Soft
Vol.04 No.02(2015), Article ID:60674,15 pages
10.4236/soft.2015.42002
Spectroscopic Investigation and Characterizations of PAM/PEO Blends Films
Gaurang Patel1,2*, Mundan B. Sureshkumar1, Purvi Patel1,2
1Department of Physics, Faculty of Science, The M.S. University of Baroda, Vadodara, India
2Science and Humanities Department, Dr. S & S S Ghandhy College, Surat, India
Email: *goru16686@gmail.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 May 2015; accepted 25 October 2015; published 28 October 2015
ABSTRACT
Polymer blends have been obtained in the form of dimensionally stable and free standing films and their properties were characterized by different techniques. FTIR analysis and Raman spectroscopic analysis cleared the hydrogen bonding intermolecular interaction between -CONH2 groups in Poly Acrylamide (PAM) and C-O-C and -CH2OH group in Poly Ethylene Oxide (PEO). From Differential Scanning Calorimeter (DSC) the study shows that crystallinity is increasing with PEO wt%. From polymer interaction parameter we also show that the polymer blend is miscible. Ther- mal stability of films is investigated by Thermo Gravimetric Analysis (TGA) and derivative Ther- mo Gravimetric Analysis (DrTG). From UV-Vis absorption spectra, absorption band edge, direct/ indirect band gap and optical activation energy have been calculated.
Keywords:
Polymer Blend, Poly Acrylamide, Poly Ethylene Oxide, Thermal Properties, Mechanical Properties

1. Introduction
Blending of polymers is an attractive as well as an important route for providing new materials with desirable properties with economically low cost [1] . The properties of the polymer blend can be controlled by blend morphology, blend composition and its processing condition [2] . The study of blend properties is very important to find its new application in the field of biomedical and pharmacy [3] [4] . Polymer compatibility and miscibility are important criterion when dealing with blend. Polymer-polymer miscibility arises due to any interaction such as hydrogen bonding, dipole-dipole interaction or charge transfer process among pure polymer mixtures [5] -[8] .
Due to increase in the applications of polymer in biomedical field, study of water soluble polymer like Poly (ethylene oxide) (PEO), poly (vinyl alcohol) (PVA), and polyacrylamide (PAM) has high interest [9] [10] . It is important to note that there are very few studies on their blends.
Polyacrylamide is used in multitude applications including water clarification, waste water treatment, oil recovery, agriculture and biomedical applications [11] -[13] . The high bio-adhesive property of acrylic polymers offers good prospects for using these polymers in controlled drug delivery systems for local applications [14] [15] . Structural and physical properties of elements doped PAM are far studied [16] .
PEO is semi crystalline synthetic polymer and because of its biocompatibility, it is used in many biomedical devices including drug delivery and tissue replacement [17] -[19] . PEO/Starch blends present great application in scaffolds for cell culture and tissue engineering [20] [21] . PEO has moderate tensile strength, and it possesses well mechanical and electrical properties [22] . Some work reported on optical and electrical properties of PEO based polymer electrolyte film [23] -[25] . Ferreiro et al. reported that when there was a change in blend ratio of PEO/PMMA, morphological transitions occurred [26] [27] . Chemical structure of PEO (presence of the ether oxygen and -OH end groups) makes some possibilities of hydrogen bonds formation as it has already confirmed for PEO/poly(vinyl alcohol) and PEO/unsaturated polyester resin systems [28] [29] .
PAM and PEO individually blend with other polymers, but to best of authors’ knowledge, there isn’t any interaction study of PAM and PEO polymer blends together. Only Vijayalakshmi et al. has studied thermal degradation characterization of blend of PEO and PAM [30] [31] . In this article, we reported on the formulation and fabrication of free standing PAM/PEO blend films. For the first time PAM-PEO polymer matrix was proposed and chosen by us to prepare polymer blends. A simple fabrication technique was adopted for the blends preparation. Miscibility is an important property of blend system, which can affect the different properties of the blends. And since miscibility depends on intermolecular interaction between polymers chain, we have investigated the miscibility of PAM and PEO blend system. Blend films were characterized through structural, thermal, optical, mechanical and morphological study of the films. The intermolecular interaction between two polymers chain of PAM and PEO was explored by the means of FTIR and Raman spectra, which had been quite helpful for the study of compatibility and miscibility of blends. Finally, these results are correlated with the results obtained from the other characterization techniques.
2. Experiment
Sample Preparation
Poly acrylamide (M. W. 50,000 g/mol) and Poly ethylene oxide (M. W. 2 × 105 g/mol) were obtained from local chemicals supplier and used as received. The polymer blend films were prepared by the solution casting method. Polymer blend films were made in three different weight percentages 30/70, 50/50 and 70/30 (wt %). PAM and PEO first dissolved in the common solvent (distilled water) and then both the polymers were homogeneously mixed using a magnetic stirrer at the constant temperature at 50˚C for 12 hours. Complete homogeneous solution was casted on Teflon petri dishes and left for the evaporation of the solvent. The resulting PAM/PEO films were dried up to 4 days at room temperature in the desiccators to remove the traces of solvent, and then lifted out of the petri dish for further analysis. The thickness of the obtained films was in the range of 60 - 120 mm.
3. Measurement
3.1. FTIR Measurement
FTIR measurements were carried out using a single beam FT-IR-4100, Jasco spectrometer in the spectral range of 600 - 4000 cm-1 with the resolution of 4 cm-1. Each of the samples was scanned 200 times in ATR mode for absorption measurement. FTIR spectra have been analyzed by spectra manager software.
3.2. UV-Vis Spectrum Measurement
Ultraviolet-visible (UV-Vis) absorption spectra were measured in the wavelength region of 200 - 800 nm using Perkin-Elmer Lambda 25 UV-VIS Spectrophotometer, USA. Free standing films were kept in the sample holder to measure UV-Vis spectra in given range.
3.3. RAMAN Spectrum Analysis
Raman spectra for 1064 nm exciting radiation were recorded on a Bruker RAM II Vertex-70 Laser Raman spectrometer, USA (Nd: YAG solid state laser) with 1 cm-1 resolution.
3.4. Differential Scanning Calorimeter
For the DSC measurements SII Exstar-6000, operating in dynamic mode (heating rate = 10˚C/min) in N2 gas environment, was employed. Samples in appropriate amount were placed in sealed aluminum pans. Prior to use the calorimeter was calibrated with metal standards; an empty aluminum pan was used as a reference.
