Case Reports in Clinical Medicine
Vol.05 No.05(2016), Article ID:66717,5 pages
10.4236/crcm.2016.55031
Hyperacute cEEG Attenuation in Aneurysmal Subarachnoid Hemorrhage
Tyson Burghardt, Gautam Sachdeva, Malathi Rao, Saqib Chaudhry, Alex Schulte, Mounzer Yassin-Kassab
Department of Neurology, Michigan State University, East Lansing, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 February 2016; accepted 21 May 2016; published 24 May 2016
ABSTRACT
We describe hyperacute generalized EEG slowing and then attenuation captured at the moment of subarachnoid hemorrhage in a monitored patient. This is the first reported cEEG capture of aneurysmal subarachnoid hemorrhage in the literature.
Keywords:
Aneurysmal Subarachnoid Hemorrhage, EEG

1. Introduction
The advent of widespread long-term continuous electroencephalographic (cEEG) recording has given rise to new issues in interpretation. New patterns [1] as well as new appreciations for known patterns [2] have both been reported.
With this level of EEG monitoring saturation, it has become likely that events rarely or never examined electrographically would become incidentally captured by cEEG. These instances can be instructive in demonstrating an electrographic “natural history” which may better inform neurophysiologists about the state of a patient monitored sometime after the index event but still, perhaps, bearing residual EEG stigmata from it. To the extent that cEEG decisions about status epilepticus [3] may still be muddled in many cases, increased knowledge of possible EEG patterns following a variety of acute events may help reduce uncertainty in the neurophysiologist.
We report a case of a 64 year-old woman with unusual electroencephalographic changes acutely in subarachnoid hemorrhage.
2. Case Report
The patient has a history of atrial myxoma and brain metastases requiring surgical resection as well as radiotherapy (focal and whole-brain). No malignant tissue was identified on pathology. The craniotomy was around the top parasagittal part of the calvarium. She was treated with steroids briefly during the course of her illness. Her medical course was complicated by a pulmonary embolism in 2006, for which she has been on warfarin.
She recently (seven years after her atrial myxoma diagnosis, and five years after her craniotomy) started to have seizures with two semeiologies: 1) characterized by jerking movements of the body and aphasia followed by loss of consciousness; and 2) characterized by nonsensical speech, word finding difficulty, and impaired comprehension lasting for several minutes. These escalated in frequency to once daily. Imaging studies did not show significant changes.
EEG showed slow posterior background rhythm (7 Hz) without definite epileptiform discharges. She had been on phenytoin in the past which did not help with her seizures. Levetiracetam at 2000 mg/day and lamotrigine at 250 mg/day appeared to be helpful only in reducing seizure frequency to three per week. Changing lamotrigine to lacosamide 400 mg/day along with increasing levetiracetam to 4000 mg/day did not help further. She was admitted to the epilepsy monitoring unit (EMU) for seizure characterization and quantification.
Her exam showed poor memory (recall 1/3 words after 1 minute and 0/3 words after 3 minutes). She did not make any speech errors. She had a left inferior quadrantanopsia, brisk reflexes in the arms and legs, and slow rapid alternating movements. The sensory examination was normal. Laboratory analyses of complete blood count, complete metabolic panel, ammonia, and urinalysis were unremarkable.
One the day of the EMU admission her baseline EEG showed a well-regulated and reactive 7.5 Hz posterior dominant rhythm (PDR). There was right parietal slowing without epileptiform discharges. No ictal events were reported or recorded during the day.
At 23:45 that night there was a paroxysmal change in the EEG (see below) and the patient was noted to have stertorous breathing without other clinical manifestations. Nursing staff tried to wake the patient. They found her unresponsive and having been incontinent of urine. Neurologic examination demonstrated withdrawal to pain in all four extremities. She had equally reactive pupils bilaterally. There was no gaze preference and oculocephalic reflexes were intact. Muscle tone was symmetric throughout. Toes were not upgoing.
