Case Reports in Clinical Medicine
Vol.06 No.04(2017), Article ID:75776,25 pages
10.4236/crcm.2017.64009
Rheumatic Mineralization of Aortic Valve and Anterior Mitral Leaflet―A Case Report
Ramachandran Muthiah
Thoothukudi Medical College Hospital, Thoothukudi, India

Copyright © 2017 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: March 23, 2017; Accepted: April 25, 2017; Published: April 28, 2017
ABSTRACT
Aim: To present the heterotopic ossification of left-sided heart valves due to rheumatic inflammation and biomineralization. Introduction: Calcification in the region of mitral-aortic continuity is significant at its origin and etio- pathogenesis. The etiology of valvular calcification may be divided into 3 groups, namely, inflammation, degeneration and metabolic disturbances. Calcification of cardiac valve leaflets is most often due to rheumatic etiology in tropical nations. Case Report: A 52-year-old male developed sudden onset of light-headedness and palpitations due to atrial fibrillation. Transthoracic 2D echocardiography revealed calcification of anterior mitral leaflet and aortic valve which resembles a bone-like structure and the patient was advised double valve replacement. Conclusion: It was known that the cellular mechanisms play an important role in its genesis and therapeutic strategies are targeted to reverse this process by understanding its biological mediators.
Keywords:
Rheumatic Mineralization, Mitral-Aortic “J-Shaped” Calcification, “Mushroom Shaped” Calcified Bicuspid Aortic Valve, Hutchinson-Gilford Progeria Syndrome, Therapeutic Strategies

1. Introduction
A calcified valve is the hardening of valve due to the deposition of calcium salts at the site of wear-and-tear and damage. It is important to remember that any valve can become calcified. However, calcification is more likely to affect the valves on the left side of the heart (mitral and aortic valves) since they experience greater pressure, more wear-and-tear damage with increased risk of deposition of calcium phosphate salts, known as dystrophic calcification. This senile valve calcification occurs on the mitral valve at the age of 50 - 70 years and on the aortic valve at 70 - 90 years of age. Calcification can occur earlier in life when associated with rheumatic heart disease and heavy calcification was seen in severely damaged valves by the rheumatic process. Since the disease process is complex and involves several different cell types, it is important to understand the cellular and matrix architecture of a normal heart valve. The valve endothelial cells (VECs) line the outer surface of the valve and maintain homeostasis by regulating permeability, paracrine signaling and function as a barrier to limit inflammatory cell infiltration and lipid accumulation. The valve interstitial cells (VICs) are the predominant cell type within the fibrosa, spongiosa and ventricularis layers of the valve and secrete extracellular matrix, including collagen, elastin, and glycosaminoglycan. The fibrosa consists of fibroblasts, circumferentially arranged fibrillar collagen fibers (type I and type III) and serves a load-bearing function. The spongiosa is predominantly found at the base of the leaflet and it is a layer of loose connective tissue containing mesenchymal cells, fibroblasts, mucopolysaccharides and it resist the compressive forces within the cusps. Ventricularis contain elastin fibers, oriented perpendicularly to the collagen and it reduces the radial strain. The normal aortic valve is made up of 3 cusps and its arrangement results in even distribution of mechanical stress to the valve ring and the aorta [1] . At their base, the valve leaflets are attached to a dense collagenous network, called the annulus, which facilitate their attachment to the aortic root and the dissipation of mechanical force. The anterior mitral leaflet is in direct continuity with left half of non-coronary cusp and the adjacent third of left coronary cusp of aortic valve. The mitral-aortic intervalvular fibrosa is the junctional tissue between the elements of mitral and aortic valves, called as mitral-aortic membrane or curtain. It is relatively avascular and offers little resistance to infection [2] .
Historically, calcification of valves has been attributed to prolonged “wear and tear” and age-related valvular degeneration, and recent evidence suggests that it is the result of active inflammatory process [3] leading to osteogenesis by involving biochemical, humoral and genetic factors. The mechanism of valvular calcification is similar to skeletal bone formation and it is the most common manifestation of rheumatic heart disease. The anterior mitral leaflet-aortic cusp continuity region was particularly affected by this mineralization process and so this case had been reported.
2. Case Report
A 52-year-old male was admitted with sudden onset of palpitations and he had a history of rheumatic fever during childhood. He developed the symptoms of exertional dyspnea at the age of 40 years and took penicillin prophylaxis for some period and then dropped it for 10 years. He married at the age of 26 years and had 5 healthy children. General examination revealed no stigmata of progeria syndrome. Pulse rate was 75 bpm and irregular. His blood pressure was 110/70 mmHg. Cardiac examination revealed a heaving apical impulse in the left 5th space at anterior axillary line, varying intensity of first heart sound with a mid- diastolic murmur at the apex and grade 4/6 mid-systolic murmur well heard at the apex as a conduction from right second intercostal space where its intensity is maximum due to “Gallavardin” phenomenon (harsh murmur of aortic stenosis change its quality and musical at the apex due to transmitted high pitched components and low pitched components were transmitted to neck vessels- as a dissociation between harsh, noisy, low frequency components at the right second space, transmitted to neck vessels as “carotid shudder” and high frequency musical components at the apex as a “musical murmur”). Blood chemistry revealed normal including the lipid profile. ECG revealed atrial fibrillation and LVH (left ventricular hypertrophy pattern) as shown in Figure 1. X-ray chest revealed concentric LVH and ascending aorta dilation as shown in Figure 2. Transthoracic 2D echocardiography revealed the dense calcification of anterior mitral leaflet and aortic valve as shown in Figures 3-6 as a result of rheumatic mineralization. The patient was treated with verapamil tablets 40 mg three times daily to keep the rhythm under control and advised early double valve replacement along with lifelong penicillin prophylaxis.
3. Discussion
3.1. Review of Literature
Lazare Riviere, a French physician in 1663, identified large caruncle-like masses

Figure 1. ECG showing the “atrial fibrillation” with LVH (left ventricular hypertrophy).

Figure 2. X-ray chest PA (postero-anterior) view showing concentric LVH (left ventricular hypertrophy-right black arrow) and dilated ascending aorta-left black arrow).
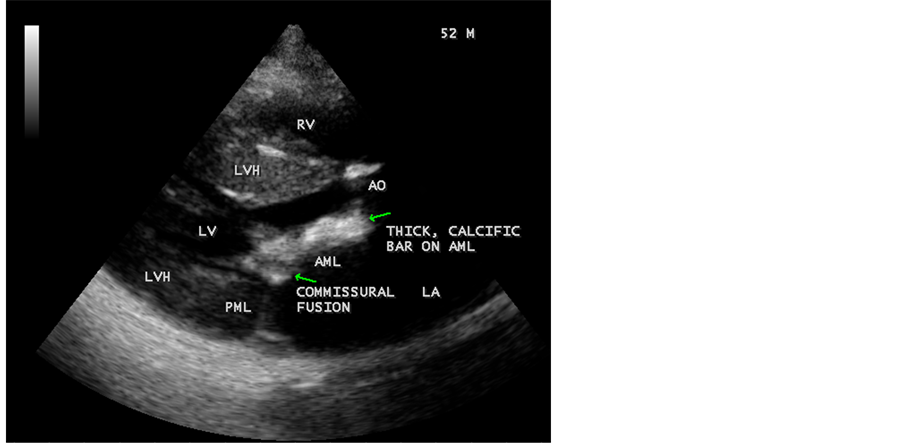
Figure 3. Parasternal long axis view showing the dense calcification of anterior mitral leaflet (AML) with commissural fusion in a 52-year-old male.
obstructing the left ventricular outflow to the aorta in necropsy findings as an earliest description of calcific aortic stenosis [4] and the calcified mass was originally attributed to endocarditis. In 1846, Hass suggested a degenerative process occurred within the valve itself to initiate ossification. In 1904, Moenckeburg found that the aortic valve calcification occurred as a result of calcium deposition on the valve cusps due to degeneration within the layers of valve leaflets or

Figure 4. Parasternal long axis view showing the calcific bar of anterior mitral leaflet and aortic valve in a 52-year-old male.