3.5. Thermogravimetry Analysis
A Perkin-Elmer, Pyris-1 TGA, USA was used for the Thermo-gravimetric analysis of the samples. Small amounts (5 - 10 mg) of the sample were taken for the study, and the samples were heated from 50˚C to 500˚C at a rate of 10˚C/min in a nitrogen atmosphere.
3.6. Mechanical Properties Measurement
Mechanical properties of pure and blend samples have been reported in this study. These measurements were made on Lloyds LR30 K (UK) instrument with a crosshead speed of 10 mm/min and a gauge length of 10 mm. All mechanical testing were conducted on pores or nicks free samples, and carried out at room temperature.
3.7. Scanning Electron Microscopy
The morphology of pure and blend polymers has been monitored by a JEOL SEM (USA), model number JSM 6380LV. As samples are non conductive, they need to be made conductive by covering the samples with a thin layer of conductive material. Coating of platinum thin film is done by a using a device called “sputter coater”. Inside the SEM chamber, the vacuum is created less than 10-7 torrs for minimizing impurities and viewing the sharp surfaces of the sample.
4. Results and Discussion
4.1. FTIR Spectroscopy
FTIR spectroscopy of blend films were carried out to detect peak shifts, which may be due to the interaction, like hydrogen bonding, between two polymers. Infrared spectroscopy is a fundamental technique to find out the presence of hydrogen bond, which is characterized by changes in absorption bands of functional groups involved in the formation of hydrogen bond [32] . The changes in absorption of the functional groups causes change in force constant of donor and acceptor groups and due to this change in frequency of stretching, deformation of these groups occurs [32] . It has been already reported that PEO is a proton accepting polymer (ether group), which is necessary for the formation of hydrogen bond between two polymers [33] [34] . PAM presents -NH2OH group as proton donor which can play important role in blend miscibility.
Figure 1 shows the spectra of pure polymers and blends films. Peak values for pure and blend polymers are shown in Table 1. PAM has two bands at 3315 cm−1 and 3212 cm−1, which indicate N-H stretching vibrations and absorption peak at 1651 cm−1 is for C=O stretching; a peak at 1607 cm−1 attributed to an N-H bending and at 1447 cm−1 is for C-N stretching vibrations [35] [36] . PEO has two strong absorption bands at 3332 cm−1 and 3177 cm−1 which indicate -OH stretching vibrations. Absorption peak at 2881 cm−1 assigns as asymmetric stretching of the -CH2 group. Peaks at 1610 cm−1 and 1655 cm−1 indicate bound H2O solvent as in crystal form [36] . 1106 cm−1 and 1454 cm−1 peaks indicate C-O-C stretching (ether oxygen) and -CH2 Scissoring vibration (connected with end -OH group) of PEO, which are highly sensitive for hydrogen bonding.
On comparing PAM/PEO blends with pure polymers, the absorption bands at 2800 - 3600 cm−1 corresponding to -CH2, -OH and -NH stretching vibrations with decreased in intensity and clearly peak shift have been observed. This peaks shifting indicate interaction between the -CONH2 group of Polyacrylamide and ethyl group and -OH group (end group of polymer chain) of Poly Ethylene Oxide. For 70/30 wt% of PAM/PEO blend, this shift is maximum on higher wave number side, so it has a strong tendency for the formation of strong bond [32]
 (a) (b)
(a) (b)
Figure 1. FTIR Spectra of pure PAM, pure PEO and blend polymers (a) 600 - 4000 cm−1 (b) 1400 - 1800 cm−1.

Table 1. Assignments of the FT-IR characterization of bands of the pure PAM, pure PEO and PAM/PEO blend [16] [18] [38] -[40] .
[37] . As increasing content of PEO in the blend, the above peak intensity starts diminishing and also shift towards lower wave number and intensity becomes very weak for 30/70 wt%. In the region, 1100 - 1800 cm-1, we observed five significant peaks, C-O-C stretching of PEO(ether oxygen) is observed at 1106 cm-1 which is shifted towards higher wave number(1106 to 1119 cm-1), C-N stretching vibration of PAM shifted to higher wave number (1447 to 1469 cm-1), -CH2 scissoring vibration of the -CH2OH group of PEO also tends to shift towards higher wave number (1454 to 1469 cm-1), N-H bending vibration of PAM is observed at 1607 cm-1which is also shifts towards higher wave number (1607 to 1620 cm-1) and peak at 1651 cm-1, due to C=O stretching vibration of the -CONH2 group of PAM also shifts to higher wave number side to 1678 cm-1. These peaks shifting observations support the formation of hydrogen bonding between the -CONH2 group of PAM and C-O-C group of PEO. In this region, maximum peak shift is observed for 70/30 wt% on higher wave number side.
From all the blend spectra, peaks shifting were observed for all blends, indicating that the intermolecular interaction occurs between two polymer chains. For 70/30 wt% of PAM/PEO blend, maximum peak shift towards higher wave number side was observed due to maximum intermolecular interaction. This indicates that the bond strength for 70/30 wt% is maximum.
We concluded from FTIR spectra that intermolecular hydrogen bonding interaction occurred between -CONH2 group of PAM and C-O-C group of PEO which increases the bond strength. This interaction is maximum for 70/30 wt%.
4.2. Raman Spectroscopy
The Raman spectroscopy is suitable and efficient method for the structural analysis of polymers. It is possible to characterize molecular bonds in various phase and conformational states with the help of Raman spectroscopy. If two polymers are fully or partially miscible then, their Raman spectra have considerable difference in band position and shapes of the spectra of blend and each of the pure polymers [41] .
We have observed distinctive differences in Raman spectra of PAM, PEO and their blends as shown in Figure 2(a) and Figure 2(b). Raman peak values for pure and blend polymers are shown in Table 2. The bands near 854 cm-1 corresponding to C-C stretching region of PAM. The band at 1107 cm-1 attributed to the C-O-C stretching modes of PAM. The band near 1400 cm-1 assigned mainly due to the C-N stretching vibration. A band near 1711 cm-1 attributed to C=O stretching mode of the PAM polymer chain.