On continuous video and EEG monitoring at 23:48:57 the rhythm changed abruptly from waking baseline to generalized, low amplitude (below 20 µV) polymorphic delta which was more apparent over the left. This rapidly led to further loss of amplitude within five seconds (Figure 1). After ten minutes there was a return of generalized, low amplitude, arrhythmic theta activity.
Computed tomography (CT) scan of the brain showed an acute subarachnoid hemorrhage (SAH) along the right lateral sulcus (Figure 2). CT Arteriography (CTA) of the head was obtained and showed a right middle cerebral artery trifurcation aneurysm at the genu measuring 1.9 × 2.1 × 3.3 mm with a neck of 1.1 × 1.7 mm.
Despite optimum medical therapy the patient later died of complications related to her SAH.
3. Discussion
Cerebral aneurysm formation is a known complication in patients with atrial myxoma.
A wide variety of EEG abnormalities are seen in SAH. EEG may detect disruption of normal PDR, slowing or disorganization, seizures (6%), lack of reactivity (14%), periodic discharges (16%), and a lack of sleep transients in up to 85% even before vasospasm onset [4] . In a study done on 151 patients on the day of SAH certain EEG patterns predicted later vasospasm, including brief frontally-predominant biphasic delta waves (called by the authors axial bursts), focal polymorphic delta overlying an area of clot, and predominance of unreactive delta activity. These patterns had good correlation with CT grades of SAH severity [5] . However, none of the cases in the literature described the primary EEG changes during our patient’s hemorrhage.
This case is unique in that it is the first report of a cerebral aneurysm rupture captured on continuous EEG recording. EEG phenomenology may provide some insights to early SAH pathophysiology.
The instant development of the generalized suppression on EEG was impressive. The left-sided predominance of this voltage attenuation is even more impressive given that the aneurysm and hemorrhage were located in the right MCA territory. We believe that these changes cannot be reconciled with readily-accepted mechanisms of brain injury in SAH.
There are several proposed mechanisms to explain early (first 72 hours) brain damage during the acute phase of SAH. These include the formation of microthrombosis, cortical spreading depression, and increases in intracranial pressure (ICP).
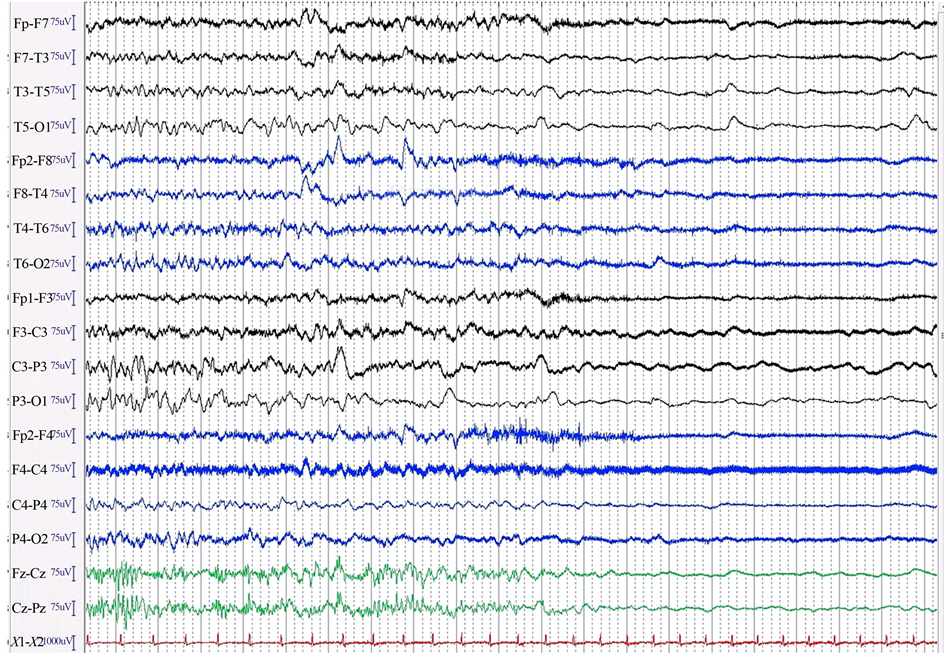
Figure 1. EEG monitoring showing hyperacute generalized and attenuation captured at the moment of subarachnoid hemorrhage.