Figure 5. Parasternal long axis view showing the J-shaped appearance of mitral-aortic calcification in a 52-year-old male.
extension of sclerotic changes of aorta to valve cusps. In 1924, Dr. Carey Coombs described the inflammatory lesion in the rheumatic valve leaflets and the presence of new vessels developing within the valves [5] . In 1931, Christian first described that aortic stenosis with calcification of the cusps was possibly of rheumatic origin [6] . In 1938, Clawson, Noble and Lugkins stated that the “calcified aortic nodular deformity” was predominantly of a rheumatic nature [7] . In 1939, Dry and Willius concluded that the etiologic factor of calcification was rheumatic [8] and Hamburger and Saphir found evidence for both a rheumatic and nonrheumatic origin [9] . In 1961, Edwards discussed the occurrence of calcification in stenotic rheumatic valves [10] .

Figure 6. Short axis view showing the dense calcific bar in the aortic valve visible as calcium stone in a 52-year-old male.
3.2. Pathogenesis
Rheumatic heart disease is the most common cause of valvular heart disease in tropical nations such as India. Rheumatic valve calcification is not a random passive process, but it is a regulated, inflammatory cellular process associated with the expression of osteoblast markers and neoangiogenesis. Early stage of calcification is in a manner akin to atherosclerosis. Once the osteoblast activity has been established, the disease progression is more likely to be regulated by the mediators of calcium homeostasis [11] . A similar osteoblast-like process that occurs in degenerative calcific valve develops in the calcific process of rheumatic valves [12] . The cellular and molecular mechanisms that involve the valve, circulating and bone marrow derived cells have been implicated in the pathogenesis of calcification. The cellular differences between the rheumatic and degenerative lesions are the intense inflammatory infiltrate and VEGF (vascular endothelial growth factor) expression in areas of neoangiogenesis found in rheumatic valves compared to degenerative valves. The cardiovascular risk factors such as exposure to biochemical (hypercholesterolemia, hyperglycemia) or mechanical (hypertension) may disrupt the normal cellular and matrix architecture of the valve, produces an inflammatory tissue milieu, atherosclerosis like lesions, osteoblastic transformation and calcification process. The key mediators of valve calcification are shown in Table 1.
These mediators promote neoangiogenesis, extracellular matrix remodeling, fibrosis and calcification of valve leaflets.
Calcification of heart valves is an immune-inflammatory disease process. The rheumatic involvement produces an inflammatory tissue environment that disrupts the endothelial layer of valve leaflets in a mechanism similar to that in atherosclerosis. The endothelial disruption may allow the lipids, including low- density lipoproteins, apolipoprotein B, (a) and E2 to penetrate and accumulate in

Table 1. Key mediators of valve calcification.
areas of inflammation and undergo oxidative modification. These oxidative lipoproteins are highly cytotoxic, capable of stimulating intense inflammatory activity and subsequent mineralization [13] . The VECs (vascular endothelial cells) are activated and the expression of adhesion molecules that facilitate recruitment and transendothelial migration of monocytes which differentiate into macrophages, and infiltration of T-lymphocytes and α-actin expressor cells. The macrophages release matrix metalloproteases and cysteine endoproteases that lead to degradation of collagen and elastin in the valve matrix to disrupt the normal valve architecture [14] . The T-cells release proinflammatory cytokines, including transforming growth factor-β1, tumor necrosis factor-α and interleukin-1-β which induce myofibroblast activation and cellular proliferation, ultimately help to stimulate and establish subsequent fibrotic and calcification processes that drive increasing valve stiffness [15] [16] . Increased levels of activated CD8 cells (cytotoxic T-cells or natural killer cells) and memory effector cells in patients with calcified valves may indicate a systemic adaptive immune response on valve calcification [17] . Lrp5, a coreceptor of the family of LDL receptors, has been found to regulate cellular proliferation and bone formation in valvular tissues [18] . Genetic bone markers have been found to be important in the development of osteoblast bone formation, a process that precedes calcification [19] . Lp(a) levels mediate the effect of SN rs10455872 in the gene LPA in valvular calcification. The kringle IV type 2 (KIV-2) repeats at LPA locus, which encode apolipoprotein (a) and its variation determines the plasma Lp(a) levels. NOTCH 1 on chromosome 9q34-35 is a susceptibility locus for bicuspid aortic valve and accelerates valvular calcification [20] . At the site of endothelial damage, the endothelial progenitor cells participate in the repair process by transiently establishing residence and secreting factors that facilitate proliferation and migration of resident endothelial cells [21] . In calcified valves, these progenitor cells are dysfunctional and express increased proapoptotic caspne-3 activity and decreased telomeric-repeat binding factor-2 (TERF2) expression, indicating progenitor cell senescence [22] . The VECs (valvular endothelial cells), lose the endothelial cell properties, no longer express endothelial-specific markers such as cadherin, pieracque-like pleomorphism characteristic of mesenchymal cells or myofibroblasts and express α-smooth muscle actin, type I collagen and vimentin [23] . The transforming growth factor-β, transcription factor Msx2 and β-catenin signaling increase the motility and migration of these “transformed” VECs into the surrounding tissues [24] . Once the endothelial-to-mesenchymal transition (EnMT) occurs, the BMP 2 and 4 (bone morphogenic proteins) secreted by the activated endothelial cells stimulate the calcification by activating Smad and Wnt/B-cate- nin signaling and upregulate the expression of osteochondrogenic transcription factor Msx2 and the faster osteoblast transcription factor Runx2 [25] . Once Runx2 is expressed, the cells are committed to an osteoblast lineage, upregulated the expression of calcification-related proteins (osteopontin, bone sialoprotein II, osteocalcin) and undergo calcification [26] . A subpopulation of VICs (valve interstitial cells) become activated in the inflammatory zone of the valve and differentiated into myofibroblasts. These myofibroblasts secrete matrix metalloproteinases which play a role in the restructuring of valve leaflet matrix and responsible for accelerated fibrosis in this process [27] . Under the influence of signaling pathways, Wnt-3-Lrp5-β catenin [28] , OPG/RANK/RANKL pathway (osteoproteogen/receptor activation of nuclear factor kappa B/RANK ligand [29] , Runx-2/NOTCH1, activated by BMPs, the myofibroblasts undergo differentiation into osteoblasts, which subsequently coordinate calcification as a part of a highly regulated process akin to new bone formation and mineralize to form calcific nodules on the valves. The local environment drives the mesenchymal progenitor cells to undergo osteogenesis differentiation, elaborate bone matrix in the valve [30] and capable of contributing to valve calcification.
The transition of valvular cells to an osteoblast-like phenotype is not a mutually exclusive process and these osteoblast-like cells may actively generate vesicles or undergo apoptosis to accumulate calcium and form microcalcifications. Matrix vesicles (100 to 300 nm) are derived from plasma membrane of smooth muscle cells, encapsulate the calcium inhibitors (MGP-vitamin K dependent matrix Gla protein, Fetuin A-a liver derived protein, osteoproteogens), sequester, render them inactive [31] and serve as a nidus for calcium deposition [32] . Calcifying nanoparticles (10 - 500 nm in size) consistent with nanobacteria [33] , [34] which nucleate, crystallize and function as a nidus for matrix calcification [35] . The activated matrix metalloproteinases (1, 2, 3 and 9) and cathepsins (S, K, V and G) degrade the collagen and elastin, compromises valve integrity, augment the inflammatory response and allows for the expansion of calcified nodules [36] . By the end stage of the disease, maturation of valve calcification occurs with lamellar bone and hemopoietic tissue and it is sustained by neoangiogenesis. The new vessels localizing to the region of inflammation and surrounds the calcific deposits. Both intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 expressions are increased in these vessels, suggesting that they act as an important portal of entry of inflammatory cells [37] . The mast cells get degranulated in calcified valves and release tryptase which degrade the angiogenesis inhibitor endostatin [38] and down regulation of local angiogenesis inhibitors such as chondromodulin-1 and pronomodulin-1 have been occurred. Once neovascularization has been established, the network of newly formed vessels, expressing the endothelial markers Tie-2, vascular endothelial growth factor receptor-2, smooth muscle α-actin, facilitate rapid transit of inflammatory cells and procalcific signaling molecules and, these inflammatory lesions in rheumatic valvular disease is associated with angiogenesis and osteogenesis which may contribute disease progression and valvular mineralization.
3.3. Echocardiographic Features
The genesis of calcification in aortic valve is always rheumatic [39] and the calcific extension to the mitral valve resembles J-shaped as shown in Figure 5 and Figure 13. Calcification resembling a string of pearls in both aortic and mitral valves are typical of endocarditis. Calcification of mitral-aortic membrane has been reported as highly suggestive of radiation-induced lesions [40] and a pathologic echo density of anterior mitral leaflet is its characteristic feature [41] . But the dense calcification of anterior mitral leaflet as a bone-like structure was observed due to rheumatic mineralization as shown in Figures 3 along with calcification of aortic valve as in Figure 4 and Figure 5. The calcified aortic valve resembling as “calcium stone” was shown in short axis view in Figure 6.
The degenerative disease, as an aging process frequently involving the aortic and mitral valves and the central fibrous body is called as ‘Lev’s disease. Lenegre’s disease primarily involves the conduction system without affecting the fibrous skeleton of the heart. Both these conditions mostly results in isolated chronic heart blocks in adults. Degenerative calcification of mitral annulus may cause annular mitral stenosis in elderly and the difference between the annular mitral stenosis and rheumatic mitral stenosis [42] was shown in Table 2.
3.4. Therapeutic Strategies
3.4.1. Statins
Lipoprotein (a) has been shown to accumulate in both early and end-stage aortic valve lesions and to co-localize with calcium deposition [43] . In atherosclerosis, inflammation and lipid deposition are the key components in both development of arterial plaque and its stability. Adverse events are predominantly related to plaque rupture, and much of the benefit from statin therapy is due to plaque stabilization and a thickening of the fibrous cap. Lowering Lp(a) levels during early - stage aortic valve disease, with the use of either niacin [44] or novel specific Lp(a) inhibitors [45] can slow the progression to aortic stenosis. Lipid-lowering with HMG-Co A reductase inhibitors (statins) have been shown to decrease inflammatory cell infiltration, osteoblast bone markers (alkaline phosphatase and Runx2) and downregulate lipoprotein receptor-related protein 5/β-catenin signaling to decrease valve calcification in rabbit models [46] . Despite these findings, 3 large-scale clinical trials (SEAS-Simvastatin and Ezetimibe in Aortic
Table 2. Difference between annular mitral stenosis and rheumatic mitral stenosis.

