SEM. Statistical evaluation of the data was by students’ t-test.
3. RESULTS
Results of our earlier studies indicated that the substrate oxidation rates and the ATP synthesizing ability of the isolated liver mitochondria were compromised in an age specific manner in the Dex-treated animals [19]. This led us to take one step forward to investigate the specific effect(s) of Dex treatment on kinetic properties of F0.F1-ATPase and membrane lipid profiles in developing and adult animals. In the initial experiments we determined the F0.F1-ATPase activity at 25˚C and at 37˚C and measured the activity ratio (Table 1). As shown in Table 1, Dex treatment significantly decreased the F0.F1-ATPase activity in all the developmental age groups (24% - 76% decrease) when measured at 25˚C and 37˚C. The adult group showed significant decrease in F0.F1-ATPase activity (21% decrease) following Dex treatment when measured at 37˚C; whereas no change in activity was observed at 25˚C. The activity ratio of 37˚C/25˚C displayed significant decrease in 4-week and adult groups (13% - 16% decrease) whereas other age-group showed no significant change in activity after Dex treatment. We further carried out rigorous kinetics analysis of F0.F1-ATPase where we ex amined the substrate and the temperature dependence of the enzyme. The typical substrate saturation plots for the control and
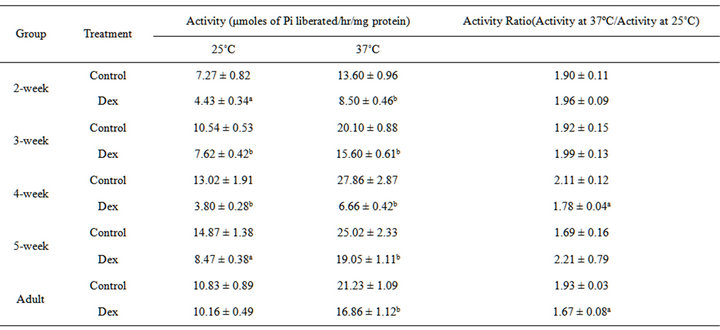
Table 1. Effect of Dex treatment on rat liver mitochondrial F0.F1-ATPase activity.
The results are given as mean ± SEM of 6 - 8 independent experiments. Activity ratio = Activity at 37˚C/Activity at 25˚C. ap < 0.01 and bp < 0.001 compared to the corresponding control.
dex treated animals belonging to all the age groups are shown in Figure 1. As can be noted, practically all the groups followed typical substrate saturation pattern except for the 3-week Dex treated animals where an allosteric pattern of substrate saturation was evident. We then analyzed our data with three different methods as outlined in the Methods Section. However, for presentation only the corresponding typical Eadie-Hofstee plots have been shown. As can be noted, for all the control groups the F0.F1-ATPase activity resolved in 3 components (Figure 2). Similar trend was noted even for Dex treated 2-week and 5-week animals. By contrast, in the 4-week and adult groups the activity resolved only in 2 components; with the third component being completely abolished following Dex treatment (Figure 2). The 3-week Dex group was unique in that it displayed only a single component (Km3 and Vmax3) as shown in Figure 2. We have calculated S0.5 which is equivalent to Km2 for the lost components by Hill plot analysis which is reported in Table 2 for comparison between age-matched 3-week control group.
We then calculated the values of Km and Vmax from the Eadie-Hofstee and Cornish-Bowden plots as described in the Methods Section. These values are shown in Table 2. It is clear that in Dex treated animals of all the age groups, the Vmax values (Vmax1,2,3) decreased significantly (30% - 75% decrease). The Km values of the three components showed mixed pattern. Thus in the 2-week group the Km1 and Km3 values increased while in the 3-week group the Km2 value has increased. For the 4-week and adult groups the Km3 has significantly increased (8% - 70% increase). In contrast, Km values of 3-week (Km3), 4-week (Km1), 5-week (Km3) and adult (Km3) have shown significant decrease (10% - 44% decrease). The Km2 equivalent (S0.5) of 3-week Dex group showed significant increase (64% increase) as compared to 3-week control group. Thus, the drastic changes in both Km and Vmax components of F0.F1-ATPase after Dex treatment may reflect on alteration in its ability to synthesize ATP in these individual groups.
Due to allosteric pattern of the 3-week Dex treated substrate saturation curve (Figure 1), we further analyzed data for this group separately using Hill plot analysis and report the values in Table 3. The typical Hill plot is shown in Figure 3. The Hill co-efficient n1 and n2, and Transition concentration (Tc) are given in Table 3. Since the 3-week Dex group only displayed an allosteric pattern, we did not have compared Hill coefficients and transition concentrations with age-matched control group. The Hill co-efficients (n1 and n2) represents the number of ATP molecules bound at active site of enzyme before or after the transition concentrations (Tc) of ATP (mM) introduced in assay system. It was interesting to note that 0.79 ATP molecules bound (Tc ≤ 0.67 mM ATP) to active site of F0.F1-ATPase. Similarly around 2.39 ATP molecules of ATP bound (Tc ≥ 0.67 mM ATP) to active site of F0.F1-ATPase. This may be the reason to see allosteric kinetics behavior of 3-week Dex substrate kinetics as shown in Figure 1.
We further carried out Arrhenius kinetics to understand the temperature-dependence of the enzyme F0.F1- ATPase. The typical temperature curves for all the groups are shown in Figure 4. To derive quantitative representation of energy of activation and phase transition


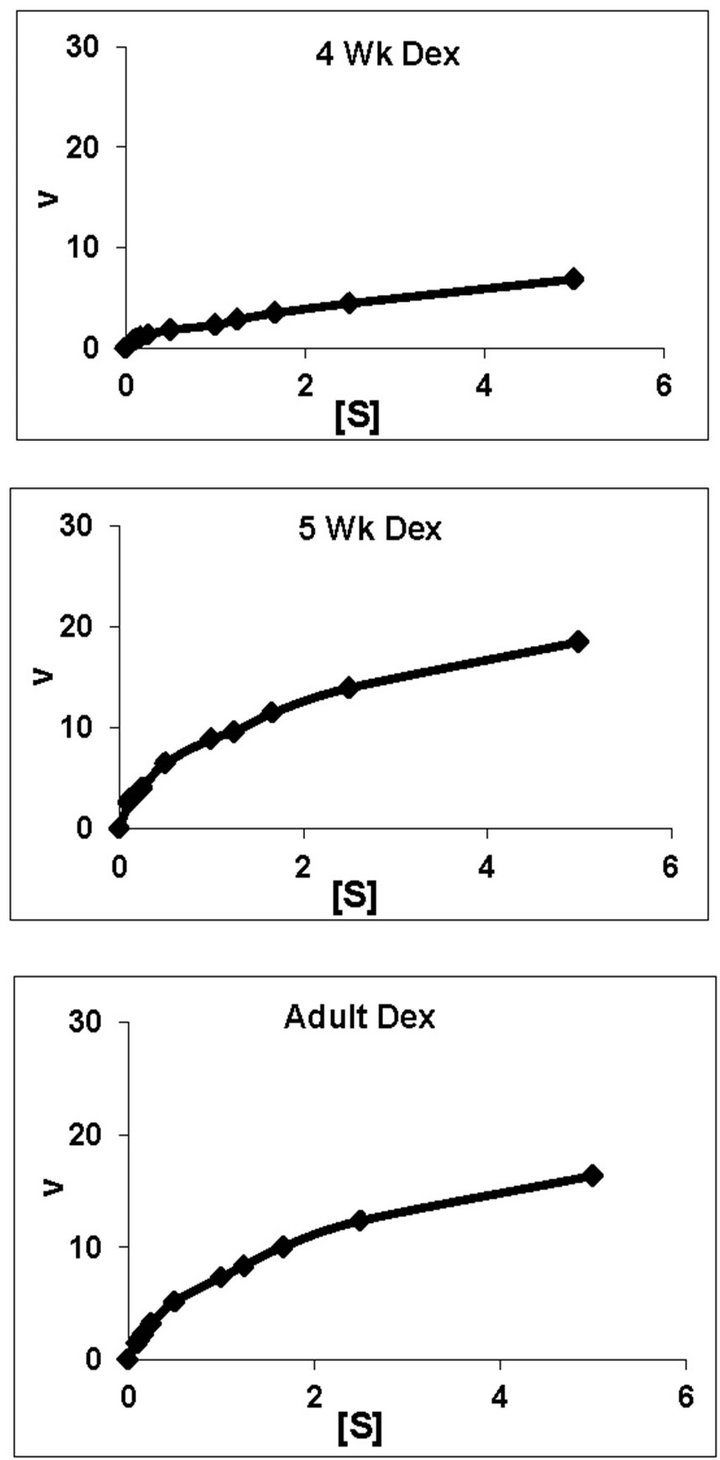
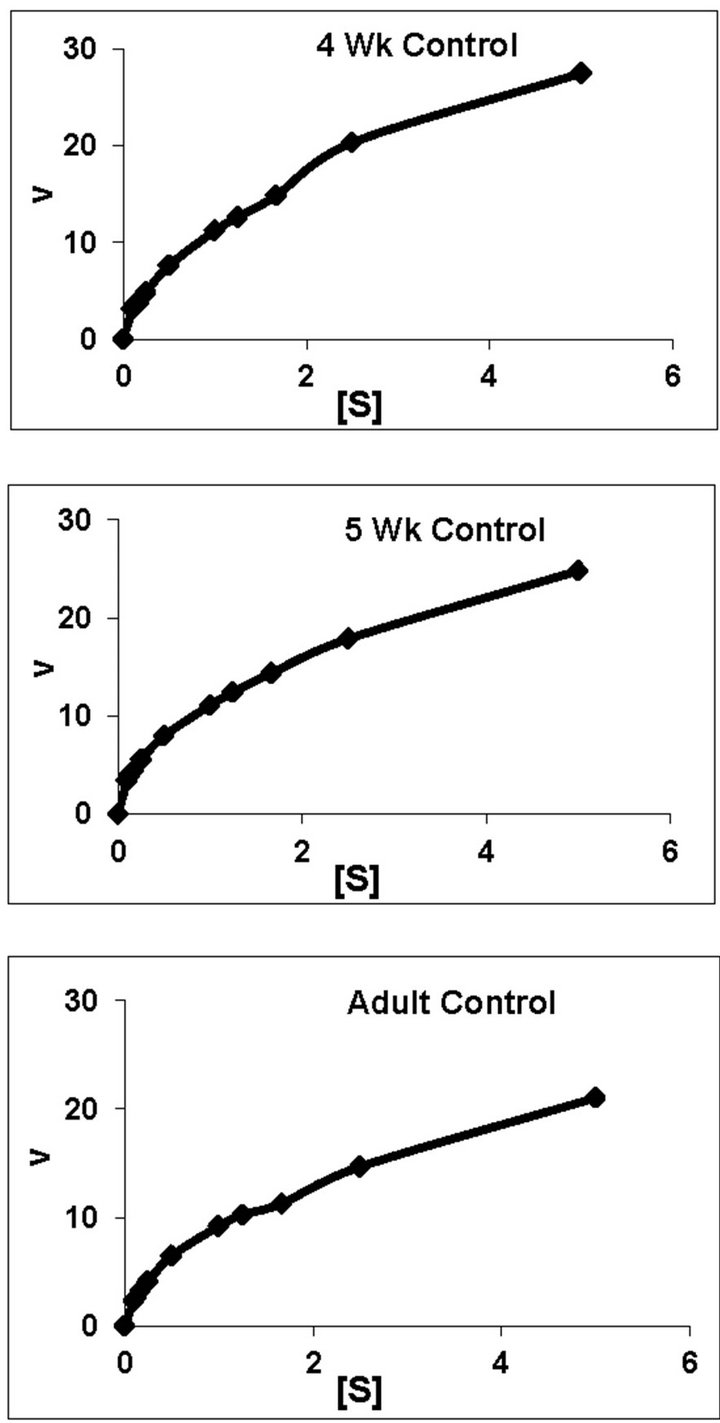
Figure 1. Representative typical substrate saturation curves for rat liver mitochondrial F0.F1-ATPase in controls (Left Panel) and Dex treated (Right Panel) rats for 2-week, 3-week, 4-week, 5-week and adult group respectively. The abscissa represents the reaction velocity v, while the ordinate represents [S]. For substrate kinetics studies, concentration of ATP was varied in the range from 0.1 mM to 5 mM with 10 datapoints. Exceptionally, 3-week Dex group represents typical allosteric sigmoidal curve. The plots are typical for eight independent observations.