Figure 2. Computed tomography (CT) scan of the brain showed an acute subarachnoid hemorrhage (SAH) (arrow) along the right lateral sulcus.
At time of ictus, blood flows from a ruptured aneurysm into subarachnoid space under high pressure causing rapid increases in ICP. Although this may create a tamponade effect to resist continuous bleeding, it also decreases cerebral blood flow (CBF) and consequently cerebral perfusion pressure (CPP). Below critical levels, decreased perfusion leads to cerebral ischemia. This will activate inflammatory pathways and the release of pro-inflammatory cytokines which produce edema [6] .
The endothelial damage at the site of vascular rupture induces the coagulation cascade. As cascade protein products diffuse, microthrombus formation with additional ischemia is the result [7] [8] .
Cerebral spreading depression (CSD) refers to the wave of depolarization and subsequent hyperpolarization in the cerebral grey matter travelling at a rate of 2 - 5 mm/min [9] . The process is initiated by different types of cortical insults that cause cation and water influx across the cell membrane [10] . Sodium and calcium pumps are activated to counterbalance the surge in intracellular sodium and calcium. This results in increased energy consumption and a fall in ATP by 50% [11] . Spreading depolarizations may be preceded by field oscillations that are propagated at distances up to 1 mm [12] . Typically there is spontaneous gradual return of non-depressed cortical activity. It is conceivable that the widespread slowing and suppression seen in our patient so rapidly may be related to this disruption in homeostasis.
Prolonged ischemia produces infarction with neuronal death. Neurological symptoms begin when CBF reaches 30 mL per 100 g of cortex tissue per minute. At 16 - 20 mL per 100 g per min evoked potentials are affected and the EEG becomes flat. At 25 - 35 mL per 100 g per min the EEG loses faster frequencies and at 17 - 18 mL per 100 g per min slower frequencies increase. At the same level, neurons lose their transmembrane concentration gradients. As CBF continues to decrease to 10 - 12 mL per 100 g per min cerebral edema results from Na+-K+ pump failure and at flows less than 10 there is complete metabolic failure: the EEG is silent and there is irreversible cellular damage.
The immediate EEG findings in our case suggest a more rapid pathophysiology than CSD or microembolic showering. In the former case, an instantaneous dropout of EEG signal would imply an extremely uncharacteristic rapidity of the wave of spreading depression. The latter case, too, is less consistent with immediate generalized suppression as it is logically more apt to produce sudden ischaemia only within a scattered region of one hemisphere.
Acute changes to ICP following hemorrhage are more consistent, as this mechanism entails widespread cerebral dysfunction and can occur very rapidly. Slowing and suppression from pressure rises in a single unilateral compartment might be expected ipsilaterally due to direct compressive effects and due to EEG attenuation from increased distance to the scalp surface. The contralateral predominance in our case, on the other hand, may reflect initial deflection of the brain into the skull table opposite the hemorrhage by the jet of arterial blood on the right. This would be followed by rapid equalization of pressure throughout due to accumulation of blood.
The immediate effect of SAH on EEG augments the available evidence that there are already early changes to cortical dynamics and function which may reflect longstanding damage long before vasospasm can further complicate the picture.