Figure 7. Apical view showing the annular mitral stenosis with basal leaflet calcification in a 65-year-old female.

Figure 8. Apical view showing the rheumatic mitral valve in a 34-year-old male (Rheumatic mitral stenosis).
Stenosis [47] , SALTIRE-Scotish Aortic Stenosis and Lipid Lowering Trial [48] and ASTRONOMER-Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin [49] ) failed to show a benefit for statins to limit the progression of aortic valve calcification. Also, statins are known to promote osteoblast differentiation and calcification [50] . Lowering of lipid levels may decrease calcium deposition and halt the progress of valve calcification, but did not reverse the established calcification or valve dysfunction [51] . When the valvular interstitial cells (VICs) become osteoblast-like cells, statins may actually have a procalcifying effect.
3.4.2. Warfarin
Warfarin is an inhibitor of γ-carboxylase and vitamin K epoxide reductase , inhibits MGP (matrix Gla protein- an inhibitor of bone morphogenic protein (BMP) signaling [52] ) activity and was shown to promote valvular calcification [53] .
3.4.3. Fetuin A
It is a potent circulating inhibitor of calcification [54] , acts by binding the cluster of calcium and phosphate to stabilize these ions and prevent uptake by cells and its deficiency was associated with valvular calcification [55] . Therapies with fetuin A show a potential in inhibition of valvular calcification [56] .
3.4.4. ACE Inhibitors
Tissue angiotensin-converting enzyme (ACE) and angiotensin II are upregulated in stenotic aortic valves and angiotensin receptors have been identified on valve myofibroblasts [57] . Areas of fibrosis are observed to co-localize with areas of myocyte apoptosis due to ischemia since the density of capillary network does not expand sufficiently as in physiologic hypertrophy and drive the decompenstation of the hypertrophic myocardial response. There is an evidence that angiotensin receptor blocker therapy inhibits the development of atherosclerosis as well as myofibroblast and osteoblast trans-differentiation in rabbit aortic valves suggests that this drug class may be helpful in slowing aortic stenosis progression [58] . The blood pressure-lowering effect of ACE inhibitors decreases the mechanical stress and strain on the aortic valve and play a role in reducing inflammation, stabilizing plaque formation and mitigating the fibrotic process [59] , an integral part of hypertrophy related to polymorphism of ACE 1/D gene.
3.4.5. Desnosumab
The OPG/RANK/RANKL axis appears to have a central regulatory role in both calcification and osteoporosis, which coordinate calcium homeostasis via the action of osteoblasts and osteoclasts. OPG (osteoprotegerin) is a decoy receptor of RANKL, a potent stimulator of osteoclast differentiation and bone resorption [60] . Increased expression of RANKL (receptor activator of nuclear factor kappa B ligand) and reduced levels of OPG have been also observed in stenotic aortic valves similar to osteoporosis. Inactivation of OPG in mice develops extensive vascular calcification [61] . The development of anti-RANKL monoclonal antibody desnosumab promising a strong rationale in the treatment of aortic stenosis.
3.4.6. Bisphosphonates
Calcification is a critical process in determining the progress of valvular stenosis and a overlap in pathophysiology between the aortic stenosis and osteoporosis provide a rationale for drugs such as bisphosphonates to reduce the valvular calcium, especially in patients with renal failure and bioprosthetic valves [62] [63] . However, it had no effect on freedom from aortic valve replacement or survival [64] .
3.5. Gene Transfer
By understanding the cell biology of valvular calcification, gene transfer or cell- based therapies are investigated to modulate the cellular mechanism of calcification. The expressing Runx2 in VICs (valvular interstitial cells) commits these cells to an osteoblast-like phenotype. Gene transfer of small interfering RNA that targets Runx2 using adenovirus or recombinant nonviral vectors have been shown to prevent calcification both in vitro and in vivo [65] [66] . Heterotopic ossification (HO), which was first identified by Dejerne and Ceillier in 1918 [67] and it is characterized by pathologic endochondral bone formation at nonskeletal sites. Angiogenesis and osteogenesis are highly coupled during bone development. In the process of osteoblast differentiation, a set of bone specific genes (ALP-alkaline phosphatase, osteocalcin) are activated. Run-related protein 2 (Runx2) is a key regulator of these genes, cooperate with BMP-specific R-smads and it is considered as a molecular switch (myoblasts to osteoblasts phenotype) in osteoblast biology. Hypoxia is able to upregulate BMP2 in osteoblasts via the activation of Hif1α signaling pathway. Inhibition of Hif1 α (hypoxia induced factor 1α) leads to reduce vessel formation, which may facilitate migration of host mesenchymal stem cells to the bone regeneration site. Synergistic inhibition of endochondral bone formation by silencing the angiogenic factor (Hif1α) and osteogenic factor (Runx2) by using the RNA interference technology, allow to design more targeted therapies to inhibit angiogenesis and osteogenesis [68] . miRNAs (microRNAs) have essential roles in osteoblast differentiation and heterotopic ossification. miR-203 is involved in these processes by repressing Runx2 expression in the post-transcriptional level. Therapeutic overexpression of miR-203 in osteoblasts may reduce heterotopic ossification and even prevent this disorder [69] . Cotransduction of small or short interfering RNA (siRNA) against Runx2 and Hif1α to inhibit heterotopic ossification have been evaluated. TNP-470 is an antiangiogenic agent that inhibits the neovessel formation and TNP-70 inhibits the biological activity of BMP2 in early stage of bone induction. A potent delivery of a nanocomposite consisting of serum-stable DOTAP/ siRNA/PPAA-g-1% Jeffamine formulation was shown to halt the osteogenic switch driven by BMP2 by silencing the Runx2, the downstream mediator of BMP2 [70] .
3.6. Investigational Therapy
Osteoclast-like cells are identified in calcified lesions , which in turn capable of resorbing mineral deposits [71] . Thus, strategies to promote osteoclast recruitment or stimulate differentiation of monocytes and macrophages toward an osteoclast phenotype are the areas of active investigation.
Strategies by delivering stem or progenitor cells via direct injection of small molecule inhibitors, osteoclasts, osteoclast-like cells or molecules that promote the transition of resident monocytes/macrophages towards an osteoclast lineage or viral vectors encoding inhibitory RNA (Runx2, Msx2) in areas of aortic valve lesions or to ischemic myocardium using a NOGA-guided catheter system [72] are under investigation.
3.7. Lonafarnib
