Case Reports in Clinical Medicine
Vol.05 No.12(2016), Article ID:72774,7 pages
10.4236/crcm.2016.512065
One Hemophagocytic Syndrome Relating to Adult Epstein-Barr Virus Infection and a Literature Review
Xiaolong Liu*, Yamin Zhang#, Zilin Cui*, Qing Tian, Dongming Duan
Department of Hepatobiliary Surgery, Tianjin First Central Hospital, Tianjin, China

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 11, 2016; Accepted: December 12, 2016; Published: December 15, 2016
ABSTRACT
Background: Adult hemophagocytic syndrome is a critical condition that is often difficult to diagnose and results in a bad prognosis because of the lack of effective and unified treatment. Aim: To investigate the clinical, diagnosis and treatment of hemophagocytic syndrome. Case Presentation: A 22-year-old female patient received a splenectomy, and the spleen was sent to the King Med Center of Medical Inspection. The results confirmed the diagnosis of hemophagocytic syndrome related to EB (Epstein-Barr) virus infection. Conclusion: Early diagnosis and early intervention are important factors for affecting the progression of the disease and improving its prognosis.
Keywords:
Hemophagocytic Syndrome, Epstein-Barr Viru, Diagnosis, Treatment

1. Introduction
Hemophagocytic syndrome (HS) is a clinically rare autoimmune disease that is characterized in clinical practice by fever, hepatomegaly, splenomegaly, pancytopenia, and hemophagocytosis in the bone marrow, liver, spleen, and lymph nodes. Scott and Rob-Smith have reported a neoplastic disorder, showing hemophagocytosing histiocytes and systemic proliferation of the precursors of histiocytes in 1939.
The diagnosis of HS includes a spectrum of inherited or acquired defects in cytotoxic lymphocyte function, often with uncontrolled infections. HS may also arise as the result of persistent antigen stimulation due to autoimmune disease or malignancy. HS is often described in binary terms as “primary,” indicating Mendelian inheritance of gene mutations resulting in cytotoxic lymphocyte dys- function, or “secondary” indicating an acquired reactive disorder. Increasing evidence describes HS as more complex phenomenon, resulting from specific immune challenges in patients with a susceptible genetic background. Early recognition of HS and evaluation of potential causes is critically important, as survival generally requires urgent treatment with immune suppression and resolution of the activating antigen. However, the diagnosis of HS is challenged by the myriad of pathways that lead to pathologic inflammation and the clinical overlap with other conditions. Further improvements in therapy will require prospective trials to define optimal strategies for each patient based on the individual paths that lead to pathologic inflammation. This paper is a case and literature review that focuses on one HS patient who was admitted to and treated in the First central hospital of Tianjin.
2. Case Report
2.1. Patient and Procedures
A 22-year-old female patient presented at the local hospital on June 30, 2013, with an intermittent fever accompanied by pancytopenia for >1.0 month. The patient’s fever of 38.5˚C began without obvious inducement; she experienced a slight intolerance to cold. After taking ibuprofen, the patient’s body temperature somewhat decreased; however, the fever returned without obvious chills and reached a high of 39.5˚C. Local treatment with infusion of antibiotics, such as penicillin, had some therapeutic effect; however, the fever returned without pha- ryngalgia, cough, phlegm, headache, nausea, vomiting, urodynia, diarrhea, abdominal pain, urgent urination, or frequent micturition.
On May 24, 2013, a bone marrow aspiration procedure and physical examinations and tests, such as a positron emission tomography-computed tomography (Graph 1), were performed on the patient in the Second People’s Hospital of Huaian; a malignant lymphoma was not excluded. On June 16, 2013, the patient


Graph 1. Positron emission tomography-computed tomography.
had diagnostic chemotherapy with a regimen comprising Cytoxan® (cyclophosphamide), Adriamycin® (hydroxy doxorubicin), vincristine (Oncovin®), and Prednisone (CHOP). On the second day of chemotherapy, the patient’s body temperature was under control; however, the fever returned 2.0 d later and a regimen of cefepime with peroral Voriconazole did not relieve the condition. The patient was given imipenem combined with vancomycin, acyclovir, and foscarnet sodium therapy for 1.0 week, but there was no obvious improvement and she was admitted into the hospital for further treatment.