Cite this paper
Tyson Burghardt,Gautam Sachdeva,Malathi Rao,Saqib Chaudhry,Alex Schulte,Mounzer Yassin-Kassab, (2016) Hyperacute cEEG Attenuation in Aneurysmal Subarachnoid Hemorrhage. Case Reports in Clinical Medicine,05,165-169. doi: 10.4236/crcm.2016.55031
References
- 1. Schmitt, S.E., Pargeon, K., Frechette, E.S., Hirsch, L.J., Dalmau, J. and Friedman, D. (2012) Extreme Delta Brush: A Unique EEG Pattern in Adults with Anti-NMDA Receptor Encephalitis. Neurology, 79, 1094-100.
http://dx.doi.org/10.1212/WNL.0b013e3182698cd8 - 2. Chong, D.J. and Hirsch, L.J. (2005) Which EEG Patterns Warrant Treatment in the Critically Ill? Reviewing the Evidence for Treatment of Periodic Epileptiform Discharges and Related Patterns. Journal of Clinical Neurophysiology, 22, 79-91.
http://dx.doi.org/10.1097/01.WNP.0000158699.78529.AF - 3. Trinka, E. and Leitinger, M. (2015) Which EEG Patterns in Coma Are Nonconvulsive Status Epilepticus? Epilepsy & Behavior, 49, 203-22.
http://dx.doi.org/10.1016/j.yebeh.2015.05.005 - 4. Claassen, J., Hirsch, L.J., Frontera, J.A., et al. (2006) Prognostic Significance of Continuous EEG Monitoring in Patients with Poor-Grade Subarachnoid Hemorrhage. Neuro-critical Care, 4, 103-112.
http://dx.doi.org/10.1385/NCC:4:2:103 - 5. Rivierez, M., Landau-Ferey, J., Grob, R., Grosskopf, D. and Philippon, J. (1991) Value of Electroencephalogram in Prediction and Diagnosis of Vasospasm after Intracranial Aneurysm Rupture. Acta Neurochirurgica, 110, 17-23.
http://dx.doi.org/10.1007/BF01402042 - 6. Claassen, J., Carhuapoma, J.R., Kreiter, K.T., Du, E.Y., Connolly, E.S. and Mayer, S.A. (2002) Global Cerebral Edema after Subarachnoid Hemorrhage: Frequency, Predictors, and Impact on Outcome. Stroke, 33, 1225-1232.
http://dx.doi.org/10.1161/01.STR.0000015624.29071.1F - 7. Frijns, C.J.M., Fijnheer, R., Algra, A., van Mourik, J.A., van Gijn, J. and Rinkel, G.J.E. (2006) Early Circulating Levels of Endothelial Cell Activation Markers in Aneurysmal Subarachnoid Haemorrhage: Associations with Cerebral Ischaemic Events and Outcome. Journal of Neurology, Neurosurgery & Psychiatry, 77, 77-83.
http://dx.doi.org/10.1136/jnnp.2005.064956 - 8. Romano, J.G., Forteza, A.M., Concha, M., et al. (2002) Detection of Microemboli by Transcranial Doppler Ultrasonography in Aneurysmal Subarachnoid Hemorrhage. Neurosurgery, 50, 1021-1026.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11950405 - 9. Leão, A.A.P. (1944) Spreading Depression of Activity in the Cerebral Cortex. The Journal of Physiology, 7, 359-390.
- 10. Adámek, S. and Vyskocil, F. (2011) Potassium-Selective Microelectrode Revealed Difference in Threshold Potassium Concentration for Cortical Spreading Depression in Female and Male Rat Brain. Brain Research, 1370, 215-219.
http://dx.doi.org/10.1016/j.brainres.2010.11.018 - 11. Dreier, J.P., Major, S., Manning, A., et al. (2009) Cortical Spreading Ischaemia Is a Novel Process Involved in Ischaemic Damage in Patients with Aneurysmal Subarachnoid Haemorrhage. Brain, 132, 1866-1881.
http://dx.doi.org/10.1093/brain/awp102 - 12. Larrosa, B., Pastor, J., López-Aguado, L. and Herreras, O. (2006) A Role for Glutamate and Glia in the Fast Network Oscillations Preceding Spreading Depression. Neuroscience, 141, 1057-1068.
http://dx.doi.org/10.1016/j.neuroscience.2006.04.005