Mutations in laminin A gene results in the production of a mutant lamin A protein, progerin, a component of nuclear envelope which becomes unstable, leading to cell death prematurally. The syndromes associated with these defects are called as “laminopathies” (Hutchinson Gilford Progeria Syndrome (HGPS), Wiedemann-Rautenstrauch syndrome, Restrictive dermopathy) [73] . In progeria, calcific aortic stenosis represents a degenerative premature aging process and principally involving the anterior mitral leaflet and proximal aortic valve cusps [74] . Systemic hypertension is common and appears before the child reaches the age of 5, and high mortality due to myocardial infarction and heart failure may occur.
Farnesylation is essential for the function of both mutant and nonmutant lamin A protein, including progerin. Lonafarnib is a farnesyl transferase inhibitor that blocks the posttranslational farnesylation of prelamin A, an experimental drug under trials [75] . Another treatment attempted is a combination of two molecules-Zoledronic acid and pravastatin.
3.8. Interventional Therapy
Balloon mitral valvotomy (BMV) is the procedure of choice for severe mitral stenosis when the valve morphology is feasible. However, BMV is contraindicated in bicommissural calcification since it results a high probability of leaflet tear with acute severe mitral regurgitation. BMV can be performed safely and successfully in unicommissuraly calcified mitral valves [76] .
An emergency percutaneous transatrial balloon mitral commissurotomy is a life-saving procedure in a valve with unfavourable morphology, presented with cardiogenic shock in critical calcific mitral stenosis [77] [78] .
3.9. Transcatheter Valve Implantation
TAVR (transcatheter aortic valve replacement) was approved by FDA (Food and drug administration) for high risk surgical candidates in June 2012. There is an increase in risk of stroke due to thrombotic embolic material from the calcified valve tissue and vascular damage results in severe paravalvular regurgitation, which is poorly tolerated in hypertrophied left ventricle leading to increase in mortality [79] . The surgical risk for mitral valve replacement is very high due to the comorbidities and technical challenges related to calcium burden. Guerrero, et al reported the first-in-human percutaneous implantation of a balloon-ex- pandable transcatheter valve in a native mitral valve [80] . The use of newer aortic THV technologies such as Lotus (Boston Scientific Corporation) and direct flow valves (direct flow medical) in the mitral position for severe MAC (mitral annular calcification) [81] with improved outcomes were observed [82] .
3.10. Surgical Therapy
Aortic valve replacement surgery remains the only established treatment for symptomatic aortic stenosis patients [83] . Hemodynamically significant asymptomatic patients will develop symptoms within 5 years, the issues of medical management and surgical correction may pay a close attention to the researchers [84] . Presence of moderate or severe valvular calcification identifies the patients with poor prognosis who may benefit from early valve replacement before the development of symptoms [85] and it is to be determined whether this benefit overweighs the risk of surgery [86] .
Calcific mitral stenosis with severe mitral annular calcification may present a different high risk to the surgeon. Extensive debridgement of calcium may increase the risk of paravalvular leakage and bleeding. Bypass of the mitral valve with a valved conduit is another option for the treatment of severe calcific mitral stenosis [87] .
Bioprosthetic valves are generally recommended for patients > 65 years and there is also greater usage in younger patients due to lack of necessity of chronic anticoagulation. The Ross operation (replacement of aortic valve with a pulmonary autograft and replacement of the pulmonary valve with a homograft) is an alternate for adolescents, young adults, including woman who wish to become pregnant and it is not favoured by the majority of surgeons due to complexity of the procedure, but excellent outcomes occurred in centers with expertise in this procedure [88] .
3.11. Screening of Population
Aortic valve calcification extending to anterior mitral leaflet was shown in Figure 9 in a 64-year-old male. A bicuspid aortic valve calcification extending to anterior mitral leaflet was shown in Figure 10 and it is “mushroom” shaped [89] or bulbous (clublike or circle) configuration [90] as in Figure 11 in a 34-year-old male. J-shaped [91] mitral-aortic calcification was shown in Figure 5 in a 52- year-old male and in Figure 13 in a 60-year-old male. Aortic valve calcification

Figure 9. Parasternal long axis view showing the extension of aortic valve calcification to anterior mitral leaflet in a 64-year-old male.

Figure 10. Parasternal long axis view showing dense calcium in aortic valve and anterior mitral leaflet in a 34-year-old male.

Figure 11. Parasternal long axis view showing the “mushroom” shaped calcification of Bicuspid aortic valve in a 34-year-old male.
resembling as “calcified amorphous tumor” [92] as in Figure 12 in a 58-year-old male. Calcification of aortic valve and anterior mitral leaflet was shown in Figure 15 in a 5-year-old child having phenotypic features of Hutchinson-Gilford Progeria syndrome as shown in Figure 14 with bicuspid aortic valve regurgitation as shown in Figure 16 and Figure 17 and AML (anterior mitral leaflet) calcification in a newborn as in Figure 18 and Figure 19 due to rheumatic etiology rather than degenerative change of premature aging.
4. Conclusion
Calcification was common in valvular heart disease of inflammatory genesis.

Figure 12. Parasternal long axis view showing the dense calcium in aortic valve mimicking as ‘calcified amorphous tumor’ in a 58-year-old male.

Figure 13. Parasternal long axis view showing the calcification of anterior mitral leaflet in a 60-year-old male and it is J-shaped.

Figure 14. Hutchinson-Gilford Progeria syndrome (short stature, prominent eyes, craniofacial disproportion, polydactyly, hypotelorism)-5 year-old male child (Photo image with consent).

Figure 15. Parasternal long axis view showing the Bicuspid Aortic valve and anterior mitral leaflet calcifications in Progeria due to rheumatic etiology in a 5-year-old child.

Figure 16. Color Doppler Imaging showing Severe aortic regurgitation in Progeria in a 5-year-old male child.
Most of them were localized to the mitral valve (47%), only in aortic valve or both in 38% of cases and it is mostly rheumatic in 80% of cases. Degenerative calcification of mitral ring was seen in 5% to 10% of cases. It is now clear that there is an active biology that leads to stiffening and calcification of valve leaflets [93] and so the valvular calcification is a biologic process. At present, therapeutic targets focused on cellular events to inhibit the process of calcification are under investigation. There is an association of rheumatic mineralization of mitral-aor- tic continuity and anterior mitral leaflet in Hutchinson-Gilford progeria Syndrome as shown in Figures 14-19 was found in this region of Thoothukudi

Figure 17. CW (continuous wave ) Doppler showing severe Aortic regurgitation in progeria in a 5-year-old child.

