American Journal of Plant Sciences
Vol.3 No.5(2012), Article ID:19473,4 pages DOI:10.4236/ajps.2012.35067
Phylogenetic Relationships in the Genus Spiraea (Rosaceae) Inferred from the Chloroplast DNA Region, trnL-trnF
![]()
Department of Molecular Biology, Dongeui University, Busan, Korea (South).
Email: mkhuh@deu.ac.kr
Received December 19th, 2011; revised January 29th, 2012; accepted February 10th, 2012
Keywords: Genus Spiraea; Chloroplast trnL-trnF; Phylogenetic Relationships
ABSTRACT
The 26 taxa of genus Spiraea (Rosaceae) in 14 Korean taxa and four taxa of them doubled in the world itself were collected and investigated phylogenetic relationships within these taxa using the chloroplast trnL-trnF region. Sequence variation within the genus Spiraea was mostly due to insertion/deletion. Total alignment sequence of trnL-trnF region in genus Spiraea was 1004 positions, of which 126 were parsimony-informative, 149 variable, 23 singleton, and 847 constant characters. The base furtherance did not show a significant difference among total taxa. Although many taxa of genus Spiraea were well separated each other, many branches were not congruent with the taxon positions with botanical names. In addition, sequences of the chloroplast trnL-trnF region for same species are different from each other according to authors. The taxonomic identification of genus Spiraea are in need of revision.
1. Introduction
Spiraea (also known as meadowsweet) is a genus with 80 - 100 species of shrubs in the subfamily Spiraeoideae (Rosaceae) [1]. They are native to the temperate Northern Hemisphere, with the greatest diversity in eastern Asia [2].
Among plant systematic and phylogeographic studies the chloroplast genome is widely used and generally accepted as an excellent source for molecular information [3,4]. There are several reasons for this. First, the uniparental inheritance ensures orthology of sequences. Second, even within an individual the possibility of recombination between genomes from individual plastids is extremely low, and there are only a few studies describing the occurrence of multimeric chloroplast DNA (cpDNA) genomes or interchromosomal cpDNA recombination [5,6]. Third, dramatic changes in gene content and structure only occurred after the chloroplast genome entered a eukaryotic cell via primary endocytobiosis [7], whereas land plant plastomes are highly conserved [8].
Chloroplast DNA (cpDNA) sequence variations has been widely used to interspecific relationships in plant systematics and phylogeography [9]. Especially, Taberlet et al. [9] introduced universal primers to amplify the region comprising the trnT (UGA) gene, the trnL (UAA) gene. Potter et al. [10] reported the relationships in Rosaceae including Spiraea densiflora using matK and trnLtrnF sequence data.
Genus Spiraea is very diverse with interspecific varieties, discriminated by differences in morphology. Spiraea has been various divided by different authors into subgenera, sections, series, and cycles [1]. This study represents a preliminary investigation of the phylogeny of the main genera of the Spiraea in Korea through the comparative analysis of cpDNA sequences. The aim of this study was to investigate the potential use of the trnL (UAA)-trnF (GAA) region to identify some Korean species and was compared them with Spiraea taxa which were reported in GenBank. These taxa will represent the foundation of an investigation on the potential to understand intraspecific genetic diversity in the world.
2. Materials and Methods
Many individuals of genus Spiraea were collected from large effective populations in Korea (Table 1) and identified according to Lee [11] and Kim and Sun [12]. Total genomic DNA was extracted from 0.5 - 1.0 g of fresh leaf material. Stephanandra incise (Thunb) Zabel of the closely related genus Stephanandra was included in the phylogenetic reconstruction as an outgroup.
DNA was extracted using the plant DNA Zol Kit (Life Technologies Inc., Grand Island, New York, USA) according to the manufacturer’s protocol.
The trnL-trnF gene regions of genus Spiraea were am-

Table 1. The collection sites and sections of genus Spiraea in Korea.
plified using the primer pair to acquire the respective entire region. The final PCR cocktail of 50 μl contained 38 μl water, 5 μl 10% buffer, 3 μl Mg+2, 1 μl dNTPs, 0.25 μl Taq polymerase, and 0.5 μl of each primer. The amplifying reactions were run for 28 cycles of denaturing for 30 s at 95˚C, primer annealing for 50 s at 57˚C, and elongation for 2 min at 72˚C.