temperature, we have analyzed temperature curve data with Arrhenius plots. The typical Arrhenius plots for all the groups are represented in Figure 5 and the quantitative representation of low and high energy of activation (EL and EH) and phase transition temperature (Tt˚C) is given in Table 4. As can be noted, in general, the energy of activation (EL) was significantly lower (14% - 57% decrease) after Dex treatment in all age groups. Similarly, the EH values also decreased significantly (26% - 33% decrease) in 5-week and adult group whereas in other groups it was unaltered. Dex treatment resulted in substantial increase (8˚C increase) in phase transition temperature Tt in the 5-week group; whereas in adults the effect was opposite in nature (5˚C decrease). No change in Tt˚C was observed in early developmental age-groups

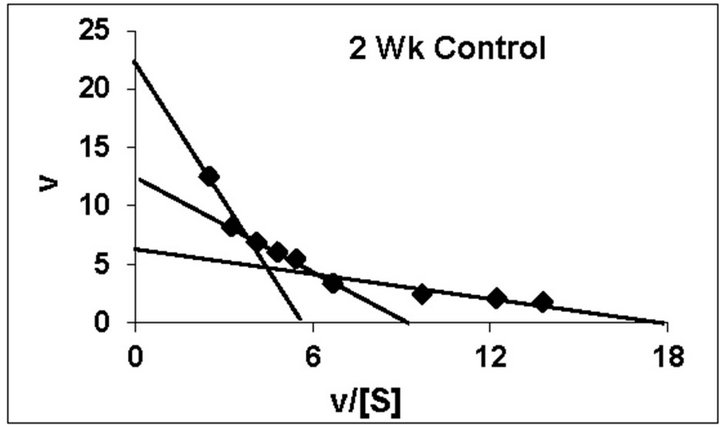
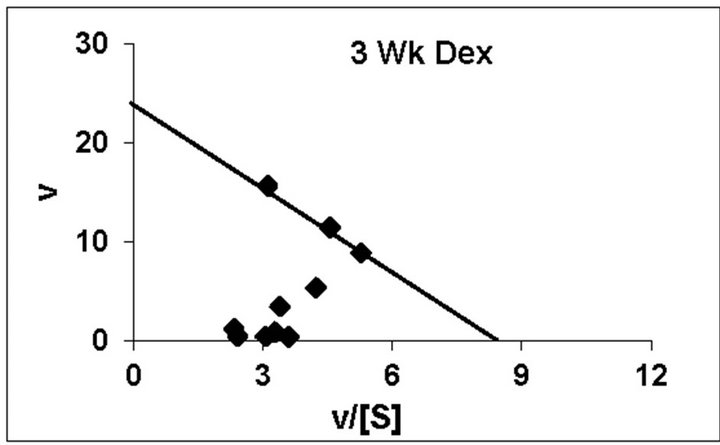
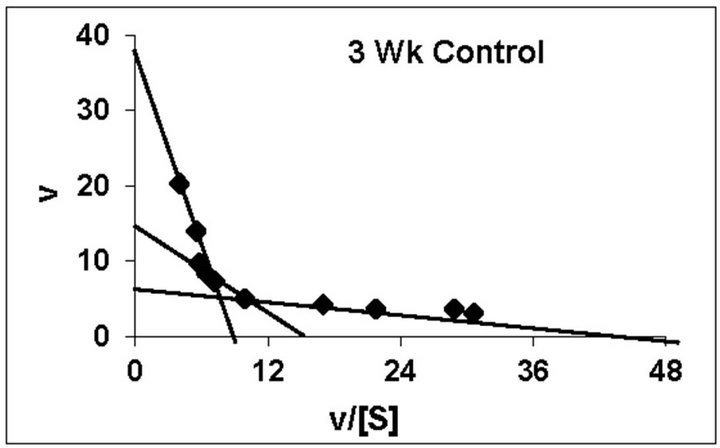
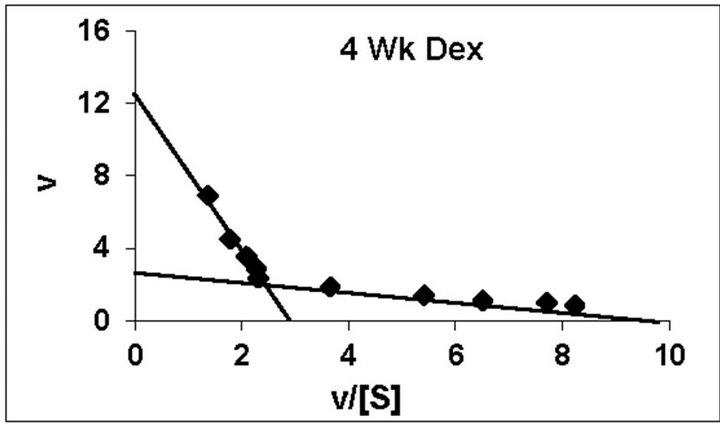

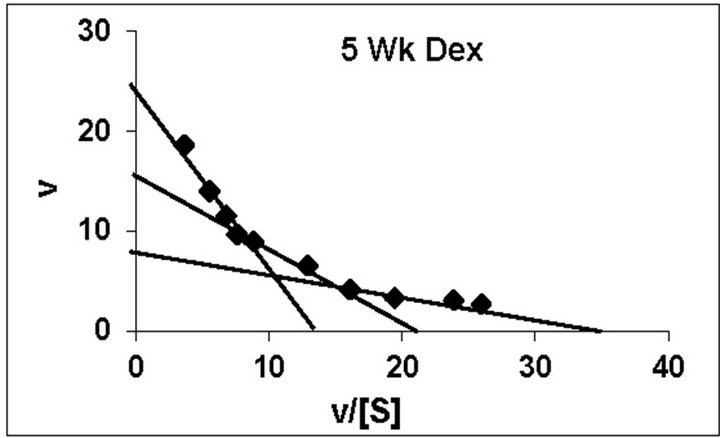

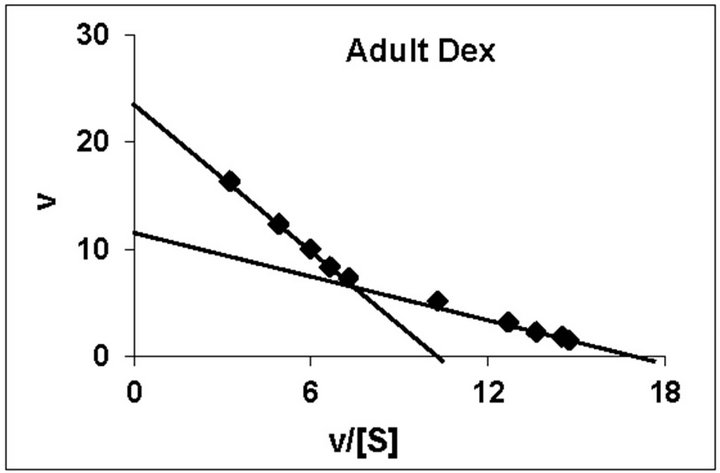
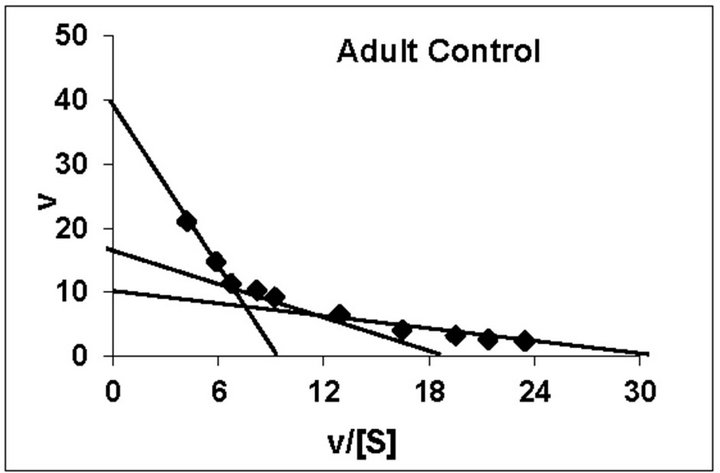
Figure 2. Representative typical Eadie-Hofstee plots for rat liver mitochondrial F0.F1-ATPase in controls (Left Panel) and Dex treated (Right Panel) animals for 2-week, 3-week, 4-week, 5-week and adult group respectively. For the Eadie-Hofstee plots, the abscissa represents the reaction velocity v, while the ordinate represents v/[S]. Reaction velocity v = m mol of Pi liberated hr-1 mg protein-1. V/[S] = reaction velocity divided by the corresponding substrate concentration. Due to typical allosteric sigmoidal curve for 3-week Dex treated group, we were unable to derive Eadie-Hofstee plots in corresponding group. All Eadie-Hofstee plots resolved in either II or III components except for 3-week Dex group. The plots are typical for eight independent observations.