Figure 18. Showing the Hutchinson-Gilford Progeria syndrome in neonate (6 days old Female-photo image with consent).
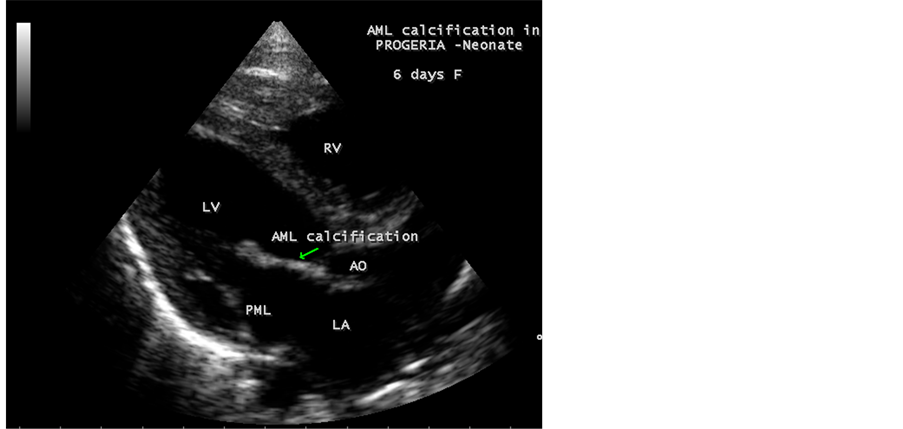
Figure 19. Showing the AML (anterior mitral leaflet) calcification in Progeria in neonate.
in Tamil Nadu state of Southern India. An mTOR (mechanistic target of rapamycin) inhibitor (rapalogs), the Sirolimus can minimize the phenotypic effects of progerin fibroblasts and may benefit in these genetic disorders [94] , the progeroid syndromes [95] .
Cite this paper
Muthiah, R. (2017) Rheumatic Mineralization of Aortic Valve and Anterior Mitral Leaflet―A Case Report. Case Reports in Clinical Medicine, 6, 95-119. https://doi.org/10.4236/crcm.2017.64009
References
- 1. Thubrikar, M.J., Nolan, S.P., Aouad, J. and Deck, J.D. (1986) Stress Sharing between the Sinus and Leaflets of Canine Aortic Valve. Annals of Thoracic Surgery, 42, 434-440.
- 2. Parashara, D.K., Jacobs, L.E., Kotler, M.N., Yazdanfar, S., Spielman, S.R., Janzer, S.F. and Bemis, C.E. (1995) Angina Caused by Systolic Compression of the Left Coronary Artery as a Result of Pseudoaneurysm of the Mitral-Aortic Intervalvular Fibrosa. American Heart Journal, 129, 417-421.
- 3. Aikawa, E., Nahrendorf, M., Figueiredo, J.L., Swiirski, F.K., Shtatland, T., Kohler, R.H., Jaffer, F.A., Aikawa, M. and Weissleder, R. (2007) Osteogenesis Associates with In-flammation in Early-Stage Atherosclerosis Evaluated by Molecular Imaging in Vivo. Circulation, 116, 2841-2850.
https://doi.org/10.1161/CIRCULATIONAHA.107.732867 - 4. Vaslef, S.N. and Roberts, W.C. (1993) Early Descriptions of Aortic Valve Stenosis. American Heart Journal, 125, 1465-1474.
- 5. Coombs, O.F. (1924) Rheumatic Heart Disease. William Wood And Co., New York.
- 6. Christian, H.A. (1931) Aortic Stenosis with Calcification of the Cusps: Distinct Clinical Entity. The Journal of the American Medical Association, 97, 158.
- 7. Clawson, B.J., Noble, J.F. and Lufkin, N.H. (1938) The Calcified Nodular Deformity of the Aortic Valve. American Heart Journal, 15, 58.
- 8. Dry, T.J. and Willius, F.A. (1939) Calcareous Disease of The Aortic Valve. American Heart Journal, 17,138.
- 9. Hamburger, W.W. and Saphir, O. (1939) Aortic Stenosis. Modern Concepts of Cardiovascular Disease, 8, No. 2.
- 10. Edwards, J.E. (1961) An Atlas of Acquired Diseases of the Heart and Great Vessels. W.B. (Walter Burns) Saunders, Co., Philadelphia.
- 11. Messika-Zeitoun, D., Bielak, L.F., Peyser, P.A., et al. (2007) Aortic Valve Calcifica-tion: Determinants and Progression in the Population. Arteriosclerosis, Thrombosis and Vascular Biology, 27, 642-648.
https://doi.org/10.1161/01.ATV.0000255952.47980.c2 - 12. Mohler, E.R., Gannon, F., Reynolds, C., Zimmerman, R., Keane, M.G. and Kaplan, F.S. (2001) Bone Formation and Inflammation in Cardiac Valves. Circulation, 103, 1522-1528.
https://doi.org/10.1161/01.CIR.103.11.1522 - 13. Parhami, F., Morrow, A.D., Balucan, J., et al. (1997) Lipid Oxidation Prod-ucts Have Opposite Effects on Calcifying Vascular Cell and Bone Cell Differentiation: A Possible Explanation for the Paradox of Arterial Calcification in Osteoporotic Patients. Arteriosclerosis Thrombosis and Vascular Biology, 17, 680-687.
https://doi.org/10.1161/01.ATV.17.4.680 - 14. Wylie-Sears, J., Aikawa, E., Levine, R.A., Yang, J.H. and Bischoff, J. (2011) Mitral Valve Endothelial Cells with Osteogenic Differentiation Potential. Arteriosclerosis, Thrombosis and Vascular Biology, 31, 598-607.
https://doi.org/10.1161/ATVBAHA.110.216184 - 15. Kaden, J.J., Dempfle, C.E., Grobholz, R., et al. (2003) Inter-leukin-1 Beta Promotes Matrix Metalloproteinase Expression and Cell Proliferation in Calcific Aortic Valve Stenosis. Atherosclerosis, 170, 205-211.
- 16. Kaden, J.J., Dempfle, C.E., Grobholz, R., et al. (2005) Inflammatory Regulation of Extracellular Matrix Re-modeling in Calcific Aortic Valve Stenosis. Cardiovascular Pathology, 14, 80-87
- 17. Winchester, R., Wiesendanger, M., O’Brien, W., Zhang, H.Z., Maurer, M.S., Gillam, L.D., Schwartz, A., Marboe, C. and Stewart, A.S. (2011) Circulating Activated and Effector Memory T Cells Are Associated with Calcification and Clonal Expansions in Bicuspid and Tricuspid Valves Calcific Aortic Stenosis. Journal of Immunology, 187, 1006-1014.
https://doi.org/10.4049/jimmunol.1003521 - 18. Rajamannan, N.M. Subramaniam, M., Caira, F., Stock, S.R. and Spelsberg, T.C. (2005) Atorvastatin Inhibits Hypercholesteremia-Induced Calcification in the Aortic Valves via the Lrp5 Receptor Pathway. Circulation, 112, 1229-1234.
- 19. Rajamannan, N.M., Subramanium, M., Rickard, D., et al. (2003) Human Aortic Valve Calcification Is Associated with an Osteoblast Phenotype. Circulation, 107, 2181-2184.
https://doi.org/10.1161/01.CIR.0000070591.21548.69 - 20. Garg, V., Muth, A.N., Ransom, J.F., et al. (2005) Mutations in NOTCH 1 Cause Aortic Valve Disease. Nature, 437, 270-274.
https://doi.org/10.1038/nature03940 - 21. Sacks, M.S. and Schoen, F.J. (2002) Collagen Fiber Disruption Occurs Independent of Calcification in Clinically Explanted Bioprosthetic Heart Valves. Journal of Biomedical Material Research, 62,359-371.
https://doi.org/10.1002/jbm.10293 - 22. Matsumoto, Y., Adam, V., Walther, C., Kleinecke, C., Brugger, P., Linke, A., Walther, T., Mohr, F.W. and Schaller, G. (2009) Reduced Number and Function of Endothelial Progenitor Cells in Patients with Aortic Valve Stenosis: A Novel Concept for Valvular Endothelial Cell Repair. European Heart Journal, 30, 346-355.
https://doi.org/10.1093/eurheartj/ehn501 - 23. Kovacic, J.C., Mercader, N., Torres, M., Boehm, M. and Fuster, V. (2012) Epithelial-to-Mesenchymal and Endothelial-to-Mesenchymal Transition from Cardiovascular Development to Disease. Circulation, 125, 1795-1808.
https://doi.org/10.1161/CIRCULATIONAHA.111.040352 - 24. Piera-Velazquez, S., Li, Z. and Jimenez, S.A. (2011) Role of Endothelial-Mesenchymal Transition (EndoMT) in the Pathogenesis of Fibrotic Disorders. American Journal of Pathology, 179, 1074-1080.