For PEO, it is possible to observe the intense bands at 847 cm-1 and 1104 cm-1, corresponding to the stretching modes of C-C and C-O respectively. The Raman band at 1336 cm-1, 1454 cm-1 and 1633 cm-1 are correspondingly assigned to the -CH2 wagging, -CH2 deformation and -CH2 twisting respectively.
Peak shifts were observed for all blends, which indicate the intermolecular interaction between two polymer chains. Maximum peak shift is observed for 70/30 wt% of PAM/PEO blend due to maximum intermolecular interaction.
In the range of the stretching vibrations of the -CH2 and -CH3 groups, an increase in PEO content causes increased intensity of the line assigned to the symmetrical vibration of the -CH2 group and a simultaneous monotonic shifting of peak position of this line occurs from 2919 cm-1 to 2879 cm-1. Maximum peak shift is observed for 30/70 wt% on lower wave no side which indicates the decreased bond strength. A simultaneous decrease in intensities observed for peaks at 3259 cm-1 and 3270 cm-1, which assigned to the -NH2 stretching of PAM and
 (a) (b)
(a) (b)
Figure 2. Raman spectra of pure PAM, pure PEO and blend films in the range (a) 600 - 2000 cm−1 (b) 2700 - 3500 cm−1.
Table 2. Assignments of Raman bands of pure PAM, pure PEO and PAM/PEO blends [39] [42] [43] .
-OH stretching region of PEO respectively. C-O stretching and -CH2 waging peak of PEO also exhibit shifting as compared to pure one. Maximum shifting and peak intensity of C-O stretching peak is observed maximum for 70/30 wt%. These confirm the interaction between -CONH2 group of PAM and C-O-C group (ether oxygen) and -CH2OH group (end group) of PEO. -OH group are present at the end of PEO chain so the number of -OH group is very less as compared to ether oxygen which is in the repeating unit of PEO. That is why interaction between hydroxyl group (end group) of PEO and amine group of PAM is less. For these groups, maximum peak shift to higher wave number side observed for 70/30 wt% (Table 2), which indicates in enhancement of bond strength.
We can see that, for 30/70 blend ratio, intensity of the -NH2 stretching of the -CONH2 group of PAM and -OH stretching of the -CH2OH group of PEO is almost disappeared, which reveals the interaction between these two groups of pure polymers. Peaks of C=O stretching and -NH2 stretching are very weak, which also confirms the prediction. From all the Raman spectra, we concluded that hydrogen bonding interaction at the molecular level occurs between -CONH2 group of PAM and C-O-C group (ether oxygen) and -CH2OH group (end group) of PEO which confirm also FTIR results.
4.3. DSC Analysis
To get the information of different phase transition temperature, DSC measurements have been carried out on the prepared samples. When a polymer blend is consisting of a crystalline polymer and an amorphous polymer, its melting temperature is observed to be decreased, from which we can observe polymer-polymer interaction which is necessary to study of miscibility of polymer blend [20] . The DSC plot of the investigated (PAM/PEO) polymer blend is shown in Figure 3(a). The melting temperature Tm of the polymer blends depends on the PEO concentration. The melting temperature (Tm) for pure PEO is observed around 67.65˚C and it was shifted to 64.88˚C, 67.20˚C and 72.09˚C for 70/30, 50/50 and 30/70 wt% of PAM/PEO blend films, respectively.
DSC provides a quick method for determining polymer crystallinity. PAM does not observed melting peak in the DSC operation range, so heat of fusion values calculated from the melting peaks were considered for PEO portions. The crystallinity (cc) of PEO in blend films was calculated from DSC data according to the following equation
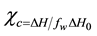 [44] Where, DH0 is heat of fusion for per gram of 100% crystalline PEO(=213.7 J/g) [45] . fw is the weight fraction of PEO and DH is heat of fusion of blend sample. The calculated melting enthalpy and the value of degree of crystallinity cc (%) are shown in Table 3. In the present investigation, the degree of crystallinity of (PAM/PEO blend) polymers increases with increment in of PEO. This increment of crystallinity of polymer blends show that the PEO interacts strongly with the PAM.
[44] Where, DH0 is heat of fusion for per gram of 100% crystalline PEO(=213.7 J/g) [45] . fw is the weight fraction of PEO and DH is heat of fusion of blend sample. The calculated melting enthalpy and the value of degree of crystallinity cc (%) are shown in Table 3. In the present investigation, the degree of crystallinity of (PAM/PEO blend) polymers increases with increment in of PEO. This increment of crystallinity of polymer blends show that the PEO interacts strongly with the PAM.
From the Flory-Huggins theory, Polymer-polymer interaction parameter can be calculated with the help of
 (a) (b)
(a) (b)
Figure 3. (a) DSC curve of pure PAM, pure PEO and blends (b) Dependence of
 with
with
 for PAM/ PEO blends.
for PAM/ PEO blends.
Table 3. Tm (˚C), DH (J/g), cc (%), of PAM/PEO Blends.
below Nishi-Wang equation [20] :
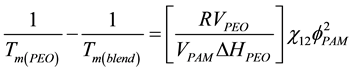
where VPEO and VPAM is the molar volume of the repeating unit of the polymer; ΔHPEO is the melting enthalpy of fully crystalline PEO; φ is the volume fraction; χ12 is the polymer-polymer interaction parameter, R is the universal gas constant. From the above parameter for the blends, the evaluation of polymer-polymer interaction was observed and hence miscibility of the system was performed. It can be shown that negatives values for the χ12 are correlated to existence of interactions between the polymers, thus resulting in the miscibility of the system.
Figure 3(b) shows the curve of
 versus
versus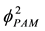 . The slope of the line is related to the value
. The slope of the line is related to the value
of χ12. If the negative slope is obtained then value of χ12 is negative which reveals the system is miscible. For PAM/PEO blend the interaction parameter χ12 was calculated using below parameter.
R = 8.314 J・K-1・mol-1,
ΔHPEO = 7.6 J・K-1・mol-1,
VPEO = 40.3 × 10-6 m3・mol-1
VPAM = 24.09 × 10-6 m3・mol-1
The value of χ12 is equal to -203.7 so the negative value for χ12 indicates miscibility of polymers.