(2-week - 4-week) after Dex treatment.
The present data on substrate and temperature kinetics of F0.F1-ATPase indicate age-specific drastic effects on ATP synthesis. Similarly, earlier studies from our laboratory indicated that chronic Dex treatment significantly alters the liver mitochondrial bioenergetics in growing and adult rats. Moreover, the mitochondrial dehydrogenases along with decreased content of cytochromes further led us to hypothesize that the observed alterations may be attributed to changes in the lipid profiles of mitochondrial membrane following Dex treatment. We therefore took further steps to analyze lipid environment of mitochondrial membrane along with fluidity
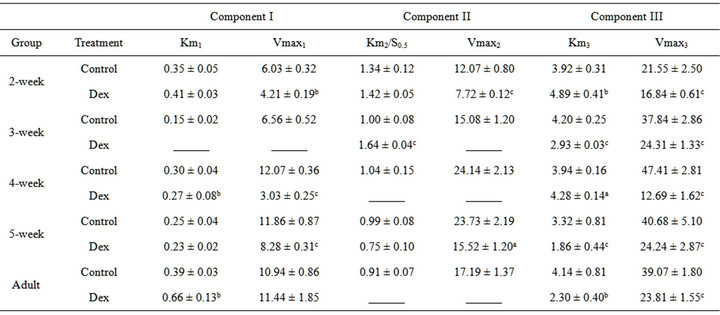
Table 2. Effect of Dex treatment on substrate kinetics properties of rat liver mitochondrial F0.F1-ATPase.
The Km and Vmax values are given as mean ± SEM of the 8 independent observations. ap < 0.05; bp < 0.002 and cp < 0.001 as compared with the corresponding age-matched control group. For the substrate ATP was used in the concentration range of 0.1 to 5 mM. The values of Km, S0.5 are expressed in mM. The Vmax is expressed as µmole of Pi liberated /hr/ mg protein.

Table 3. Effect of Dex treatment on hill plot analysis of rat liver mitochondrial F0.F1-ATPase.
The results are given as mean ± SEM of 6 - 8 independent experiments. The experimental details are as given in the text. ATP was used as substrate in the concentration range of 0.01 to 5 mM. n1 and n2 are Hill co-efficient. The n1 and n2 represent the number of ATP molecules bound to active site over the given transition concentration (Tc) of ATP (mM).
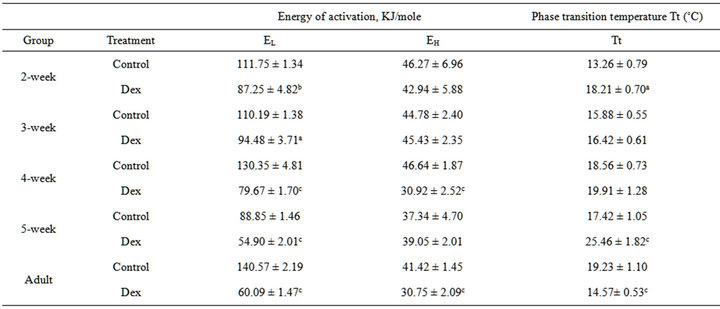
Table 4. Effect of Dex treatment on arrhenius kinetics properties of rat liver mitochondrial F0.F1-ATPase.
The results are given as mean ± SEM of the 8 independent numbers of observations. ap < 0.05, bp < 0.01 and cp < 0.001 compared with the corresponding control.
measurement to understand the effect of Dex treatment on lipid environment in mitochondrial membrane which could directly or indirectly influence the kinetics properties of F0.F1-ATPase.

Figure 3. Typical Hill plot for rat liver mitochondrial F0.F1- ATPase for 3-week Dex treated group. Hill plot analysis were made for sigmoidal curves where the abscissa represents log (v/V-v), while the ordinate represents log [S]. Both of the abscissa and ordinate units were derived from substrate kinetic of F0.F1-ATPase units, v = mmol of Pi liberated hr-1 mg protein-1 and [S0.5] = mM equivalent to Km for Eadie-Hofstee plots. The plot is typical for eight independent observation of 3-week Dex group.
Our lipid profile data in Table 5, indicate that Dex treatment resulted in significant increase in the total phospholipid (TPL) content (42% - 139% increase) in the initial stages of development. Paradoxically, in the adults the TPL content decreased by 66%. Similarly, Dex treatment significantly increased the cholesterol (CHL) content (46% - 186% increase) in growing animals whereas reverse is true with significant decrease (43% decrease) in adults. The TPL and CHL contents directly influenced the