- 25. Bostrom, K.I., Rajamannan, N.H. and Towler, D.A. (2001) The Regulation of Valvular and Vascular Sclerosis by Osteogenic Morphogens. Circulation Research, 109, 564-577.
https://doi.org/10.1161/CIRCRESAHA.110.234278 - 26. Johnson, R.C., Leopold, J.A. and Loscalzo, J. (2006) Vascular Cal-cification: Pathobiological Mechanisms and Clinical Implications. Circulation Research, 99, 1044-1059.
https://doi.org/10.1161/01.RES.0000249379.55535.21 - 27. Liu, A.C., Joag, V.R. and Gotlieb, A.I. (2007) The Emerging Role of Valve Interstitial Cell Phenotypes in Regulating Heart Valve Pathobiology. American Journal of Pathology, 171, 1407-1418.
https://doi.org/10.2353/ajpath.2007.070251 - 28. Caira, F.C., Stock, S.R., Gleason, T.G., et al. (2006) Human Degenerative Valve Disease Is Associated with Up-Regulation of Low-Density Lipoprotein Receptor-Related Protein 5 Receptor-Mediated Bone Formation. Journal of American College of Cardiology, 47, 1707-1712.
- 29. Kaden, J.J., Bickelhaupt, S., Grobholz, R., et al. (2004) Receptor Activator of Nuclear Factor Kappa B Ligand and Osteoprotegerin Regulate Aortic Valve Calcification. Journal of Molecular And Cellular Cardiology, 36, 57-66.
- 30. Chen, J.H., Yip, C.Y., Sone, E.D. and Simmons, C.A. (2009) Identification and Char-acterization of Aortic Valve Mesenchymal Progenitor Cells with Robust Osteogenic Calcification Potential. American Journal of Pathology, 174, 1109-1119.
https://doi.org/10.2353/ajpath.2009.080750 - 31. Schoppet, M., Kavurma, M.M., Hofbauer, J.L.C. and Shanahan, C.M. (2011) Crystallizing Nanoparticles Derived from Vascular Smooth Muscle Cells Contain the Calcification Inhibitor Osteoprotegerin. Biochemical and Biophysical Research Communications, 407, 103-107.
- 32. Wuthis, R.E. and Lipscomb, G.F. (2011) Matrix Vesicles: Structure, Composition, Formation and Function in Calcification. Frontiers in Bioscience, 17, 2812-2902.
- 33. Miller, V.M., Rodgers, G., Charlesworth, J.A., Kirkland, B., Severson, S.R., Rasmussen, T.E., Yagubyan, M., Rodgers, J.C., Cockerill, F.R. III, Folk, R.T., Rzewuska-Lech, E., Kumar, V., Farell-Bard, G. and Lieska, J.C. (2004) Evidence of Nanobacterial-Like Structures in Calcified Human Arteries and Cardiac Valves. American Journal of Physiology: Heart and Circu-latory Physiology, 287, H1115-H1124.
- 34. Domingo, A., Carrascal, Y., Mota, P., Martin-Luengo, C., Bermejo, J., San Roman, J.A., Rodriguez-Torres, A. and Fernandez-Aviles, F. (2008) Association between Self-Replicating Calcifying Nanoparticles and Aortic Stenosis. A Possible Link to Valve Calcification. European Heart Journal, 29, 371-376.
https://doi.org/10.1093/eurheartj/ehm592 - 35. Schlieper, G., Kruger, T., Heiss, A. and Jahnen-Dechent, W. (2011) A Red Herring in Vascular Calcification: “Nanobacteria” Are Protein-Mineral Complexes Involved in Biomineralization. Nephrology Dialysis Transplantation, 26, 3436-3439.
https://doi.org/10.1093/ndt/gfr521 - 36. Chen, J.H. and Simmons, C.A. (2011) Cell-Matrix Interaction S in the Pathobiology of Calcific Aortic Valve Disease: Critical Roles for Matricellular, Matricrine, and Matrix Mechanic Cues. Circulation Research, 108, 1510-1524.
https://doi.org/10.1161/CIRCRESAHA.110.234237 - 37. Mazzone, A., Epistolato, M.C., De Caterina, R., et al. (2004) Neoangiogenesis, T-Lymphocyte Infiltration and Heat Shock Protein-60 Are Biological Hallmarks of An Immunomediated Inflammatory Process in End-Stage Calcified Aortic Valve Stenosis. Journal of American College of Cardiology, 43, 1670-1676.
- 38. Syvaranta, S., Helske, S., Laine, M., Lappalainen, J., Kupari, M., Mayranpaa, M.I., Lindstedt, K.A. and Kovanen, P.T. (2010) Vascular Endothelial Growth Factor-Secreting Mast Cells and Myofibroblasts: A Novel Self-Perpetuating Angiogenic Pathway in Aortic Valve Stenosis. Arteriosclerosis, Thrombosis and Vascular Biology, 30, 1220-1227.
https://doi.org/10.1161/ATVBAHA.109.198267 - 39. Hall, E.M. and Ichioka, T. (1940) Etiology of Calcified Nodular Aortic Stenosis. American Journal of Pathology, 16, 761.
- 40. Hering, D., Faber, L. and Horstkotte, D. (2003) Echocardiographic Features of Radiation-Associated Valvular Disease. American Journal of Cardiology, 92, 226-230.
- 41. Feigenbaum, H. and Armstrong, W.F. (2005) Feigenbaum’s Echocardiography. 6th Edition, Mitral Valve Disease, Chapter 11, 350.
- 42. Akram, M.R., Chan, T., McAuliffe, S. and Chenzbraun, A. (2009) Non-Rheumatic Annular Mitral Stenosis: Prevalence and Characteristics. European Journal of Echocardiography, 10, 103-105.
- 43. Gotoh, T., Kuroda, T., Yamasawa, M., et al. (1995) Correlation between Lipoprotein (a) and Aortic Valve Sclerosis Assessed by Echocardiography (The JMS Cardiac Echo and Cohort Study). American Journal of Cardiology, 76, 928-932.
- 44. Capuzzi, D.M., Guyton, J.R., Morgan, J.M., et al. (1998) Efficacy and Safety of an Extended-Release Niacin (Niaspan): A Long-Term Study. American Journal of Cardiology, 82, 74U-81U.
- 45. Merki, E., Graham, M., Taleb, A., et al. (2011) Antisense Oligonucleotide Lowers Plasma Levels of Apolipoprotein (a) and Lipoprotein (a) in Transgenic Mice. Journal of American College of Cardiology, 57, 1611-1621.
- 46. Rajamannan, N.M., Subramaniam, M., Springett, M., Sebo, T.C., Niekrasz, M., McConnell, J.P., Singh, R.J., Stone, N.J., Bonow, R.D. and Spelsberg, T.C. (2002) Atorvastatin Inhibits Hypercholesterole-mia-Induced Cellular Proliferation and Bone Matrix Production in the Rabbit Aortic Valve. Circulation, 105, 2660-2665.
https://doi.org/10.1161/01.CIR.0000017435.87463.72 - 47. Rossebo, A.B., Pedersen, T.R., Boman, K., Brudi, P., Chambers, J.B., Egstrup, K., Gerdts, E., Gohlke-Barwolf, C., Holme, I., Kesaniemi, Y.A., Malbecq, W., Nienaber, C.A., Ray, S., Skjaerpe, T., Wachtell, K. and Willenheimer, R. (2008) Intensive Lipid Lowering with Simvastatin and Ezetimibe in Aortic Stenosis. New England Journal of Medicine, 359, 1343-1356.