4.4. UV-Vis Spectroscopy
The absorption of light energy by polymeric materials in the UV-Vis regions involves transition of electrons in s, p and n-orbital from the ground state to higher energy states [46] -[48] . The optical absorption method can be used for the investigation of the optically induced transitions. It provides information about the band structure and energy gap in crystalline and non-crystalline materials [49] . The absorbance process plays an important role in the optical properties of the polymers. The absorption coefficient was determined from the UV-VIS spectra using the formula:
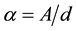 (1)
(1)
where A is the absorbance and d is the thickness of the film. The Tauc relation for dependence of absorbance on photon energy is [50] :
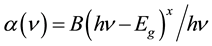 (2)
(2)
where α(ν) is the absorption coefficient, Eg is the optical energy gap of the substance, h is plank’s constant, ν is the corresponding frequency, x is the parameter that gives the type of electron transition. Two distinct linear relations were observed, one for x = 1/2 (Direct transition) and other was x = 2 (Indirect transition). Factor B depends on the transition probability and can be assumed to be constant within the optical frequency range [51] [52] . Eg is the optical energy gap. On the basis of Equation (2), direct and indirect band gap and absorption edge can be determined [53] .
4.5. Absorbance and Band Edges
Figure 4(a) shows the absorption spectra of the Pure and blended polymer films. From the spectra, we can see that peak height increases, band edge increases and the absorption band shifts towards shorter wavelengths with
 (e)
(e)
Figure 4. Plot of (a) Absorption coefficient (α) vs. Wavelength (λ); (b) Absorption coefficient (α) vs. Photon Energy (hν); (c) (αhν)2 vs. hν; (d) (αhν)1/2 vs. hν; (e) ln α vs. Photon Energy (hν).
vs. Photon Energy (hν); (c) (αhν)2 vs. hν; (d) (αhν)1/2 vs. hν; (e) ln α vs. Photon Energy (hν).
increasing the weight percentage of PEO. The optical absorption coefficient (α) was determined from the absorption spectra using Equation (1). The plot of absorption coefficients (α) versus photon energy (hν) of the pure and blended polymer films is shown in Figure 4(b). The position of the absorption edge were calculated by extrapolating the linear portions of this plots to zero absorption values which are shown in Table 4.
4.6. Direct and Indirect Optical Band Gap
The optical band gap of the Pure and blend samples was determined from the UV-Vis spectra [38] [54] . The value of the optical direct and indirect energy gap is determined from the intersection of the extrapolated line of the curves with the photon energy axis at zero absorption value. In an allowed direct transition, the electron is simply transferred vertically from the top of the valence band to the bottom of the conduction band, without a change in momentum (wave vector) [55] . The direct optical band gap can be determined from the plot of (αhν)2 as a function of photon energy (hν) as shown in Figure 4(c).
In indirect band gap, a transition from the valence to the conduction band should always be associated with a phonon of the right magnitude of crystal momentum [51] . For indirect transition, photon assistance requires. Indirect band gap can be obtained from the plots of (αhν)1/2 versus photon energy (hν) as shown in Figure 4(d).
The values of direct and indirect band gap for the pure and blended films are listed in Table 4. From the table, direct and indirect band gap increases with increasing PEO percentage. This increase in the optical band gap values is due to the formation of defects created during the blending, and the interaction between the polymer chains [56] -[58] and the formation of some bonds [59] . As the amorphous nature of the films increases, the charge carrier finds a continuous chain to travel [51] . But here natures of blend films tend towards crystallinity, so we found band gap were increase. Our result is also match with above results.
4.7. Activation Energy
The optical activation energy can be determined using the Urbach rule [60] as
 (3)
(3)
where B is a constant and Ea is the activation energy, i.e. the inverse slopes of the exponential edge. The latter interprets as the width of the tail of localized states extending into the forbidden band gap from either the valence or conduction band [52] .
The values of activation energy Ea has been determined by taking the reciprocals of the slopes of the linear portions of plots of ln α versus photon energy (hν) as shown in Figure 4(e). The values of Ea for the Pure and blended doped films are listed in Table 4.
4.8. Mechanical Properties
Mechanical properties of PAM/PEO were taken to study the Max load, Ultimate tensile strength (UTS), young’s modulus (Y.M.), stress at break, stiffness and elongation at break for pure and blends films and are shown in Figure 5. The mechanical properties in blends are changing because pure polymer matrix provided different cross linking density with different weight % of blended polymer [59] . Polymers have crosslinking density or rigid chain gain a higher strength and lower extensions and hence, polymers with higher young’s modulus and ul
Table 4. Absorption edge, optical band gap (both direct and indirect) and activation energy values of pure PAM, pure PEO and PAM/PEO polymer blend films.

Figure 5. Variation in Max load, Ultimate tensile strength, Young’s Modulus, stress at break, Elongation at break, Stiffness as a function of PAM/PEO content.
timate tensile strength value will have lower elongation value [61] -[63] . When we introduce PEO into PAM polymer matrix, mechanical properties of blends greatly influence. From the graph, results also agree with the above conclusion. For 70/30 blend ratio, the YM and UTS values are higher but have lower elongations value. Mechanical properties of blend are improving successfully when we blend PEO with PAM. When PEO blends with PAM, interaction at the molecular level occurs, which cause the enhancement in mechanical properties. Enhancement in mechanical properties is due to the strong hydrogen bonding between -CONH2 groups in PAM and C-O-C (ether oxygen) group in PEO. This interaction becomes maximum for 70/30 wt%. Therefore, we obtain maximum value of mechanical properties. This can also be correlated with IR analysis. The maximum peak shift observes for 70/30 wt%, which indicates strong bond interaction between two polymers. Because of this interaction, increment in mechanical properties occurs.
4.9. Thermo Gravimetric Analysis
Thermogravimetric analysis (TGA) is most suitable methods for studying the thermal properties of polymers. The TGA and derivative TGA (DrTG) curve provide information about the nature and extent of degradation of the polymers. The effect of blend weight percentage on the TGA and DrTG of PAM/PEO blends is shown in Figure 6(a) and Figure 6(b). Detailed information of thermograms is given in Table 5 and Table 6.
An important thermal property is the temperature corresponding to the maximum rate of weight loss (Tp), which is defined as the peak value of the first derivative of the TGA curve. Tp is used as a mark of thermal stability. The first derivative curves for pure PAM, pure PEO and their blends are shown in Figure 6(b), and their Tp values are listed in Table 5.