molar ratio of TPL/CHL which decreased significantly (35% - 51% decrease) for 3-week to 5- week groups and in the adults whereas 2-week group shown significant increase (50% increase) after Dex exposure. Our data are thus indicative of significant alterations in the phospholipids profiles of mitochondrial membrane. To ascertain further the mitochondrial membrane changes in depth, we additionally measured the mitochondrial membrane fluidity. Our fluidity data indicate that after Dex treatment the mitochondrial membrane became more fluidized as indicated by significant decrease (13% - 17% decrease) in fluorescence polarization (p) values in developing age groups (2-week, 4-week and 5-week); whereas the 3 week and adults have not significantly altered structure of the membrane.
The increased TPL content in growing animals after Dex treatment prompted us to look into the percentage changes in the individual phospholipid class. As shown in Table 6, there were significant changes in the percent lipid composition of membrane. As can be noted in the control groups the major percentage being shared by phosphatidylcholine (PC) (40% - 50%), phosphatidylethanolamine (PE) (30%) and diphosphatidylglycerol (DPG) (7% - 10%) which are the major phospholipids. Thus the changes in PC, PE and DPG composition could confer the highest effect on membrane composition. As
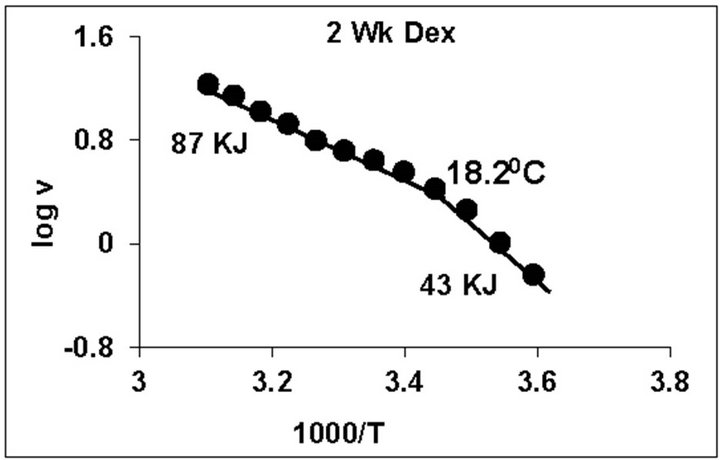
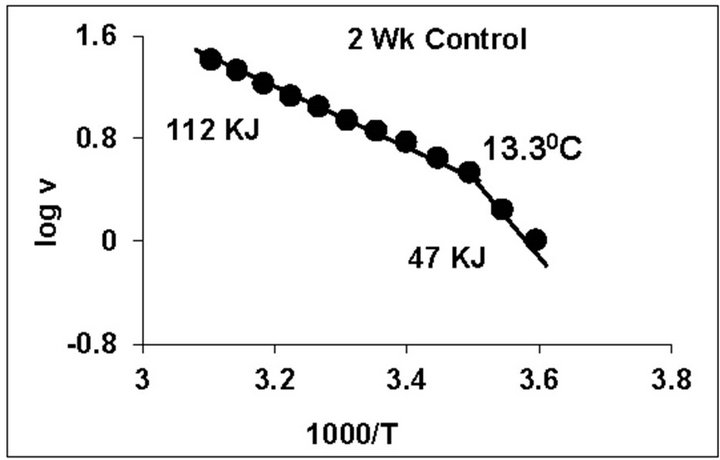
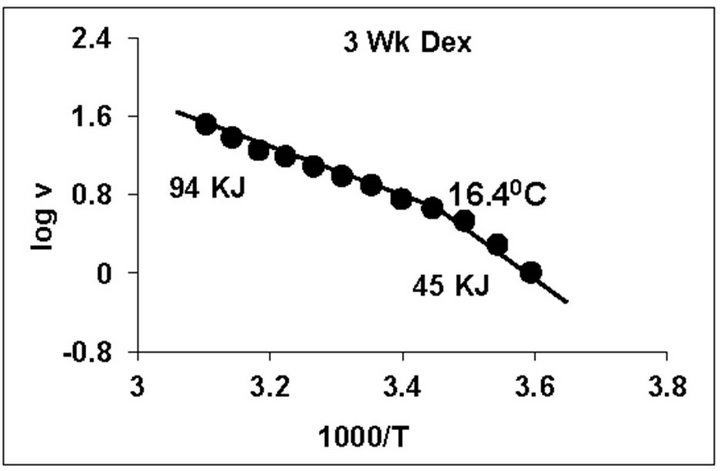
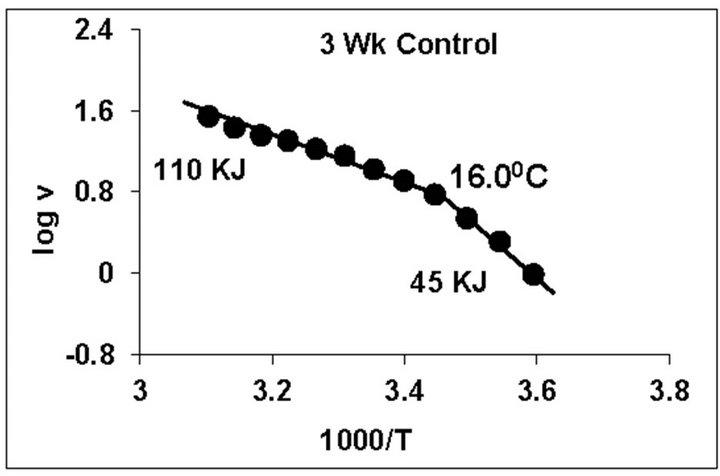

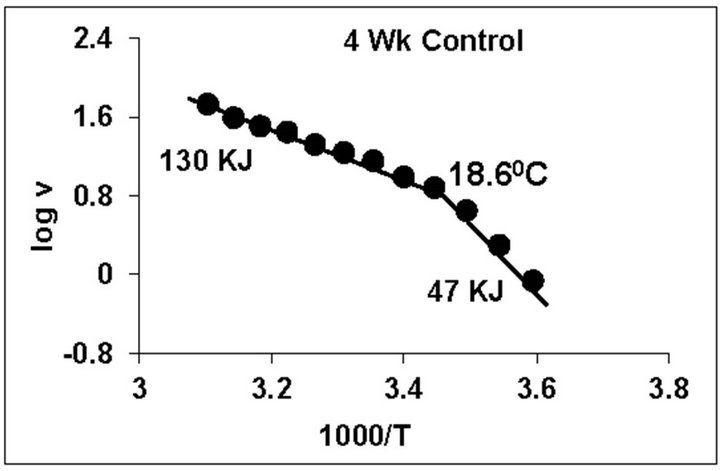
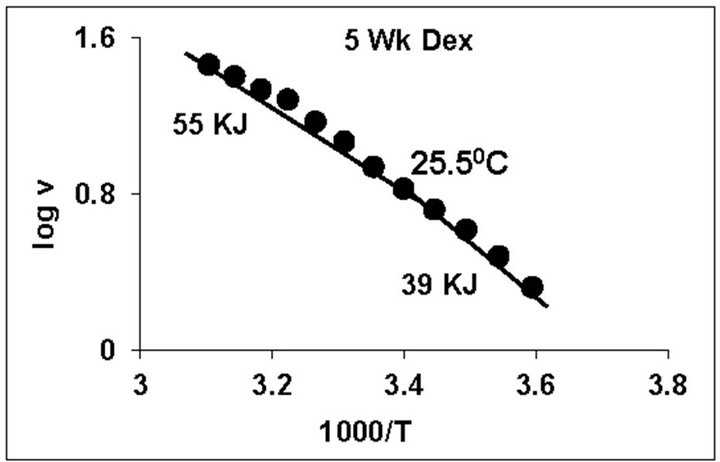
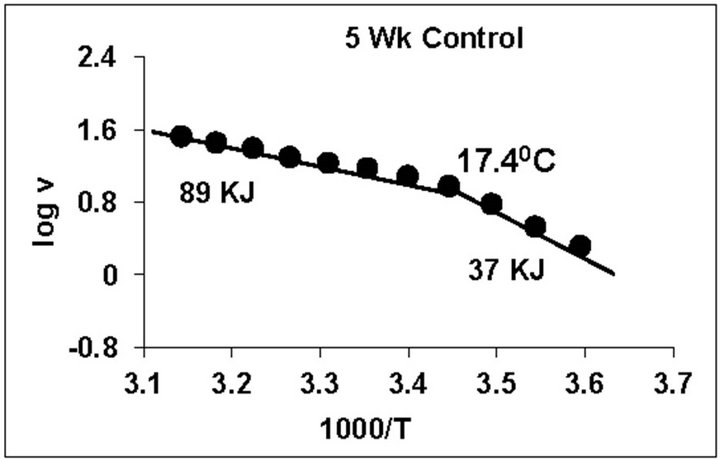
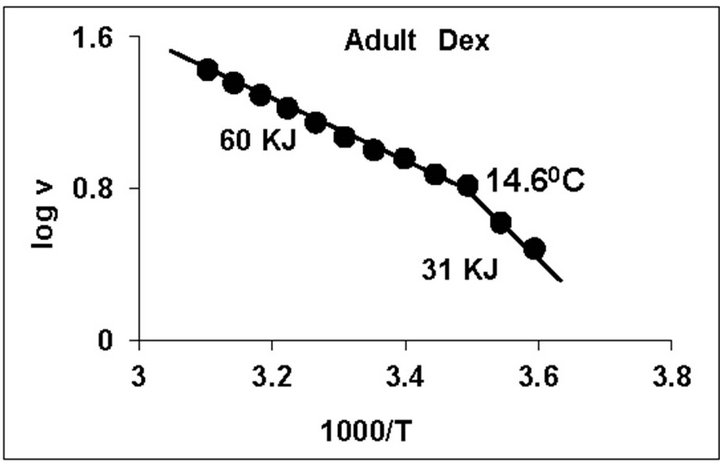
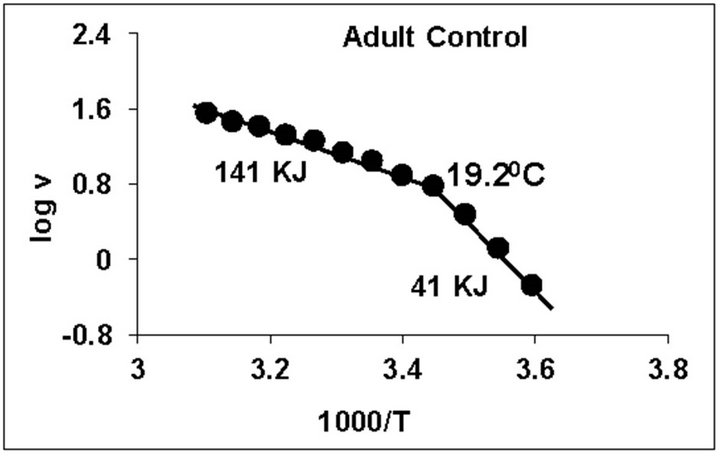
Figure 4. Typical temperature curves of rat liver mitochondrial F0.F1-ATPase in controls (Left Panel) and Dex treated (Right Panel) animals for 2-week, 3-week, 4-week, 5-week and adult group respectively. Abscissa represents the reaction velocity v, while the ordinate represents temperature range ˚C. Reaction velocity v = mmol of Pi liberated hr-1 mg protein-1. For temperature kinetics studies, experiments were carried out with fixed ATP concentration of 5 mM and the temperature was varied from 5˚C - 53˚C with an increment of 4˚C with 13 data points. The plots are typical for eight independent observations.
can be noted, after Dex treatment, the PC changes were age-specific. The PC percentage in 2-week showed significant increase and the opposite was observed for 4-week and adult groups which showed significant decrease. The 3-week and 5-week groups remained unaltered. The PE was elevated in 4-week, 5-week and adults whereas 3-week group display decrease and no changes were observed in 2-week group. For a DPG pattern opposite to that of PE was noted in an age-specific manner. The other components of phospholipids-lysophospholipid. (Lyso) and sphingomyelin (SPM) shared (4% - 8% each) as the basic and neutral phos pholipids respectively, show significant increase in 3-week, 5-week and adult
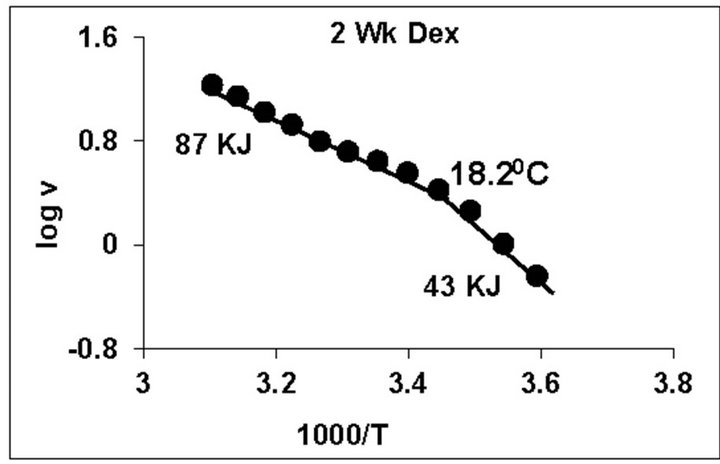
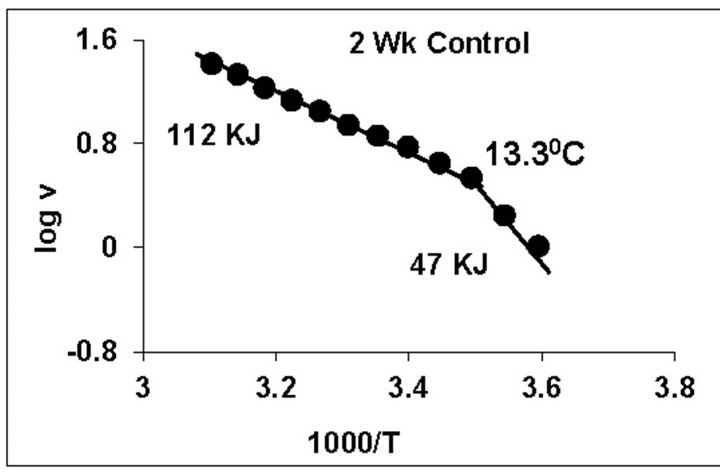

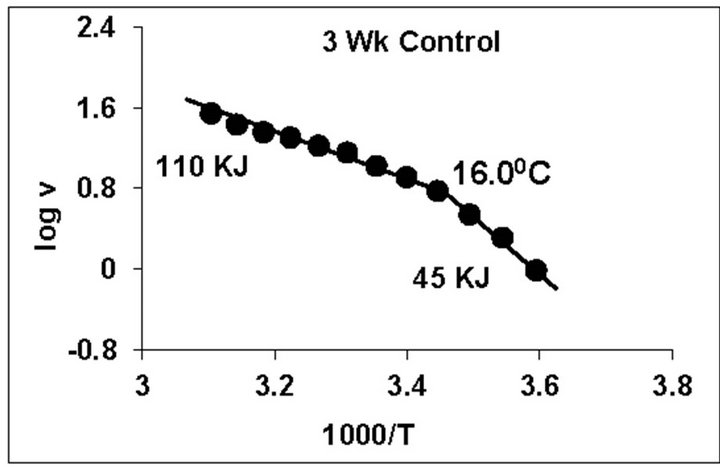