2.2. Patient Data
On admission, the patient received a full physical examination. She appeared to have mild anemia with pale skin and mucosae, but no yellow staining, rash, or hemorrhagic spots. There were several movable, swollen lymph nodes with a maximum diameter of ~1.0 cm in the middle on the neck and under the armpit but no tenderness. The sclera was not yellow. The abdomen was flat without tenderness or rebound tenderness. The liver was felt 7.0 cm below the subcostal margin and the spleen was felt 11 cm below the subcostal margin.
Routine blood tests showed the following data: white blood cell count, 2.98 × 109/L, hemoglobin (Hb), 74.00/L, platelet count (PLT), 23.00 × 109/L, C-reactive protein, 7.57 mg/dL, plasma thromboplastin antecedent, 73%, thrombin time, 23.00 s, fibrinogen, 1.17 g/L, D-dimer 4004.16 μg/L. The bone marrow examination showed the presence of was erythrocytosis, guanulocytosis, and thrombocytosis accompanied by unidentifiable cells that were further inspected. Epstein- Barr (EB) virus DNA 4.63 × 105 copies/ml.
2.3. Pathology Reports
The results from pathological examinations showed that hyperplasia of the hemopoietic tissue was active and guanulocytosis was obvious (Graph 2). The
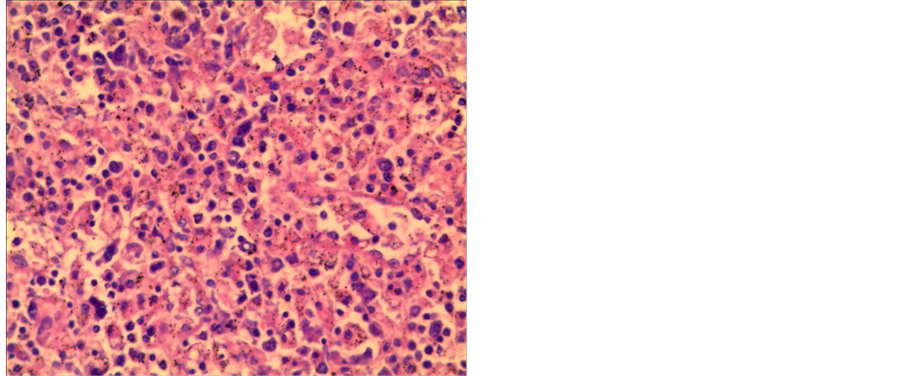
Graph 2. The results from pathological examinations.
patient’s anemia was corrected by an infusion of red blood cells. After receiving hormonotherapy for 1.0 week, the blood platelet count became normal and the spleen obviously retracted. After the condition was stable, a splenectomy was performed under general anesthesia on August 2, 2013. The extracted tissue was sent to the King Med Center of Medical Inspection. The results from the pathology tests indicated that hemophagocytic syndrome related to EB virus infection was the first consideration, and a T-cell receptor (TCR) examination was recommended to check for extranodal lymphoma, which was later ruled out from the TCR results.
The diagnostic criteria for hemophagocytic lymphohistiocytosis (HLH) from the International Tissue and Cell Society drafted in 2004 are as follows [1] : 1) a fever ≥38.5˚C for >7.0 d; 2) splenomegaly (≥3.0 cm below the costal margin); 3) peripheral blood cytopenia (reduction of more than two items in the blood routine examination); Hb < 90 g/L, ANC < 1.0 × 109/L, PLT < 100 × 109/L and not caused by bone marrow hematopoietic function reduction; 4) hypertriglyceridemia ≥ 3.0 mmol/L or hypofibrinogenemia ≤ 1.5 g/L; 5) increase in hematophages in the bone marrow, spleen, and lymph nodes; no evidence of a malignant tumor; 6) serum ferritin ≥ 500 µg/L; 7) reduction or deficiency in the activity of natural killer cells; and 8) soluble CD25 ≥ 2400 U/mL. If a patient meets five of the above criteria, he or she can be diagnosed with secondary HLH. This patient met items one through seven; therefore, a clear diagnosis was made. The patient’s family was informed of her condition and the patient was advised to undergo further standard treatment; however, she refused because of economic factors and was discharged from the hospital. She was never reexamined in our hospital and there was no follow up.
3. Discussion
HS, also known as HLH, is reactive histiocytosis [2] that is characterized mainly by fever, hepatomegaly, splenomegaly, pancytopenia, and hemophagocytosis occurring in the bone marrow, liver, spleen, and lymph nodes [3] [4] . Its pathogenesis is not completely clear but it is now considered an immunological derangement characterized by out-of-control activation of T lymphocytes and macrophages.