https://doi.org/10.1056/NEJMoa0804602 - 48. Cowell, S.J., Newby, D.E., Prescott, R.J., Bloomfield, P., Reid, J., Northridge, D.B. and Boon, N.A. (2005) A Randomized Trial of Intensive Lipid-Lowering Therapy in Calcific Aortic Stenosis. New England Journal of Medicine, 352, 2389-2397.
https://doi.org/10.1056/NEJMoa043876 - 49. Chan, K.L., Teo, K., Dumesnil, J.G., Ni, A. and Tam, J. (2010) Effect of Lipid Lowering with Rosuvastatin on Progress of Aortic Stenosis: Results of the Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin (Astronomer) Trial. Circulation, 121, 306-314.
https://doi.org/10.1161/CIRCULATIONAHA.109.900027 - 50. Mundy, G., Garretlt, R., Harris, S., Chan, J., Chen, D., Rossini, G., Boyce, B., Zhao, M. and Gutierrez, G. (1999) Stimulation of Bone Formation in Vitro and in Rodents by Statins. Science, 286, 1946-1949.
https://doi.org/10.1126/science.286.5446.1946 - 51. Miller, J.D., Weiss, R.M., Serrano, K.M., Brooks, R.M. II, Berry, C.J., Zimmerman, K., Young, S.G. and Heistad, D.D. (2009) Lowering Plasma Cholesterol Levels Halts Progression of Aortic Valve Disease in Mice. Circulation, 119, 2693-2701.
https://doi.org/10.1161/CIRCULATIONAHA.108.834614 - 52. Yao, Y., Bennett, B.J., Wang, X., Rosenfeld, M.E., Giachelli, C., Lusis, A.J. and Bostrom, K.I. (2010) Inhibition of Bone Morphogenetic Proteins Protects against Atherosclerosis and Vascular Calcification. Circulation Research, 107, 485-494.
https://doi.org/10.1161/CIRCRESAHA.110.219071 - 53. Lemer, R.G., Aronow, W.S., Sekhri, A., Palaniswamy, C., Ahn, C., Singh, T., Sandhu, R. and McClung, J.A. (2009) Warfarin Use and the Risk of Valvular Calcification. Journal of Thrombosis and Haemostasis, 7, 2023-2027.
https://doi.org/10.1111/j.1538-7836.2009.03630.x - 54. Jahnen-Dechent, W., Heiss, A., Schafer, C. and Ketteller, M. (2011) Fetuin-A Regulation of Calcified Matrix Metabolism. Circulation Research, 108, 1494-1509.
https://doi.org/10.1161/CIRCRESAHA.110.234260 - 55. Kaden, J.J., Reinohl, J.O., Blesch, B., Brueckmann, M., Haghi, D., Borggrefe, M., Schmitz, F., Klomfass, S., Pillich, M. and Ortlepp, J.R. (2007) Systemic and Local Levels of Fetuin-A in Calcific Aortic Valve Stenosis. International Journal of Molecular Medicine, 20, 193-197.
https://doi.org/10.3892/ijmm.20.2.193 - 56. Ix, J.H., Chesrow, G.M., Shlipak, M.G., Brandenburg, V.M., Ketteler, M. and Whooley, M.A. (2007) Association of Fetuin-A with Mitral Annular Calcification and Aortic Stenosis among Persons with Coronary Heart Disease: Data from the Heart and Soul Study. Circulation, 115, 2533-2539.
https://doi.org/10.1161/CIRCULATIONAHA.106.682450 - 57. O.Brien, K.D., Shavelle, D.M., Caulfield, M.T., et al. (2002) Association of Angiotensin-Converting Enzyme with Low-Density Lipoprotein in Aortic Valvular Lesions and in Human Plasma. Circulation, 106, 2224-2230.
https://doi.org/10.1161/01.CIR.0000035655.45453.D2 - 58. Arishiro, K., Hoshiga, M., Negoro, N., et al. (2007) Angiotensin Receptor-1 Blocker Inhibits Atherosclerotic Changes and Endothelial Disruption of the Aortic Valve in Hypercholesterolemic Rabbits. Journal of American College of Cardiology, 49, 1482-1489.
- 59. Newby, D.E., Cowell, S.J. and Boon, N.A. (2006) Emerging Medical Treatments for Aortic Stenosis: Statins, Angiotensin Converting Enzyme Inhibitors, or Both? Heart, 92, 729-734.
https://doi.org/10.1136/hrt.2005.066852 - 60. Sattler, A.M., Schoppet, M., Schaefer, J.R. and Hofbauer, L.C. (2004 ) Novel Aspects on Rank Ligand and Osteoproteogerin in Osteoporosis and Vascular Disease. Calcified Tissue International, 74, 103-106.
https://doi.org/10.1007/s00223-003-0011-y - 61. Bucay, N., Sarosi, I., Dunstan, C.R., et al. (1998) Osteoprotegerin-Deficient Mice Develop Early Onset Osteoporosis and Arterial Calcification. Genes & Development, 12, 1260-1268.
https://doi.org/10.1101/gad.12.9.1260 - 62. Price, P.A., Faus, S.A. and Williamson, M.K. (2001) Bisphosphonates: Alendronate and Ibandronate Inhibit Artery Calcification at Doses Comparable to Those That Inhibit Bone Resorption. Arteriosclerosis, Thrombosis, And Vascular Biology, 21, 817-824.
https://doi.org/10.1161/01.ATV.21.5.817 - 63. Rapoport, H.S., Connolly, J.M., Fulmer, J., et al. (2007) Mechanisms of the in Vivo Inhibition of Calcification of Bioprosthetic Porcine Aortic Valve Cusps and Aortic Wall with Triglycidylamine/Mercapto Bisphosphonate. Biomaterials, 28, 690-699.
- 64. Aksoy, O., Cam, A., Goel, S.S., Houghtaling, P.L., Williams, S., Ruiz-Rodriguez, E., Menon, V., Kapadia, S.R., Tuzcu, E.M., Blackstone, E.H. and Griffin, B.P. (2012) Do Bisphosphonates Slow the Progression of Aortic Stenosis? Journal of American College of Cardiology, 59, 1452-1459.
- 65. Xue, T., Mao, Z., Lin, L., Hou, Y., Wei, X., Fu, X., Zhang, J. and Yu, C. (2010) Non-Virus-Mediated Transfer of siRNAs against Runx2 and Smad 4 Inhibit Heterotopic Ossification in Rats. Gene Therapy, 17, 370-379.
https://doi.org/10.1038/gt.2009.154 - 66. Lin, L., Chen, L., Wang, H., Wei, X., Fu, X., Zhang, J., Ma, K., Zhou, C. and Yu, C. (2006) Adenovirus-Mediated Transfer of siRNA against Runx2/Cbfa1 Inhibits the Formation of Heterotopic Ossification in Animal Model. Biochemical and Biophysical Research Communications, 349, 564-572.
- 67. Shehab, D., Elgazzar, A.H. and Collier, B.D. (2002) Heterotopic Ossification. Journal of Nuclear Medicine, 43, 346-353.
- 68. Lin, L., Shen, Q., Leng, H., Duan, X., Fu, X. and Yu, C. (2011) Synergistic Inhibition of Endochondral Bone Formation by Silencing Hif1α and Runx2 in Trauma-Induced Heterotopic Ossification. Molecular Therapy, 19, 1426-1432.
https://doi.org/10.1038/mt.2011.101 - 69. Tu, B., Liu, S., Yu, B., Zhu, J., Ruan, H., Tang, T. and Fan, C. (2016) MiR-203 Inhibits the Traumatic Heterotopic Ossification by Targeting Runx2. Cell Death & Disease, 7, e2436.