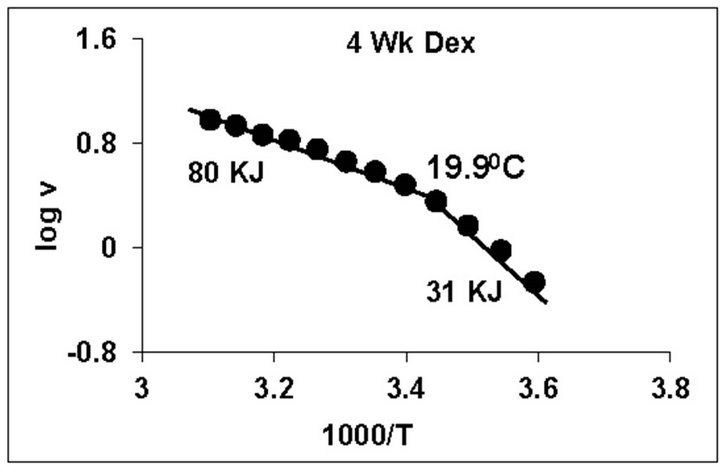
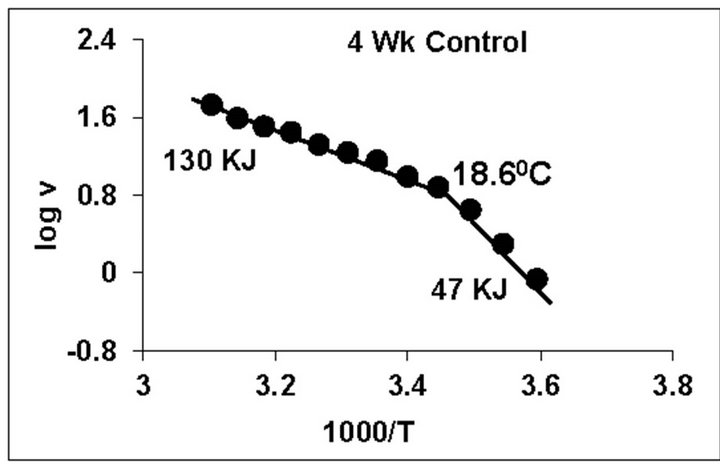
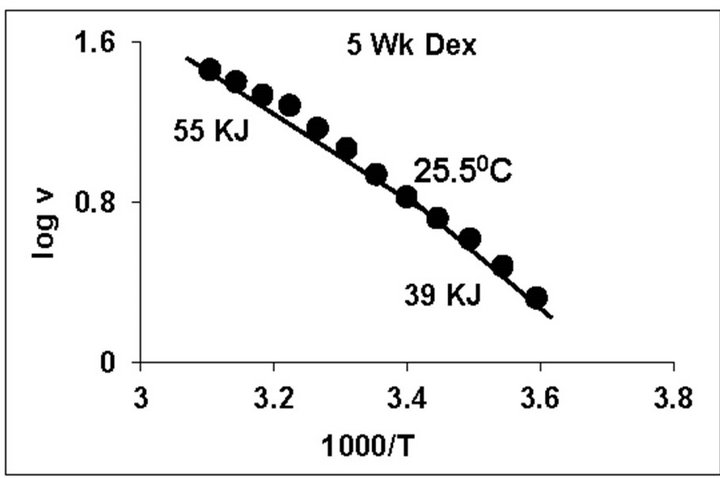
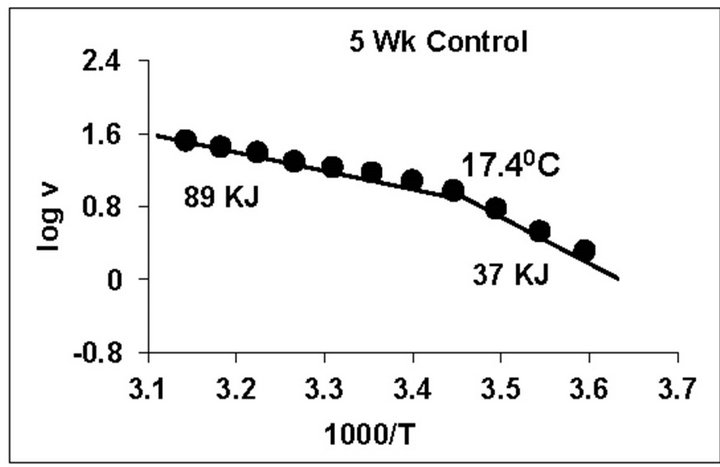
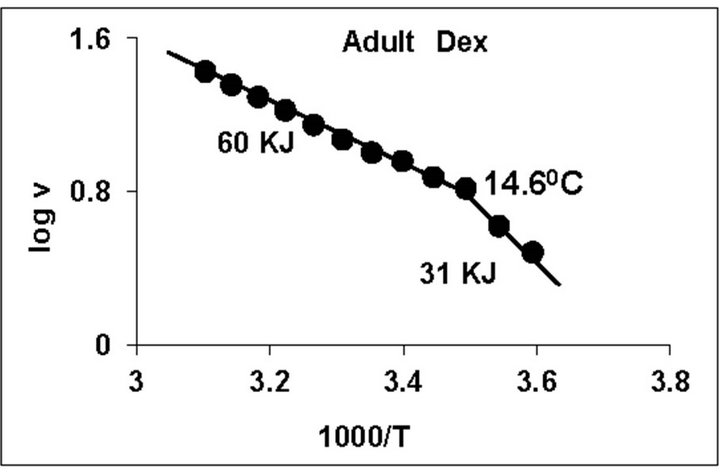
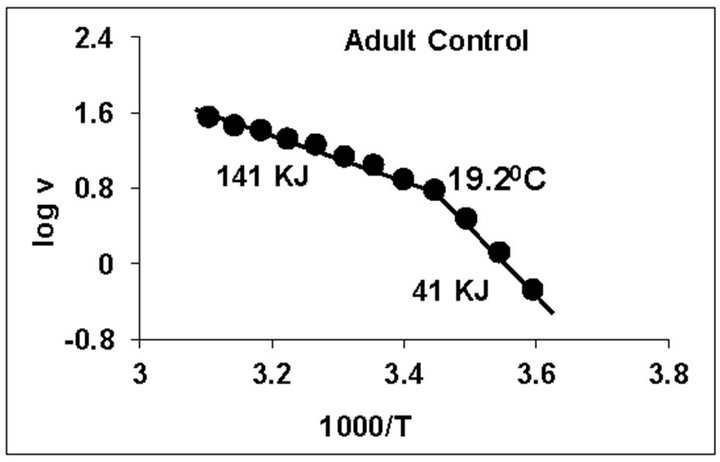
Figure 5. Typical Arrhenius plots for temperature kinetics of rat liver mitochondrial F0.F1-ATPase in controls (Left Panel) and Dex treated (Right Panel) animals for 2-week, 3-week, 4- week, 5-week and adult group respectively. Abscissa represents the log of reaction velocity v, while the ordinate represents reciprocal of absolute temperature T*1000. Reaction velocity v = m mol of Pi liberated hr-1 mg protein-1. Absolute temperature T = Kelvin (˚C +273.2). For temperature kinetics studies, experiments were carried out with fixed ATP concentration of 5 mM and the temperature was varied from 5˚C - 53˚C with an increment of 4˚C with 13 data points. The plots are typical for eight independent observations.
groups whereas 4-week group shows significant decrease. The two acidic phospholipids-phosphatidylinositol (PI) and phosphatedylserine (PS) which have minor share (2% - 6% each) in membrane composition changed in age-specific manner.
Due to alteration in percentage and total phospholipid content, it is possible to evaluate the changes in the contents of the individual phospholipids class. Thus the data in Table 7 indicate that Dex treatment significantly increased all the individual phospholipids in growing animal group; whereas by contrast, in the adults there was significant decrease in the contents of individual phospholipids. As can be noted, the PI, PS, PE and DPG were among the ones which increased significantly in all the postnatal developmental age groups. More specifically, Dex treatment significantly increased PI (33% - 210% increase), PS (13% - 230% increase), PE (30% - 140% increase) and DPG (29% - 129% increase) in 2- to 5- week group. The other two phospholipids Lyso and SPM had increased in 2-week to 4-week group by 29% - 160% and ~2 - 4 fold respectively following Dex treatment. The PC content had increased by 70% - 160% in 2-, 3- and 5- week group whereas 4-week group remained unaffected. In the adults, Dex treatment drastically decreased all the phospholipids classes (30% - 86% decrease) except for Lyso which was unchanged. Due to high impact on content of TPL, CHL as well as on individual phospholipid composition and content; it is possible that mitochondrial functional properties i.e. ATP synthesis capacity may have been affected due to direct or indirect interaction with F0.F1-ATPase activity domain.
Based on overall results of the present studies it may be concluded that the developmental and adult group responded differently with respect to both kinetics properties of the F0.F1-ATPase as well as lipid profile of mitochondria. Moreover, the significant increase in TPL, CHL and individual profile during developmental period may be involved in changing the F0.F1-ATPase ability to generate ATP as it is evident from observed decrease in the F0.F1-ATPase activity and changes in properties of substrate and temperature dependence of F0.F1-ATPase.