HS is divided into either the primary and acquired condition [5] . Primary HS, namely familial hemophagocytic lymphohistiocytosis, is an autosomal recessive disorder that often attacks infants; some HS cases are triggered by a viral infection and most result in an unfavorable prognosis. Its 5.0-year survival rate is only 7.0%. HS was first reported by Farquhar et al. [6] in 1952. HLH includes infection-associated hemophagocytic syndrome (IAHS) and malignant tumor hemophagocytic syndrome (MAHS). Most IAHS cases are caused by viruses such as EB, cytomegaloviruses, hepatitis E, and hepatitis B, and bacteria, such as Escherichia coli, Streptococcus pneumoniae, Salmonella typhi, and a serotype of S. enterica [7] [8] . Most MAHS cases are caused by malignant lymphoma.
The progression of HS is swift and violent, is often misdiagnosed, and has fatality rates. In particular, it is difficult to distinguish primary from acquired HS. The initial diagnosis of primary HS requires a complete patient history, including childhood diseases and any family members who tested positive for the condition; however, in recent years with continuous improvement of diagnostic criteria these requirements have changed. Individual cases of primary adult HS are regularly reported. The oldest patient on record is a 62-year-old male reported by Nagafuji et al. [9] , and the Beijing Friendship Hospital reported a 48-year-old male patient with primary HS [10] . The HLH-2004 Diagnostic Guide formulated by the International Tissue and Cell Society clearly points out that a genetic defect is the standard by which to distinguish primary from acquired HS. An infection, especially a viral infection, can not only be the pathogeny of acquired HS, but can also coexist with primary HS [11] ; therefore, primary and acquired HS should not be distinguished only by age, positive family history, infections, and diseases such as tumors, and gene screening is the gold standard by which to diagnose and distinguish the condition. Because gene screening was not performed on this patient, the subtype of her disease could not be determined. In the past 10 years, five gene mutations relating to familial hemophagocytic lymphohistiocytosis (FHL) have been successively authenticated and recognized (Table 1) [12] .
There is no effective treatment for HS. It is dormant in its early stages, but critical in the acute stages with a bad prognosis. About one-half of HS patients die mainly from hemorrhage, infection, multiple organ failure, and disseminated intravascular coagulation. The key to HS treatment is early diagnosis and early inhibition of the uncontrollable activity of lymphocytes and macrophages. Janka et al. [13] advised that HS be treated with dexamethasone + etoposide + cyclosporine + immunoglobulin. Shin et al. [14] reported that treatment with the CHOP scheme is also effective. The therapeutic schedule of HS revised by the International Tissue and Cell Society in 2004 is large doses of dexamethasone, etoposide, and ciclosporin A. If there is a central nervous system symptom that cannot be relieved with dexamethasone treatment for 2.0 weeks, methotrexate

Table 1. OMIM classifications of familial hemophagocytic lymphohistiocytosis.
FHL: familial hemophagocytic lymphohistiocytosis; OMIM: online Mendelian inheritance in man.
can be injected into the spinal column [15] . Some researchers suggest that if there is a bad curative effect or an adverse drug reaction administering the HLH- 2004 therapeutic protocol, the treatment should be properly modified. For example, VP-16 can be replaced with 150 mg/m2 teniposide per week and cyclosporine A (CsA) with peroral mycophenolate mofetil, or VP-16 can be replaced with mycophenolate mofetil plus CsA. If the disease is related to EB infection, ganciclovir can be added as a treatment. For cases without remission, treatment as follows was reported to be a remedial measure: anti-human thymocyte globulin (ATG), antagonists against tumor necrosis factor alpha (TNF-α) directly, anti CD52 monoclonal antibody, anti CD20 monoclonal antibody, and fludarabine. Because most HS cases are seen in children, most therapeutic regimens are recommended based on clinical research on children; therefore, the sensitivity and specificity of those regimens to adults need further research. The lack of diagnostic criteria of adult HLH makes the difficult diagnosis even harder; therefore, it is necessary to formulate HLH diagnosis and treatment standards.
4. Conclusion
Adult HLH is a critical condition that is often difficult to diagnose and results in a bad prognosis because of the lack of effective and unified treatment. Early diagnosis and early intervention are important factors for affecting the progression of the disease and improving its prognosis. In clinical practice, when children or adults have a fever, hepatosplenomegaly, lymphadenectasis, and a significantly abnormal ferritin level, this disease should be considered.