Thermal stability of the blend is higher than the pure PAM in the major weight loss region because Tp has shifted towards higher temperature. Tp was maximum for 70/30 wt %, so this blend ratio is more thermally stable. This higher thermal stability is observed for 70/30 wt % blend sample from TGA and DrTG are due to the intermolecular cross linking reaction which gave highly compatible impact blend system [63] -[65] .
Table 6 gives an idea about the effect of the blend ratio on the temperature corresponding to different weight losses (T30 = temperature corresponding to 30 wt % degradation, and so on). It is seen from the Table 6 that 70/30 wt% had maximum temperature value for different weight losses. So we can conclude that the 70/30 wt% have greater thermal stability as compared to pure component. R500 indicate the residue value of polymer content at 500˚C. This value is also higher for the blend ratio of 70/30 wt%. From TGA, we conclude that the thermal stability in major weight loss region of the blended samples were higher than the PAM and thermal stability is enhanced by increment of PEO content in PAM polymer matrix, and it becomes more stable for 70/30 wt%. This indicates the strong bonding between PAM and PEO due to -CONH2 groups in PAM and C-O-C (ether oxygen) group in PEO, which is also confirmed by obtained FTIR study.
 (a) (b)
(a) (b)
Figure 6. (a) TG of pure PAM, pure PEO and blends (b) Dr TG of pure PAM, pure PEO and blends.
Table 5. TG and DrTG data of Pure PAM, PEO and their blended samples.
Table 6. Effect of the blend ratio on the temperatures corresponding to different percentage weight losses in PAM/PEO Blends.





Figure 7. Scanning eectron micrograph of (a) pure PAM; (b) 70/30; (c) 50/50; (d) 30/70; (e) pure PEO.
4.10. Morphological Characterization
Figures 7(a)-(e) shows scanning electron micrographs of the fracture surfaces of PAM and PEO and PAM/PEO blend with different composition. SEM micrographs clearly show the changed in surface morphologies of the different blends as compared to pure polymers. As shown in Figure 7(a) and Figure 7(e), surface of PAM and PEO are very smooth, showing only a limited number of small particles dispersed along the micrograph. As we increase the fraction of PEO in the PAM polymer matrix, surface appears heterogeneous due to enhanced volume fraction of polymer.
From the blend’s micrographs (Figures 7(b)-(d)), we concluded that as we added PEO in PAM polymer matrix, due to PEO nature, blend films form irregular shaped clusters. But for 70/30 wt% quite homogeneous smooth surface was observed which indicate the miscibility of polymers. This result also confirmed by DSC analysis. Miscibility for 70/30 wt % may be attributed to the formation of hydrogen bonding between two polymers [66] . From Figure 7(c) and Figure 7(d), surface appears packed in, separated domains and porous for 50/50 wt % and 30/70 wt %. So 70/30 wt% shows the maximum strong intermolecular interaction between PAM and PEO. Hence 70/30 wt% had more thermal stability and higher mechanical properties. These results are also correlated with FTIR conclusion.
5. Conclusion
Blends of PAM and PEO were obtained as semi-transparent, flexible and free standing films with good thermal, optical and mechanical properties. FTIR and Raman analysis showed that when PEO blended with PAM, blend components exhibited significant interaction with each other via hydrogen bonding between -CONH2 groups in PAM and C-O-C group (ether oxygen) and -CH2OH group (end group) of PEO. This intermolecular interaction is maximum for 70/30 wt% which exhibites the strong bond interaction and due to this; it has maximum thermal and mechanical properties. So we directly correlate enhancement of thermal stability and mechanical properties by noticing the blue shift of IR and Raman peak, which are contributed to intermolecular interaction between polymers. Optical spectra provide proof for interaction between PAM and PEO. The shift of the absorption edge in the blends reflects the difference in the energy gap which arises due to the intermolecular interaction between PAM and PEO, which shows that structural variation occurs in blends system. This variation increases with increasing concentration of PEO, which reflectes in the form of decrease in the energy gap of the blends. So from this study we conclude that the blend of PAM/PEO with 70/30 wt% is the most suitable and compatible with most enhancing properties.
Acknowledgements
The authors are thankful to Prof. C. F. Desai for providing financial support and facilities under DRS-Phase-III and also grateful to Prof. N. L. Singh for fruitful discussion. Thanks are also due to Department of Zoology, The M.S. University of Baroda for providing experimental facilities. Patel Gaurang is also grateful to UGC, New Delhi for providing Research fellowship (UGC-RFSMS).
Cite this paper
GaurangPatel,Mundan B.Sureshkumar,PurviPatel, (2015) Spectroscopic Investigation and Characterizations of PAM/PEO Blends Films. Soft,04,9-24. doi: 10.4236/soft.2015.42002
References
- 1. Sauchez, I.C. (1983) Bulk and Interface-Thermodynamics of Polymer Alloys. Annual Review of Material Science, 13, 387-412. http://dx.doi.org/10.1146/annurev.ms.13.080183.002131
- 2. Utracki, L.A. (2003) Polymer Blends Handbook, Vol. 2. Kluwer Academic Publishers, USA. http://dx.doi.org/10.1007/0-306-48244-4
- 3. Florence, C. and Christine, J. (2013) Chitosan-Based Biomaterials for Tissue Engineering. European Polymer Journal, 49, 780-792. http://dx.doi.org/10.1016/j.eurpolymj.2012.12.009
- 4. Caykara, T. and Demirci, S. (2007) Thermal, Spectroscopic, and Mechanical Properties of Blend Films of Poly(N- Vinyl-2-Pyrrolidone) and Sodium Alginate. Polymer-Plastic Technology & Engineering, 46, 737-741. http://dx.doi.org/10.1080/03602550701273971
- 5. Yuk, S.H., Cho, S.H. and Lee, H.B. (1995) pH-Sensitive Drug Delivery System Using O/W Emulsion. Journal of Controlled Release, 37, 69-74. http://dx.doi.org/10.1016/0168-3659(95)00065-G
- 6. Araya-Hermosilla, R., Broekhuis, A.A. and Picchioni, F. (2014) Reversible Polymer Networks Containing Covalent and Hydrogen Bonding Interactions. European Polymer Journal, 50, 127-134. http://dx.doi.org/10.1016/j.eurpolymj.2013.10.014
- 7. Dean, K., Yu, L., Bateman, S. and Dong, Y.W. (2007) Gelatinized Starch/Biodegradable Polyester Blends: Processing, Morphology, and Properties. Journal of Applied Polymer Science, 103, 802-811. http://dx.doi.org/10.1002/app.25149
- 8. Mruthyunjaya, S. and Swamy, T.M. (2007) Studies on Miscibility of Sodium Alginate/Polyethylene Glycol Blends. Journal of Macromolecular Science, Part A: Pure and Applied Chemistry, 44, 321-327. http://dx.doi.org/10.1080/10601320601077492
- 9. Imre, B. and Pukánszky, B. (2013) Compatibilization in Bio-Based and Biodegradable Polymer Blends. European Polymer Journal, 49, 1215-1233. http://dx.doi.org/10.1016/j.eurpolymj.2013.01.019
- 10. Caulfield, M.J., Qiao, G.G. and Solomon, D.H. (2002) Some Aspects of the Properties and Degradation of Polyacrylamides. Chemical Reviews, 102, 3067-3084. http://dx.doi.org/10.1021/cr010439p
- 11. Lopatin, V.V., Askadskii, A.A., Peregudov, A.S. and Vasilev, V.G. (2005) Structure and Relaxation Properties of Medical-Purposed Polyacrylamide Gels. Journal of Applied Polymer Science, 96, 1043-1058. http://dx.doi.org/10.1002/app.21477
- 12. Abdelhak, M., Abdelkarim, H. and Barbara, I. (2012) Effect of Plant Fiber-Polyacrylamide Blend on Retention and Evaporation Water at Arid and Semi-Arid Soils of Algeria. Journal of Chemistry and Chemical Engineering, 6, 7-17.