4. DISCUSSION
Results of our present study caution against the multiple use of Dex in the treatment of patients. Our study indicates the undesired side-effects of Dex treatment on rat liver mitochondrial lipid profiles and F0.F1- ATPase kinetics during developmental period and in adult life. The data on the F0.F1-ATPase activity and its kinetics properties (Figures 1-5 and Tables 1-4) suggest that Dex treatment results in dysregulation which eventually has deleterious effects on mitochondrial ability to synthesize ATP in age-specific manner. Briefly, (Table 1), it is evident that there is generalized significant decrease in the mitochondrial F0.F1-ATPase activity in all the developing and adult groups. The substrate and temperature kinetics data also point out to the age-specific alteration or abolishment of components after Dex treatment. As can be noted (Figure 1), all groups except for Dex treated 3-week group follow typical substrate saturation pattern. The 3-week group follows allosteric pattern of substrate saturation. While the kinetics analysis data for all the experimental groups resolved the F0.F1-ATPase
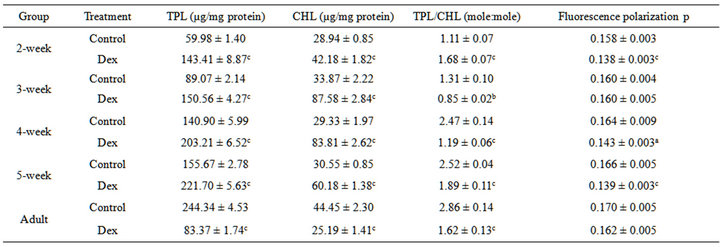
Table 5. The effect of Dex treatment on total phospholipids (TPL), cholesterol (CHL) and fluorescence polarization (p) of rat liver mitochondria.
The results are given as mean ± SEM of 16 - 24 independent experiments. The experimental conditions are as described in the text. ap < 0.05; bp < 0.002 and cp < 0.001 as compared with the corresponding control.
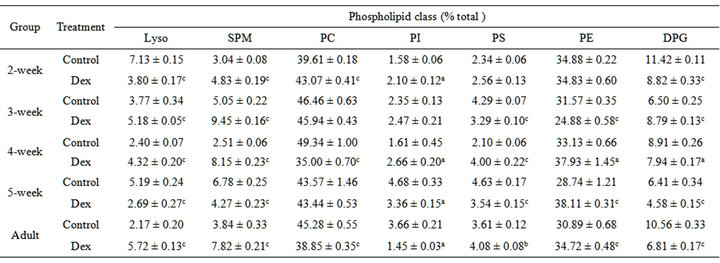
Table 6. Effect of Dex treatment on percentage composition of individual phospholipid class in rat liver mitochondria.
The results are given as mean ± SEM of 12 - 14 independent experiments. ap < 0.05; bp < 0.002 and cp < 0.001 as compared with the corresponding age-matched control group
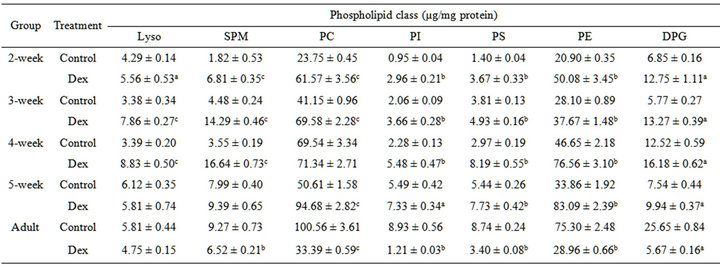
Table 7. Effect of Dex treatment on contents of individual phospholipid class in rat liver mitochondria.
The results are given as mean ± SEM of 12 - 14 independent experiments. ap < 0.05; bp < 0.002 and cp < 0.001 as compared with the corresponding age-matched control group.
activity in 3 components (Figure 2); with the exception of the 4-week and adult groups in which the activity resolved in only 2 components where one of the component was completely abolished due to Dex treatment.
In general, the Km and Vmax values of the developmental groups decreased significantly (Table 2) which is indicative of decrease in ability to synthesize ATP. This is consistent with our earlier observations [19]. The 3-week Dex treated group did not show normal substrate saturation pattern but displayed an allosteric pattern. Hill plot analysis (Figure 3) revealed that below transition concentration of 0.67 mM, 1 molecule of ATP was bound while beyond this concentration, 3 molecules were bound (Table 3). This may possibly be a compensatory mechanism to overcome constraints caused by decreased Vmax as well as simultaneous uncoupling which we have reported earlier [19].
The significantly altered activity and kinetics properties (Figures 1-5 and Tables 1-4) of F0.F1-ATPase in all the age group is consistent with our previously reported observation that, Dex treatment caused significant reduction in oxygen consumption of complex I and II substrates in all age-groups as well as site-specific uncoupling in 3- and 5- week and adults [19]. In similar studies it was reported that treatment with Dex for 5 days resulted in the uncoupling in liver mitochondria which was attributed to proton leaking and consequent decrease in the thermodynamic efficiency of the mitochondrial oxidative phosphorylation [39-41]. Literature survey indicates that, glucocorticoids directly influence the energy status of the cell by regulating mitochondrial respiration and oxidative phosphorylation [42]. The glucocorticoids modulate nuclear and mitochondrial genes involved in biosynthesis of respiratory enzyme thereby acting as key regulators of energy metabolism [43].
In our earlier studies, we observed that there was significant reduction in activities of mitochondrial dehydrogenases and loss of cytochromes [19]. In similar line of studies it was shown that in adult rats treatment with Dex for 4 days resulted in decrease in total pyruvate dehydrogenase (PDH) activity [44]. The decreased dehydrogenase activities would obviously lead to decreased rates of substrate oxidation thereby lowering ATP synthesis ability of liver mitochondria. It may also likely that the substrate pool for synthesize ATP may become limited after Dex treatment.
Survey of the literature suggests that mitochondrion is now considered as a primary site of action of glucocorticoids [43,45]. It was reported that glucocorticoid receptor (GR) translocates into mitochondria and mediates apoptosis by inducing mitochondria-dependent apoptotic pathway [46,47]. In brain, Dex directly activates cell death by NMDA receptor-induced excitotoxic events [17]. In other studies on human cell lines, it has been reported that Dex induces depolarization of mitochondrial membrane, releases cytochrome c and causes DNA fragmentation [48]. It is also known that mitochondrial permeability transition is a central coordinating event of apoptosis [49]. Thus all of the foregoing observations indicate that Dex may induce activation of mitochondrial regulated apoptotic cell death. Results of our present studies also support indirectly a postulate that possible defects in mitochondrial bioenergetics may thus one of the factors that may leads to apoptotic cell death event after Dex treatment. However, we have not elucidated cell death mechanism directly in our current study.
The typical temperature kinetics by Arrhenius plots (Figures 4 and 5) and its quantitative representation (Table 4), indicate that Dex treatment in general significantly lowered the energies of activation (EL) in all the age groups whereas, the EH values were significant decreased in only 5-week and adult group. Additionally, we observed substantial increase (8˚C increase) in phase transition temperature Tt˚C in the 5-week group along with opposite effect (5˚C decrease) in adult after Dex treatment. To our knowledge, this is a novel observation for testing developing and adult group substrate and temperature kinetic of F0.F1-ATPase in relation to Dex treatment.
As is clear from the results section (Table 5), the mitochondrial TPL and CHL contents in the developing animals increased significantly after Dex treatment. The phospholipids composition and contents of the individual phospholipid classes showed age-specific changes (Tables 6 and 7). Briefly, from the data in Table 7, it is evident that the contents of PI, PS, PE and DPG significantly increased for 2-week to 5-week groups; whereas Lyso and SPM increased in 2-week to 4-week group. Moreover, PC increased in 2-, 3- and 5-week group. The data on fluidity (Table 5) indicate that after Dex treatment membrane became more fluidized in the developing age groups (2-week, 4-week and 5-week). The reverse pattern was observed for adult group. Since both TPL and CHL contents decreased significantly, there was no apparent change in membrane fluidity parameters (Table 5). In adults, Dex treatment significantly decreased contents of all the phospholipid classes except Lyso which was unchanged (Table 7). Thus, in general, our results indicate that the Dex treatment significantly increased TPL, CHL and individual phospholipids content during developmental period whereas adult group displayed an opposite effect.
Our observations on the increase in phopholipid content during developmental period have been indirectly supported by finding of other investigators. Several indirect observations support the possibility of upregulation of lipid synthesis or accumulation in liver during developmental period after chronic Dex treatment in neonates. Yan Liu et al. have reported significantly increased triglyceride accumulation within liver after Dex treatment in neonatal age after 14 days [50]. Dex treatment (100 mg/kg) for 5 days increased triglycerides retention in liver by decreasing their secretion [51]. In this study, the authors used fifty times higher dose of Dex. The regulatory role of glucocorticoids in gluconeogenic and insulin resistance during development has known [52, 53]. In hepatocytes, the effect of induction of gluconeogenesis after Dex treatment was shown [54]. This may possibly indicate the increased turnover of fatty acids and their metabolism in liver mitochondria during developmental period. Foster and Bailey, demonstrated upregulation of activities of several mitochondrial enzymes involved in fatty acid oxidation during developmental period immediately after birth which decreased after weaning [55]. They speculated that this could be due to increased fatty acid supply to the liver from the mother’s milk and also due to hormonal changes during developmental period. Similarly, it has been reported that the enzyme activity and gene expression of acyl-CoA dehydrogenase, the enzymes that catalyzes β-oxidation follows similar developmental pattern [56,57]. Kapitulnik et al. have shown increased fluidity in developing fetal rat liver microsomal membranes with significant increase in contents of polyunsaturated fatty acids after early prenatal exposure [58]. This may support our observation of fluidized membrane during developmental period.
In the adult group, the decreased contents of phospholipids and CHL of mitochondrial membrane is also supported by the observations of other investigators who reported decreased contents of lipid classes after chronic administration of Dex [59]. Although Kaur et al., have used ten time higher dose than one which we used in our study. Mangiapane et al., have shown decrease in the triglyceride and PC content of the cultures of rat hepatocytes with Dex pre-incubation [60]. Other researchers have shown decreased glycosylated PC levels in hepatocyte [53]. All of the above reports either indicate or directly state the involvement of Dex in lipid metabolism. Moreover, these reports may also suggest the possibility of the differences in the properties of lipid profiles between developmental group and adults. However, one study reported by Arvier et al., indicates no changes in lipid composition in adults hepatic mitochondria using Dex treatment which remains unexplained but may possibly be due to strain specific differences in phospholipid composition [39].