- 70. Mishra, S., Vaughn, A.D., Devore, D.I. and Roth, C.M. (2012) Delivery of siRNA Silencing Runx2 Using a Multifunctional Polymer-Lipid Nanoparticle Inhibits Osteogenesis in a Cell Culture Model of Heterotopic Ossification. Integrative Biology, 4, 1498-1507.
https://doi.org/10.1039/c2ib20200j - 71. Steitz, S.A., Speer, M.Y., Mckee, M.D., Liaw, L., Almeida, M., Yang, H. and Giachelli, C.M. (2012) Osteopontin Inhibits Mineral Deposition and Promotes Regression of Ectopic Calcification. American Journal of Pathology, 161, 2035-2046.
- 72. Smits, P.C., Van Geuns, R.J., Poldermans, D., Bountioukeios, M., Onderwates, E.E., Lee, C.H., Maat, A.P. and Serruys, P.W. (2003) Catheter-Based Intramyocardial Injection of Autologous Skeletal Myoblasts as a Primary Treatment of Ischemic Heart Failure: Clinical Experience with Six-Month Follow-Up. Journal of American College of Cardiology, 42, 2063-2069.
- 73. Csoka, A.B., Cao, H., Sammak, P.J., Constantinescu, D., Schatten, G.P. and Hegele, R.A. (2004) Novel Lamin A/C Gene(LMNA) Mutations in Atypical Progeroid Syndrome. Journal of Medical Genetics, 41, 304-308.
https://doi.org/10.1136/jmg.2003.015651 - 74. Natesh, B., Hanumanthappa, G.M., Jeyaranganath, M. and Cholenahally, M.N. (2011) Hutchinson-Gilford Progeria Syndrome with Severe Calcific Aortic Valve Stenosis. Annals of Pediatric Cardiology, 4, 204-206.
https://doi.org/10.4103/0974-2069.84670 - 75. Merideth, M.A., Gordon, L.B., Clauss, S., Sachdev, V., Smith, A.C., Perry, M.B., et al. (2008) Phenotype and Course of Hutchinson-Gilford Progeria Syndrome. New England Journal of Medicine, 358, 592-604.
https://doi.org/10.1056/NEJMoa0706898 - 76. Ravindranath, et al. (2009) Balloon Mitral Valvotomy (BMV) for Calcific Mitral Stenosis, Cardiovascular Interventions. Journal of American College of Cardiology, 2, 263-264.
- 77. Dugal, J.S., Jetley, V., Sabharwal, J.S., Sofat, S. and Singh, C. (2003) Life-Saving PTMC for Critical Calcific Mitral Stenosis in Cardiogenic Shock with Balloon Impasse. International Journal of Cardiovascular Interventions, 5, 172-174.
- 78. Goldman, J.H., Slade, A. and Clague, J. (1998) Cardiogenic Shock Secondary to Mitral Stenosis Treated by Balloon Mitral Valvuloplasty. Catheterization and Cardiovascu-lar Diagnosis, 43, 195-197.
https://doi.org/10.1002/(SICI)1097-0304(199802)43:2<195::AID-CCD19>3.0.CO;2-N - 79. Webb, J.G., Altwegg, L., Boone, R.H., Cheung, A., Ye, J., Lichtenstein, S., Lee, M., Masson, J.B., Thompson, C., Moss, R., Carere, R., Munt, B., Nietlispach, F. and Humphries, K. (2009) Transcatheter Aortic Valve Implantation: Impact on Clinical and Valve-Related Outcomes. Circulation, 119, 3009-3016.
https://doi.org/10.1161/CIRCULATIONAHA.108.837807 - 80. Guerrero, M., Greenbaum, A. and O’Neill, W. (2014) First in Human Percutaneous Implantation of a Balloon Expandable Transcatheter Heart Valve in a Severely Stenosed Native Mitral Valve. Catheterization and Cardiovascular Interventions, 4, E287-E291.
https://doi.org/10.1002/ccd.25441 - 81. Lim, Z.Y., Boix, R., Prendergast, B., et al. (2015) First Reported Case of Transcatheter Mitral Valve Implantation in Mitral Annular Calcification with a Fully Repositionable and Self-Expanding Valve. Circulation: Cardiovascular Interventions, 8, e003031.
https://doi.org/10.1161/CIRCINTERVENTIONS.115.003031 - 82. Guerrero, M., Dvir, D., Himbert, D., et al. (2016) Transcatheter Mitral Valve Replacement in Native Mitral Valve Disease with Severe Mitral Annular Calcification: Results from the First Multicenter Global Registry. Journal of American College of Cardiology: Cardiovascular Interventions, 9, 1361-1371.
- 83. Carabello, B.A. and Crawford, F.A. (1997) Valvular Heart Disease. New England Journal of Medicine, 337, 32-41.
https://doi.org/10.1056/NEJM199707033370107 - 84. Pellika, P.A., Sarano, M.E., Nishimura, R.A., et al. (2005) Outcome of 622 Adults with Asymptomatic, Hemodynamically Significant Aortic Stenosis during Prolonged Follow-Up. Circulation, 11, 3290-3295.
https://doi.org/10.1161/CIRCULATIONAHA.104.495903 - 85. Chaliki, H.P., Brown, M.L., Sundt, T.M. and Tajik, A.J. (2007) Timing of Operation in Asymptomatic Severe Aortic Stenosis. Expert Review of Cardiovascular Therapy, 5, 1065-1071.
https://doi.org/10.1586/14779072.5.6.1065 - 86. Pai, R.G., Kapoor, N., Bansal, R.C. and Varadarajan, P. (2006) Malignant Natural History of Asymptomatic Severe Aortic Stenosis: Benefit of Aortic Valve Replacement. Annals of Thoracic Surgery, 82, 2116-2122.
- 87. Van Meeteren, J. and Schaff, H. (2017) Extracardiac Valved Conduit for Calcific Mitral Stenosis, CTS Net.
- 88. El-Hamamsy, I., Eryigit, Z., Stevens, L.M., Sarang, Z., George, R., Clark, L., Melina, G., Takkenberg, J.J. and Yacoub, M.H. (2010) Long-Term Outcomes after Autograft versus Homograft Aortic Root Replacement in Adults with Aortic Valve Disease: A Randomised Controlled Trial. The Lancet, 376, 524-531.
- 89. Contractor, S.G. and Lin, E.C. (2015) Valvular Calcifications, Overview, Imaging of Cardiac Calcifications. Medscape, August 5.
- 90. Perloff, J.K. (2003) Congenital Aortic Stenosis: Congenital Aortic Regurgitation. In: Perloff, J.K., Ed., Clinical Recognition of Congenital Heart Disease, 5th Edition, Chapter 7, Saunders, Philadelphia, 98.
- 91. Bartley, O. (1958) Cardiac Valve Calcifications and Registration of Their Movements. Acta Radiologica, 49, 329-347.
https://doi.org/10.3109/00016925809170999 - 92. Teshnizi, M.A., Ghorbanzadeb, A., Zirak, N., Manafi, B. and Moeinipour, A. (2017) Cardiac Calcified Amorphous Tumor of the Mitral Valve Presenting as Transient Ischemic Attack. Case Reports in Cardiology, 2017, Article ID: 2376096.
- 93. Miller, J.D., Weiss, R.M. and Heistad, D.D. (2011) Calcific Aortic Valve Stenosis: Methods, Models, and Mechanisms. Circulation Research, 108, 1392-1412.
https://doi.org/10.1161/CIRCRESAHA.110.23413