- 13. Wever, D.A., Picchioni, F. and Broekhuis, A.A. (2013) Branched Polyacrylamides: Synthesis and Effect of Molecular Architecture on Solution Rheology. European Polymer Journal, 49, 3289-3301. http://dx.doi.org/10.1016/j.eurpolymj.2013.06.036
- 14. Gavrilin, M.V. (2001) Application of Polymers and Copolymers Based on Acrylic Acid and Ethylene Oxide in Pharmacy (A Review). Pharmaceutical Chemistry Journal, 35, 35-39. http://dx.doi.org/10.1023/A:1010402826818
- 15. Suzuki, H., Miyemoto, N., Masad, T., Hayakawa, E. and Ito, K. (1996) Solid Dispersion of Benidipine Hydrochloride. I. Preparation Using Different Solvant System and Dissolution Properties. Chemical and Pharmaceutical Bulletin (Tokyo), 44, 364-371. http://dx.doi.org/10.1248/cpb.44.364
- 16. Abderlrazek, E.M. and Ibrahim, H.S. (2010) Effect of Heprin Calcium Different Concentration on Some Physical Properties and Structure in PAM Matrix. Physica B, 405, 4339-4343. http://dx.doi.org/10.1016/j.physb.2010.07.038
- 17. Lee, K.Y. and Yuk, S.H. (2007) Polymeric Protein Delivery Systems. Progress in Polymer Science, 32, 669-697. http://dx.doi.org/10.1016/j.progpolymsci.2007.04.001
- 18. Parmar, A.V., Bahadur, A., Kuperkar, K. and Bahadur, P. (2013) PEO-PPO Based Star-Block Copolymer T904 as pH Responsive Nanocarriers for Quercetin: Solubilization and Release Study. European Polymer Journal, 49, 12-21. http://dx.doi.org/10.1016/j.eurpolymj.2012.10.009
- 19. Salmaso, S., Semenzato, A., Bersani, S., Matricardi, P., Rossi, F. and Caliceti, P. (2007) Cyclodextrin/PEG Based Hydrogels for Multi-Drug Delivery. International Journal of Pharmaceutics, 345, 42-50. http://dx.doi.org/10.1016/j.ijpharm.2007.05.035
- 20. Pereira, A.G., Paulino, A.T., Rubira, A.F. and Muniz, E.C. (2010) Polymer-Polymer Miscibility in PEO/Cationic Starch and PEO/Hydrophobic Starch Blends. eXPRESS Polymer Letters, 4, 488-499. http://dx.doi.org/10.3144/expresspolymlett.2010.62
- 21. Pereira, A.G., Paulino, A.T., Nakamura, C.V., Britta, E.A., Rubira, A.F. and Muniz, E.C. (2011) Effect of Starch Type on Miscibility in Poly(ethylene oxide) (PEO)/Starch Blends and Cytotoxicity Assays. Materials Science and Engineering C, 31, 443-451. http://dx.doi.org/10.1016/j.msec.2010.11.004
- 22. Sim, L.H., Gan, S.N., Chan, C.H., Kammer, H.W. and Yahya, R. (2009) Compatibility and Conductivity of LiClO4 Free and Doped Polyacrylate-Poly(ethylene oxide) Blends. Materials Research Innovations, 13, 278-281. http://dx.doi.org/10.1179/143307509X440523
- 23. Kiran Kumar, K., Ravi, M., Pavani, Y., Bhavani, S., Sharma, A.K. and Narasimha Rao, V.V. (2011) Investigations on the Effect of Complexation of NaF Salt with Polymer Blend (PEO/PVP) Electrolytes on Ionic Conductivity and Optical Energy Band Gaps. Physica B, 406, 1706-1712. http://dx.doi.org/10.1016/j.physb.2011.02.010
- 24. Reddeppa, N., Sharma, A.K., Narsimha Rao, V.V. and Chen, W. (2013) Preparation and Characterization of Pure and KBr Doped Polymer Blend (PVC/PEO) Electrolyte Films. Microelectronic Engineering, 112, 57-62. http://dx.doi.org/10.1016/j.mee.2013.05.015
- 25. Elashmawi, I.S., Abdelrazek, E.M., Hezma, A.M. and Rajeh, A. (2014) Modification and Development of Electrical and Magnetic Properties of PVA/PEO Incorporated with MnCl2. Physica B, 434, 57-63. http://dx.doi.org/10.1016/j.physb.2013.10.038
- 26. Ferreiro, V., Douglas, J.F., Warren, J. and Karim, A. (2002) Nonequilibrium Pattern Formation in the Crystallization of Polymer Blend Films. Physical Review E, 65, Article ID: 042802. http://dx.doi.org/10.1103/physreve.65.042802
- 27. Ferreiro, V., Douglas, J.F., Warren, J. and Karim, A. (2002) Growth Pulsations in Symmetric Dendritic Crystallization in Thin Polymer Blend Films. Physical Review E, 65, Article ID: 051606. http://dx.doi.org/10.1103/physreve.65.051606
- 28. Sawatari, C.H. and Kondo, T. (1999) Inter Chain Hydrogen Bonds in Blend Films of Poly(vinyl alcohol) and Its Derivatives with Poly(ethylene oxide). Macromolecules, 32, 1949-1955. http://dx.doi.org/10.1021/ma980900o
- 29. Zheng, H., Zheng, S. and Guo, Q. (1997) Thermosetting Polymer Blends of Unsaturated Polyester Resin and Poly(ethylene oxide). II. Hydrogen-Bonding Interaction, Crystallization Kinetics, and Morphology. Journal of Polymer Science A—Polymer Chemistry, 35, 3169-3179. http://dx.doi.org/10.1002/(SICI)1099-0518(19971115)35:15<3169::AID-POLA10>3.0.CO;2-9