It has been reported that in the liver microsomes, Dex treatment only marginally influenced phospholipids composition but altered the proportion of the arachidonic, oleic and palmatic acid in PC [61]. Another study by Holloway et al. has shown the involvement of Dex in lipid properties of rat hepatic microsomal membranes [62]. The increased TPL and CHL content up to the 5th week which we observe here suggest that Dex treatment may enhance the transfer of this component form microsomes to mitochondria. By the same token the differential changes in the individual phospholipids composition would suggest that their transfers from microsomes may be differentially affected by Dex treatment. Since the major phospholipids of mitochondria originate from microsomes, undoubtly the fatty acid composition of the phospholipids might have been changed. However, this possibility has not been examined in the present studies. It may be anticipated that the lipid/phospholipids changes would influence function and kinetics properties of membrane bound enzymes such as F0.F1-ATPase [21].
One another possibility of glucocorticoids action may be postulated based on our observations. It has been reported that the free fatty acids (FFA) play an important role in uncoupling of mitochondria [63,64]. It is possible that the increased contents of phospholipids in developmental groups may somehow increase the turnover of the FFA and may thereby increase mitochondrial uncoupling during the developmental period. It has also been known for a long time that mitochondrial endogenous uncoupling proteins (UCPs) mediate uncoupling event in mitochondria thereby playing an important role in proton cycling and thermogenesis [65,66]. Recently, the regulatory role of UCPs in co-ordination with FFA has been reviewed [67,68]. It may thus be possible that increase in phospholipids and CHL during developmental period may trigger UCPs expression thereby effecting mitochondrial uncoupling. This may be the reason for observing decrease in F0.F1-ATPase activity in our developmental group study. Paradoxically, the increased phospholipids contents may influence on inhibitory effect on F0.F1-ATPase activity in adults. It is possible that in adults, the UCPs profile may have been regulated differently by fatty acids or by UCPs. At this stage, role of FFA and endogenous UCPs in developmental and adult period have not been explored. However, it is possible that phospholipids contents may indirectly regulate FFA induced uncoupling events and thereby may indirectly inhibit F0.F1-ATPase activity.
Lastly, it is possible that excess glucocorticoids may increase the formation of ketone bodies in liver as Dex regulates ketogenesis as reported in hepatocyte culture [69]. Another group has reported that HMG-CoA synthase protein plays a modulatory role in increasing the ketogenic pathway in liver mitochondria after single dose of Dex (5 mg/kg) treatment in adult rats [70]. Moreover, acetoacetate has been shown to over-express uncoupling protein-2 (UCP2) under in vitro conditions in cancer cell line [71]. These observations prompt us to hypothesize that Dex induced ketone bodies formation may indirectly regulate the endogenous UCPs; thereby influencing ATP production in cells. Once again this possibility has not been tested in our present study.
In summary, our data suggest direct involvement of lipid profile on regulation of activity and kinetics properties of F0.F1-ATPase in liver mitochondria after Dex treatment in developmental and adult groups. Our data also warns against excessive repeated use of antenatal DEX in treatments in all growing and adult group.
5. ACKNOWLEDGEMENTS
Authors would like to thank Shri Navdurga High School Educational Trust, Rajpipla, Gujarat, India for providing financial support for carrying out this work.
REFERENCES
- Schimmer, B.P. and Parker, K.L. (2001) Adrenocorticotropic hormone; adrenocortical steroids and their synthetic analogs; inhibitors of the synthesis and actions of adrenocortical hormones. In: Gilman, A.G., Hardman, J.G. and Limbird, L.E., Eds., The Pharmacological Basis of Therapeutics, 10th Edition, The McGraw-Hill Companies, New York, 1635-1648.
- Whitelaw, A. and Thoresen, M. (2000) Antenatal steroids and the developing brain. Archives of Disease in Childhood, Fetal and Neonatal Edition, 83, F154-F157. doi:10.1136/fn.83.2.F154
- Gerrard, G.E. and Franks, K.N. (2004) Overview of the diagnosis and management of brain, spine, and meningeal metastases. Journal of Neurology, Neurosurgery and Psychiatry, 75, ii37-ii42. doi:10.1136/jnnp.2004.040493
- Hagberg, H. and Jacobsson, B. (2005) Brain injury in preterm infants—What can the obstetrician do? Early Human Development, 81, 231-235. doi:10.1016/j.earlhumdev.2005.01.003
- Kumar, P. (2005) Effect of decreased use of postnatal corticosteroids on morbidity in extremely low birthweight infants. American Journal of Perinatology, 22, 77-81. doi:10.1055/s-2005-837274
- Rastogi, A., et al. (1996) A controlled trial of dexamethasone to prevent bronchopulmonary dysplasia in surfactant-treated infants. Pediatrics, 98, 204-210.
- Romagnoli, C., et al. (1999) Effect on growth of two different dexamethasone courses for preterm infants at risk of chronic lung disease. Pharmacology, 59, 266-274. doi:10.1159/000028329
- Tsuneishi, S., et al. (1991) Effects of dexamethasone on the expression of myelin basic protein, proteolipid protein, and glial fibrillary acidic protein genes in developing rat brain. Development Brain Research, 61, 117-123. doi:10.1016/0165-3806(91)90121-X
- Brocklehurst, P., et al. (1999) Are we prescribing multiple courses of antenatal corticosteroids? A survey of practice in the UK. An International Journal of Obstetrics and Gynaecology, 106, 977-979. doi:10.1111/j.1471-0528.1999.tb08440.x
- Leung, T.N., et al. (2003) Repeated courses of antenatal corticosteroids: Is it justified? Acta Obstetricia et Gynecologica Scandinavica, 82, 589-596. doi:10.1034/j.1600-0412.2003.00204.x
- Wapner, R. (2004) Antenatal corticosteroids: We continue to learn. American Journal of Obstetrics and Gynecology, 190, 875. doi:10.1016/j.ajog.2004.01.045
- Flagel, S.B., et al. (2002) Effects of tapering neonatal dexamethasone on rat growth, neurodevelopment, and stress response. American Journal of Physiology, Regul atory, Integrative and Comparative Physiology, 282, R55-R63.
- Barrington, K.J. (2001) The adverse neuro-developmental effects of postnatal steroids in the preterm infant: A systematic review of RCTs. BMC Pediatrics, 1, 1. doi:10.1186/1471-2431-1-1
- Doyle, L. and Davis, P. (2000) Postnatal corticosteroids in preterm infants: Systematic review of effects on mortality and motor function. Journal of Paediatrics and Child Health, 36, 101-107. doi:10.1046/j.1440-1754.2000.00481.x
- Neal Jr., C.R., et al. (2004) Effect of neonatal dexamethasone exposure on growth and neurological development in the adult rat. American Journal of Physiology, Regulatory, Integrative and Comparative Physiology, 287, R375-R385. doi:10.1152/ajpregu.00012.2004
- Haouzi, D., et al. (2000) Cytochrome P450-generated reactive metabolites cause mitochondrial permeability transition, caspase activation, and apoptosis in rat hepatocytes. Hepatology, 32, 303-311. doi:10.1053/jhep.2000.9034
- Jacobs, C.M., et al. (2006) Dexamethasone induces cell death which may be blocked by NMDA receptor antagonists but is insensitive to Mg2+ in cerebellar granule neurons. Brain Research, 1070, 116-123. doi:10.1016/j.brainres.2005.10.093
- Pandya, J.D., Agarwal, N.A. and Katyare, S.S. (2007) Dexamethasone treatment differentially affects the oxidative energy metabolism of rat brain mitochondria in developing and adult animals. International Journal of Developmental Neuroscience, 25, 309-316. doi:10.1016/j.ijdevneu.2007.05.001
- Pandya, J.D., Agarwal, N.A. and Katyare, S.S. (2004) Effect of dexamethasone treatment on oxidative energy metabolism in rat liver mitochondria during postnatal developmental periods. Drug and Chemical Toxicology, 27, 389-403. doi:10.1081/DCT-200039778
- Ferguson, S.A. and Holson, R.R. (1999) Neonatal dexamethasone on day 7 causes mild hyperactivity and cerebellar stunting. Neurotoxicology and Teratology, 21, 71-76. doi:10.1016/S0892-0362(98)00029-4
- Brown, R.E. and Cunningham, C.C. (1982) Negatively charged phospholipid requirement of the oligomycinsensitive mitochondrial ATPase. Biochimica Biophysica Acta, 684, 141-145. doi:10.1016/0005-2736(82)90059-1
- Hoch, F.L. (1992) Cardiolipins and biomembrane function. Biochimica Biophysica Acta, 1113, 71-133. doi:10.1016/0304-4157(92)90035-9
- Ahlersova, E., Ahlers, I. and Smajda, B. (1992) Influence of light regimen and time of year on circadian oscillations of insulin and corticosterone in rats. Physiology Research, 41, 307-314.
- Swegert, C.V., Dave, K.R. and Katyare, S.S. (1999) Effect of aluminium-induced Alzheimer like condition on oxidative energy metabolism in rat liver, brain and heart mitochondria. Mechanisms of Ageing and Development, 112, 27-42. doi:10.1016/S0047-6374(99)00051-2
- Katyare, S.S., et al. (2007) Thyroid hormone-induced alterations in membrane structure-function relationships: Studies on kinetic properties of rat kidney microsomal Na(+),K (+)-ATPase and lipid/phospholipid profiles. Journal of Membrane Biology, 219, 71-81. doi:10.1007/s00232-007-9063-7
- Fiske, C.H. and Subbarow, Y. (1925) Colorimetric determination of phosphorus. Journal of Biological Chemistry, 66, 375-400.
- Dixon, M. and Webb, E.C. (1979) Enzymes, Longman, London.
- Patel, H.G., et al. (2000) Kinetic attributes of Na+, K+ ATPase and lipid/phospholipid profiles of rat and human erythrocyte membrane. Z Naturforsch C, 55, 770-777.
- Raison, J.K. (1972) The influence of temperature-induced phase changes of the kinetics of respiration and other membrane-associated enzyme system. Bioenergetics, 4, 559-583.
- Folch, J., Lees, M. and Sloane Stanley, G.H. (1957) A simple method for the isolation and purification of total lipides from animal tissues. Journal of Biological Chemistry, 226, 497-509.
- Zlatkis, A., Zak, B. and Boyle, A.J. (1953) A new method for the direct determination of serum cholesterol. Journal of Laboratory and Clinical Medicine, 41, 486-492.