Cite this paper
Liu, X.L., Zhang, Y.M., Cui, Z.L., Tian, Q. and Duan, D.M. (2016) One Hemophagocytic Syndrome Re- lating to Adult Epstein-Barr Virus Infection and a Literature Review. Case Reports in Clinical Medicine, 5, 511-517. http://dx.doi.org/10.4236/crcm.2016.512065
References
- 1. Henter, J.I., Horne, A., Arico, M., et al. (2007) HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatric Blood & Cancer, 48, 124-131.
https://doi.org/10.1002/pbc.21039 - 2. Janka, G. (2009) Hemophagocytic Lymphohistiocytosis: When the Immune System Runs Amok. Klinische Pädiatrie, 221, 278-285.
https://doi.org/10.1055/s-0029-1237386 - 3. Henter, J.I., Tondini, C. and Pritchard, J. (2004) Histiocyte Disorders. Critical Reviews in Oncology/Hematology, 50, 157-174.
https://doi.org/10.1016/j.critrevonc.2004.01.002 - 4. Janka, G.E. (2012) Familial and Acquired Hemophagocytic Lymphohistiocytosis. Annual Review of Medicine, 63, 233-246.
https://doi.org/10.1146/annurev-med-041610-134208 - 5. Saxena, P., Bihari, C., Rastogi, A., et al. (2014) Hepatitis A Induced Hemophagocytic Syndrome. Tropical Gastroenterology, 35, 122-124. https://doi.org/10.7869/tg.195
- 6. Farquhar, J.W. and Claireaux, A.E. (1952) Familial Haemophagocytic Reticulosis. Archives of Disease in Childhood, 27, 519-525.
https://doi.org/10.1136/adc.27.136.519 - 7. Singha, A., Mukherjee, A., Dasgupta, R., et al. (2014) A Case of Hemophagocytic Syndrome Due to Tuberculosis: Uncommon Manifestation of a Common Disease. Case Reports in Medicine, 2014, 613845.
https://doi.org/10.1155/2014/613845 - 8. Kanhere, S., Bhagat, M., Kadakia, P., et al. (2014) Hemophagocytic Lymphohistiocytosis Associated with Cytomegalovirus Infection in an Immunocompetent Infant: A Diagnostic and Therapeutic Challenge! Indian Journal of Hematology and Blood Transfusion, 30, 299-302.
https://doi.org/10.1007/s12288-014-0366-4 - 9. Nagafuji, K., Nonami, A., Kumano, T., et al. (2007) Perforin Gene Mutations in Adult-Onset Hemophagocytic Lymphohistiocytosis. Haematologica, 92, 978-981.
https://doi.org/10.3324/haematol.11233 - 10. Wang, Y.N., Wang, Z. and Wang, X. (2012) A Case Report of Adult Onset of Primary Hemophagocytic Syndrome with Literature Review. Chinese Journal of Hematology, 33, 291-293.
- 11. Okur, H., Balta, G., Akarsu, N., et al. (2008) Clinical and Molecular Aspects of Turkish Familial Hemophagocytic Lymphohistiocytosis Patients with Perforin Mutations. Leukemia Research, 32, 972-975. https://doi.org/10.1016/j.leukres.2007.11.033
- 12. Cetica, V., Pende, D., Griffiths, G.M., et al. (2010) Molecular Basis of Familial Hemophagocytic Lymphohistiocytosis. Haematologica, 95, 538-541.
https://doi.org/10.3324/haematol.2009.019562 - 13. Janka, G.E. (2007) Familial and Acquired Hemophagocytic Lymphohistiocytosis. European Journal of Pediatrics, 166, 95-109.
https://doi.org/10.1007/s00431-006-0258-1 - 14. Shin, H.J., Chung, J.S., Lee, J.J., et al. (2008) Treatment Outcomes with Chop Chemotherapy in Adult Patients with Hemophagocytic Lymphohistiocytosis. Journal of Korean Medical Science, 23, 439-444. https://doi.org/10.3346/jkms.2008.23.3.439
- 15. Suresh, N., Uppuluri, R., Geetha, J., et al. (2014) Hemophagocytic Lymphohistiocytosis Masking the Diagnosis of Lymphoma in an Adolescent Male. Indian Journal of Hematology and Blood Transfusion, 30, 135-137.
https://doi.org/10.1007/s12288-013-0291-y
NOTES

*These authors contributed equally to this work.