- 30. Vijayalakshmi, S.P. and Madras, G. (2006) Thermal Degradation of Water Soluble Polymers and Their Binary Blends. Journal of Applied Polymer Science, 101, 233-240. http://dx.doi.org/10.1002/app.23246
- 31. Vijayalakshmi, S.P., Raichar, A. and Madras, G. (2006) Thermal Degradation of Poly(ethylene oxide) and Poly Acrylamide with Ascorbic Acid. Journal of Applied Polymer Science, 101, 3067-3072. http://dx.doi.org/10.1002/app.24115
- 32. Pielichowska, K., Glowinkowski, S., Lekki, J., Binias, D., Pielichowski, K. and Jenczy, J. (2008) PEO/Fatty Acid Blends for Thermal Energy Storage Materials-Structural/Morphological Features and Hydrogen Interactions. European Polymer Journal, 44, 3344-3360. http://dx.doi.org/10.1016/j.eurpolymj.2008.07.047
- 33. Coleman, M.M. and Moskala, E.J. (1983) FTi.r. Studies of Polymer Blends Containing the Poly(hydroxyl ether of bisphenol A) and Poly(ε-caprolactone). Polymer, 24, 251-257. http://dx.doi.org/10.1016/0032-3861(83)90258-6
- 34. Fernandez-Berridi, M.J., Valero, M., Deilarduya, A.M., Espi, E. and Iruin, J.J. (1993) Phenoxy Blends: An Approach to the Miscibility by FTi.r. and Chemical Modification of the Interacting Sites. Polymer, 34, 38-42. http://dx.doi.org/10.1016/0032-3861(93)90280-N
- 35. Sowwan, M., Faroun, M., Musa, I., Ibrahim, I., Makharza, S., Sultan, W. and Dweik, H. (2008) Study on the Morphology of Polyacrylamide—Silica Fumed Nanocomposite Thin Films. International Journal of Physical Science, 3, 144-147.
- 36. Colthup, N.B., Daly, L.H. and Berley, S.E. (1990) Introduction to Infrared and Raman Spectroscopy. 3rd Edition, Academic Press Inc., Boston.
- 37. Chen, N. and Zhang, J. (2010) The Role of Hydrogen-Bonding Interaction in Poly(vinyl alcohol)/Poly(acrylic acid) Blending Solutions and Their Films. Chinese Journal of Polymer Science, 28, 903-911. http://dx.doi.org/10.1007/s10118-010-9167-x
- 38. Patel, G. and Sureshkumar, M.B. (2013) Preparation of PAM/PVA Blending Films by Solution Cast Technique and Its Characterization: A Spectroscopic Study. Iranian Polymer Journal, 23, 153-162. http://dx.doi.org/10.1007/s13726-013-0211-x
- 39. Guo, C., Liu, H., Wang, J. and Chen, J. (1999) Conformational Structure of Tri-Block Copolymers by FT-Raman and FTIR Spectroscopy. Journal of Colloid and Interface Science, 209, 368-373. http://dx.doi.org/10.1006/jcis.1998.5897
- 40. Bostan, M.S., Mutlu, E.C., Kazak, H., Keskin, S.S., Oner, E.T. and Eroglu, M.S. (2014) Comprehensive Characterization of Chitosan/PEO/Levan Ternary Blend Films. Carbohydrate Polymers, 102, 993-1000. http://dx.doi.org/10.1016/j.carbpol.2013.09.096
- 41. Dong, J., Fredericks, P.M. and George, G.A. (1997) Studies of the Structure and Thermal Degradation of Poly(viny1 chloride)-Poly(N-vinyl-2-pyrrolidone) Blends by Using Raman and FTIR Emission Spectroscopy. Polymer Degradation and Stability, 58, 159-169. http://dx.doi.org/10.1016/S0141-3910(97)00040-2
- 42. Sundaraganesan, N., Puviarasan, N. and Mohan, S. (2001) Vibrational Spectra, Assignments and Normal Coordinate Calculation of Acrylamide. Talanta, 54, 233-241. http://dx.doi.org/10.1016/S0039-9140(00)00585-3
- 43. da costa, A.M.A. and Amada, A.M. (2000) Molecular Interaction/Lithium Perchlorate Hydrogel Composites. Polymer, 41, 5361-5365. http://dx.doi.org/10.1016/S0032-3861(99)00732-6
- 44. Jurkin, T. and Pucic, I. (2012) Poly(ethylene oxide) Irradiated in the Solid State, Melt and Aqueous Solution—A DSC and WAXD Study. Radiation Physics and Chemistry, 81, 1303-1308. http://dx.doi.org/10.1016/j.radphyschem.2011.12.021
- 45. Shin, J.H., Kim, K.W., Ahn, H.J. and Ahn, J.H. (2002) Electrochemical Properties and Interfacial Stability of (PEO)10 LiCF3SO3-TinO2n-1 Composites Polymer Electrolytes for Lithium/Sulfur Battery. Journal of Material Science Engineering B, 95, 148-156. http://dx.doi.org/10.1016/S0921-5107(02)00226-X
- 46. John, R.D. (1965) Applications of Absorption Spectroscopy of Organic Compounds. Prentice-Hall Inc., Englewood Cliffs.
- 47. Srivastava, A.K. and Virk, H.S. (2000) Modification of Optical Response of Polyvinyl Acetate Induced by 250 keV D Exp+ Ion Bombardment. Journal of Polymer Materials (Netherlands), 17, 325-328.