- Bartlett, G.R. (1959) Phosphorus assay in column chromatography. Journal of Biological Chemistry, 234, 466- 468.
- Pandya, J.D., Dave, K.R. and Katyare, S.S. (2001) Effect of long-term aluminum feeding on lipid/phospholipid profiles of rat brain synaptic plasma membranes and microsomes. Journal of Alzheimers Disease, 3, 531-539.
- Stahl, E. (1969) Apparatus and general techniques. In: Stahl, E., Ed., TLC in thin layer chromatography: A Laboratory Handbook, 2nd Edition, Springer-Verlag, New York.
- Bangur, C.S., Howland, J.L. and Katyare, S.S. (1995) Thyroid hormone treatment alters phospholipid composition and membrane fluidity of rat brain mitochondria. Biochemical Journal, 305, 29-32.
- Mehta, J.R., et al. (1991) Lipid fluidity and composition of the erythrocyte membrane from healthy dogs and Labrador retrievers with hereditary muscular dystrophy. Neurochemical Research, 16, 129-135. doi:10.1007/BF00965700
- Van Blitterswijk, W.J., Van Hoeven, R.P. and Van der Meer, B.W. (1981) Lipid structural order parameters (reciprocal of fluidity) in biomembranes derived from steady-state fluorescence polarization measurements. Biochimica Biophysica Acta, 644, 323-332. doi:10.1016/0005-2736(81)90390-4
- Lowry, O.H., et al. (1951) Protein measurement with the Folin phenol reagent. Journal of Biological Chemistry, 193, 265-275.
- Arvier, M., et al. (2007) Adenine nucleotide translocator promotes oxidative phosphorylation and mild uncoupling in mitochondria after dexamethasone treatment. American Journal of Physiology, Endocrinology and Metabolism, 293, E1320-E1324. doi:10.1152/ajpendo.00138.2007
- Dumas, J.F., et al. (2003) Mitochondrial energy metabolism in a model of undernutrition induced by dexamethasone. British Journal of Nutrition, 90, 969-977. doi:10.1079/BJN2003980
- Roussel, D., et al. (2003) Dexamethasone treatment specifically increases the basal proton conductance of rat liver mitochondria. FEBS Letters, 541, 75-79. doi:10.1016/S0014-5793(03)00307-7
- Desquiret, V., et al. (2008) Mitochondrial effects of dexamethasone imply both membrane and cytosolicinitiated pathways in HepG2 cells. International Journal of Biochemistry and Cell Biology, 40, 1629-1641. doi:10.1016/j.biocel.2007.12.010
- Tsiriyotis, C., Spandidos, D.A. and Sekeris, C.E. (1997) The mitochondrion as a primary site of action of glucocorticoids: Mitochondrial nucleotide sequences, showing similarity to hormone response elements, confer dexamethasone inducibility to chimaeric genes transfected in LATK-cells. Biochemical and Biophysical Research Communications, 235, 349-354. doi:10.1006/bbrc.1997.6787
- Begum, N., Tepperman, H.M. and Tepperman, J. (1984) Effect of dexamethasone on adipose tissue and liver pyruvate dehydrogenase and its stimulation by insulingenerated chemical mediator. Endocrinology, 114, 99-107. doi:10.1210/endo-114-1-99
- Demonacos, C., et al. (1995) The mitochondrion as a primary site of action of glucocorticoids: The interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. The Journal of Steroid Biochemistry and Molecular Biology, 55, 43-55. doi:10.1016/0960-0760(95)00159-W
- Rambal, A.A., et al. (2009) MEK inhibitors potentiate dexamethasone lethality in acute lymphoblastic leukemia cells through the pro-apoptotic molecule BIM. Leukemia, 23, 1744-1754. doi:10.1038/leu.2009.80
- Sionov, R.V., et al. (2006) Role of mitochondrial glucocorticoid receptor in glucocorticoid-induced apoptosis. Journal of Experimental Medicine, 203, 189-201. doi:10.1084/jem.20050433
- Nuutinen, U., et al. (2006) Inhibition of PI3-kinase-Akt pathway enhances dexamethasone-induced apoptosis in a human follicular lymphoma cell line. Experimental Cell Research, 312, 322-30.
- Marchetti, P., et al. (1996) Mitochondrial permeability transition is a central coordinating event of apoptosis. Journal of Experimental Medicine, 184, 1155-1160. doi:10.1084/jem.184.3.1155
- Liu, Y., et al. (2007) Postnatal treatment with dexamethasone perturbs hepatic and cardiac energy metabolism and is associated with a sustained atherogenic plasma lipid profile in suckling rats. Pediatric Research, 61, 165-170. doi:10.1203/pdr.0b013e31802d89ff
- Letteron, P., et al. (1997) Glucocorticoids inhibit mitochondrial matrix acyl-CoA dehydrogenases and fatty acid beta-oxidation. American Journal of Physiology, 272, G1141-G1150.
- Amin, S.B., et al. (1999) Lipid intolerance in neonates receiving dexamethasone for bronchopulmonary dysplasia. Archives Pediatric in Adolescent Medicine, 153, 795-800.
- Cabello, M.A., et al. (1990) Effect of adrenalectomy and glucocorticoid treatment on the levels of an insulinsensitive glycosyl-phosphatidylinositol in isolated rat hepatocytes. Molecular and Cell Endocrinology, 68, R1-R5. doi:10.1016/0303-7207(90)90173-6
- Allan, E.H. and Titheradge, M.A. (1984) Effect of treatment of rats with dexamethasone in vivo on gluconeogenesis and metabolite compartmentation in subsequently isolated hepatocytes. Biochemical Journal, 219, 117-123.
- Foster, P.C. and Bailey, E. (1976) Changes in the activities of the enzymes of hepatic fatty acid oxidation during development of the rat. Biochemical Journal, 154, 49-56.
- Carroll, J.E., et al. (1989) Acyl-CoA dehydrogenase enzymes during early postnatal development in the rat. Biology of Neonate, 55, 185-190. doi:10.1159/000242915
- Nagao, M., Parimoo, B. and Tanaka, K. (1993) Developmental, nutritional, and hormonal regulation of tissuespecific expression of the genes encoding various acylCoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. Journal of Biological Chemistry, 268, 24114-24124.
- Kapitulnik, J., Weil, E. and Rabinowitz, R. (1986) Glucocorticoids increase the fluidity of the fetal-rat liver microsomal membrane in the perinatal period. Biochemical Journal, 239, 41-45.
- Kaur, N., Sharma, N. and Gupta, A.K. (1989) Effects of dexamethasone on lipid metabolism in rat organs. Indian Journal of Biochemistry and Biophysics, 26, 371-376.
- [61] Mangiapane, E.H. and Brindley, D.N. (1986) Effects of dexamethasone and insulin on the synthesis of triacylglycerols and phosphatidylcholine and the secretion of very-low-density lipoproteins and lysophosphatidylcholine by monolayer cultures of rat hepatocytes. Biochemical Journal, 233, 151-160.
- [62] Brenner, R.R., Ayala, S. and Garda, H.A. (2001) Effect of dexamethasone on the fatty acid composition of total liver microsomal lipids and phosphatidylcholine molecular species. Lipids, 36, 1337-1345. doi:10.1007/s11745-001-0850-1
- [63] Holloway, C.T. and Margolis, R.N. (1980) Dynamic lipid changes in rapidly proliferating hepatic smooth endoplasmic reticulum during acute dexamethasone treatment of adrenalectomized rats. Lipids, 15, 1037-1043. doi:10.1007/BF02534320
- [64] Di Paola, M. and Lorusso, M. (2006) Interaction of free fatty acids with mitochondria: Coupling, uncoupling and permeability transition. Biochimica et Biophysica Acta, 1757, 1330-1337. doi:10.1016/j.bbabio.2006.03.024
- [65] Wojtczak, L. and Wieckowski, M.R. (1999) The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. Journal of Bioenergetics and Biomembranes, 31, 447-455. doi:10.1023/A:1005444322823
- [66] Lardy, H. and Copenhaver, Jr., J.H. (1954) Efficiency of oxidative phosphorylation. Nature, 174, 231-232. doi:10.1038/174231b0
- [67] Loomis, W.F. and Lipmann, F. (1949) Inhibition of phosphorylation by azide in kidney homogenate. Journal of Biological Chemistry, 179, 503.
- [68] Davis, L.M., Rho, J.M. and Sullivan, P.G. (2008) UCPmediated free fatty acid uncoupling of isolated cortical mitochondria from fasted animals: Correlations to dietary modulations. Epilepsia, 49, 117-119. doi:10.1111/j.1528-1167.2008.01854.x
- [69] Sluse, F.E., et al. (2006) Mitochondrial UCPs: New insights into regulation and impact. Biochimica et Biophysica Acta, 1757, 480-485. doi:10.1016/j.bbabio.2006.02.004
- [70] Agius, L., Chowdhury, M.H. and Alberti, K.G. (1986) Regulation of ketogenesis, gluconeogenesis and the mitochondrial redox state by dexamethasone in hepatocyte monolayer cultures. Biochemical Journal, 239, 593-601.
- [71] Serra, D., et al. (1993) Regulation of mitochondrial 3- hydroxy-3-methylglutaryl-coenzyme a synthase protein by starvation, fat feeding, and diabetes. Archives of Biochemistry and Biophysics, 307, 40-45. doi:10.1006/abbi.1993.1557
- [72] Fine, E.J., et al. (2009) Acetoacetate reduces growth and ATP concentration in cancer cell lines which over-express uncoupling protein 2. Cancer Cell International, 9, 14. doi:10.1186/1475-2867-9-14
ABBREVIATIONS
CHL, Cholesterol; Dex, Dexamethasone;DNP, 2,4- Din-i trophenol; DPG, Diphosphatidylglycerol; DPH, 1,6-Diphenyl-1,3,5-hexatriene; FFA, Free Fatty Acids; MPT, Membrane Permeability Transition; PL, Phospholipids;
Lyso, Lysophospholipid; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PI, Phosphatidylinositol; PS, Phosphatidylserine; SPM, Sphingomyelin; TPL, Total phospholipids; UCPs, Uncoupling Proteins.