- 48. Patel, G., Sureshkumar, M.B. and Patel, P. (2011) An Optical Characterization of PVA/PAM Polymer Blend. AIP Conference Proceedings, 1349, 166-167. http://dx.doi.org/10.1063/1.3605788
- 49. Kumar, R., Asad Ali, S., Mahur, A.K., Virk, H.S., Singh, F., Khan, S.A., Avasthi, D.K. and Prasad, R. (2008) Study of Optical Band Gap and Carbonaceous Clusters in Swift Heavy Ion Irradiated Polymers with UV-Vis Spectroscopy. Nuclear Instruments and Methods in Physics Research B, 266, 1788-1792. http://dx.doi.org/10.1016/j.nimb.2008.01.010
- 50. Tauc, J., Grigorovici, R. and Vanku, A. (1966) Optical Properties and Electronic Structure of Amorphous Germanium. Physica status Solidi (b), 15, 627-637. http://dx.doi.org/10.1002/pssb.19660150224
- 51. Shahada, L., Kassem, M.E., Abdelkader, H.I. and Hassan, H.M. (1997) Optical and Electrical Properties of a New Polymer. Journal of Applied Polymer Science, 65, 1653-1657. http://dx.doi.org/10.1002/(SICI)1097-4628(19970829)65:9<1653::AID-APP1>3.0.CO;2-E
- 52. Mohan, V.M., Raja, V., Balaji, B.P., Sharma, A.K. and Narasimha Rao, V.V.R. (2007) Structural, Electrical and Optical Properties of Pure and NaLaF4 Doped PEO Polymer Electrolyte Films. Journal of Polymer Research, 14, 283-290. http://dx.doi.org/10.1007/s10965-007-9108-8
- 53. Balasubramanyam, V.A., Reddy, T.J.R., Sharma, A.K. and Narasimha Rao, V.V.R. (2007) Electrical, Optical, and Structural Characterization of Polymer Blend (PVC/PMMA) Electrolyte Films. Ionics, 13, 349-354. http://dx.doi.org/10.1007/s11581-007-0124-9
- 54. Mott, N.F. and Davis, E.A. (1979) Electronic Processes in Non Crystalline Materials. 2nd Edition, Oxford University Press, London.
- 55. Kilarkaje, S., Manjunatha, V., Raghu, S., Ambika Prasad, M.V. and Devendrappa, H. (2011) Effect of 8 MeV Electron Irradiation on the Optical Properties of Doped Polymer Electrolyte Films. Journal of Physics D: Applied Physics, 44, Article ID: 105403. http://dx.doi.org/10.1088/0022-3727/44/10/105403
- 56. Nouh, S.A. (2004) Physical Changes Associated with Gamma Doses of PM-555 Solid-State Nuclear Track Detector. Radiation Measurement, 38, 167-172. http://dx.doi.org/10.1016/j.radmeas.2003.11.004
- 57. Singh, N.L., Sharma, A. and Avasthi, D.K. (2003) Effects of High Energy (MeV) Ion Beam Irradiation on Polyethylene Terephthalate. Nuclear Instruments and Methods in Physics Research B, 206, 1120-1123. http://dx.doi.org/10.1016/S0168-583X(03)00935-2
- 58. Buttafava, A., Consolati, G., Di Landro, L. and Mariani, M. (2002) γ-Irradiation Effects on Polyethylene Terephthalate Studied by Positron Annihilation Lifetime Spectroscopy. Polymer, 43, 7477-7481. http://dx.doi.org/10.1016/S0032-3861(02)00708-5
- 59. Mishra, R., Tripathy, S.P., Sinha, D., Dwivedi, K.K., Ghosh, S., Khathing, D.T., Muller, M., Fink, D. and Chung, W.H. (2000) Optical and Electrical Properties of Some Electron and Proton Irradiated Polymers. Nuclear Instruments and Methods in Physics Research B, 168, 59-64. http://dx.doi.org/10.1016/S0168-583X(99)00829-0
- 60. Lakshmi, G.B.V.S., Ali, V., Siddiqui, A.M., Kulriyac, P.K. and Zulfequar, M. (2009) Studies on Structural, Optical and Cluster Size of Poly(m-toluidine)-Polyvinyl Chloride Blends. Radiation Effect and Defects in Solids, 164, 162- 169. http://dx.doi.org/10.1080/10420150902764186
- 61. Patel, G., Sureshkumar, M.B., Singh, N.L. and Bhattacharya, S.S. (2010) Spectroscopic Correlation of Mechanical Properties of PVC/PMMA Polymer Blend. Journal of International Academy of Physical Sciences, 14, 91-100.
- 62. Chi, S.K. and Seung, M.O. (2001) Performance of Gel-Type Polymer Electrolytes According to the Affinity between Polymer Matrix and Plasticizing Solvent Molecules. Electrochimica Acta, 46, 1323-1331. http://dx.doi.org/10.1016/S0013-4686(00)00727-1
- 63. Patel, G., Sureshkumar, M.B. and Patel, P. (2011) Structural, Optical and Mechanical Properties of PVC/PMMA Polymer Blend by Spectroscopic Techniques. AIP Conference Proceedings, 1391, 645-648. http://dx.doi.org/10.1063/1.3643636
- 64. El-Kader, F.H., Gafer, S.A., Basha, A.F., Bannan, S.I. and Basha, M.A.F. (2010) Thermal and Optical Properties of Gelatin/Poly(vinyl alcohol) Blends. Journal of Applied Polymer Science, 118, 413-420. http://dx.doi.org/10.1002/app.30841
- 65. Aggour, Y.A. (1996) Thermal Degradation Studies of Poly(ethylene glycol allenyl-methyl ether)s and Their Copolymers with Styrene. Polymer Degradation and Stability, 51, 265-269. http://dx.doi.org/10.1016/0141-3910(95)00205-7
- 66. Mishra, S., Bajpai, R., Katare, R. and Bajpai, A.K. (2006) Preparation, Characterization and Microhardness Study of Semi Interpenetrating Polymer Networks of Polyvinyl Alcohol and Crosslinked Polyacrylamide. Journal of Material Science: Materials in Medicine, 17, 1305-1313. http://dx.doi.org/10.1007/s10856-006-0605-9
NOTES

*Corresponding author.