PCR products were separated on 1.5% agarose gels and purified using the QIAquick Gel Extraction Kit (QIAGEN Inc., Chatsworth, CA) following the manufacture’s protocols. The amplified fragments were cloned into a bluescript vector and sequenced using ABI Prism 377 Sequencer (Applied Biosystems Inc., Foster City, CA). At least ten individuals’ clones of each taxon were analyzed. The sequencing data were analyzed and edited using the Sequencher software program (Gene Codes Corporation, Ann Arbor, MI, USA).
A pairwise alignment was calculated using the Clustal X program. Phylogenetic relationship were estimated by MEGA5 version 5.0 [13] treating all alignment gaps as missing. The quality of these alignments was assessed with homoplasy indicators [consistency index (CI) [14, 15], retention index (RI) [16], and rescaled consistency index (RC) [16]. The alignments were then checked, and apparent alignment errors were corrected by hand to minimize homoplasy.
Models with the lowest BIC scores (Bayesian Information Criterion) are considered to describe the substitution pattern the best. For each model, AICc value (Akaike Information Criterion, corrected), Maximum Likelihood value (lnL), and the number of parameters (including branch lengths) are also presented. Non-uniformity of evolutionary rates among sites may be modeled by using a discrete Gamma distribution (+G) with 5 rate categories and by assuming that a certain fraction of sites are evolutionarily invariable (+I). Whenever applicable, estimates of gamma shape parameter and/or the estimated fraction of invariant sites are shown. Assumed or estimated values of transition/transversion bias (R) are shown for each model, as well. Codon-based tests of neutrality for analysis between trnL-trnF sequences of genus Spiraea were conducted using the Nei-Gojobori method.
The DNA sequence data of the trnL (UAA)-trnF (GAA) region were obtained from two different sources. A first source is focusing on this study. All these sequences had been already deposited in GenBank. The second source is GenBank. The sequences of 27 accessions have been selected from previously published studies on Spiraea systematics (Table 2).
A maximum parsimony tree (MP) was inferred using heuristic search, branch-swapping options and tree bisection-reconnection using MEGA5 [13]. Confidence values for individual branches were determined by a bootstrap analysis with 100 repeated sampling of the data.
3. Results
Sequencings of trnL-trnF region for seventeen taxa of Spiraea in Korea were successful in all of the species. Aligned nucleotides of trnL-trnF were varied within Spiraea varying from 963 bp in S. cantoniensis to 975 bp

Table 2. Accession numbers and trnL-trnF sequences of genus Spiraea from GenBank.
Sequencings of trnL-trnF region for seventeen taxa of Spiraea in Korea were successful in all of the species. Aligned nucleotides of trnL-trnF were varied within Spiraea varying from 963 bp in S. cantoniensis to 975 bp in S. chinensis, S. japonica, and S. miyabei (Table 3). The mean nucleotide frequencies for seventeen taxa of genus Spiraea in Korea are A = 36.1%, C = 16.2%, G = 16.4%, and T = 31.3%. Total alignment length of genus Spiraea in Korea is 1004 positions, of which 126 are parsimony-informative, 149 variable, 26 singleton, and 847 constant characters. Number of segregating sites of genus Spiraea in Korea was 143 and nucleotide diversity (π) was 0.034. Under the neutral mutation hypothesis, the probability that D is negative (–1.030) is less than 0.5 (Table 4). The negative value of D was observed in the case of deletion/insertion. Thus, deletion/insertion increased the genetic variation. All trnL-trnF trees generated in Korea exhibited well solved topology with high bootstrap support irrespective of the methods (parsimony) and the setting used. S. pubescens and S. media were very distinct in MP tree (Figure 1). S. fritschiana was also very distinct. S. microgyna was also very distinct. This result confirmed monophyletic group for all species.
In 27 world accessions, 14 overlapped in Korean taxa and four taxa had doubled in the world itself. Thus, 26 taxa were true. Total alignment length in 27 world accessions is 1004 positions, of which 126 are parsimonyinformative, 149 variable, 23 singleton, and 847 constant characters. The base composition did not showed a significant difference among total taxa. The estimated value of the shape parameter for the discrete Gamma Distribution is 1.7129. Substitution pattern and rates were estimated under the Tamura-Nei’ model (+G). A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories, [+G]). Mean evolutionary rates in these categories were 0.22, 0.51, 0.82, 1.23, 2.22 substitutions per site. The nucleotide frequencies are A = 36.3%, T = 31.3%, C = 16.2%, and G = 16.2%. For estimating ML values, a user-specified topology was used. The maximum Log likelihood for this computation was –2063.296. The analysis involved 42 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions containing gaps and missing data were eliminated. There were a total of 871 positions in the final dataset.
The seven parsimonious trees were recovered from the
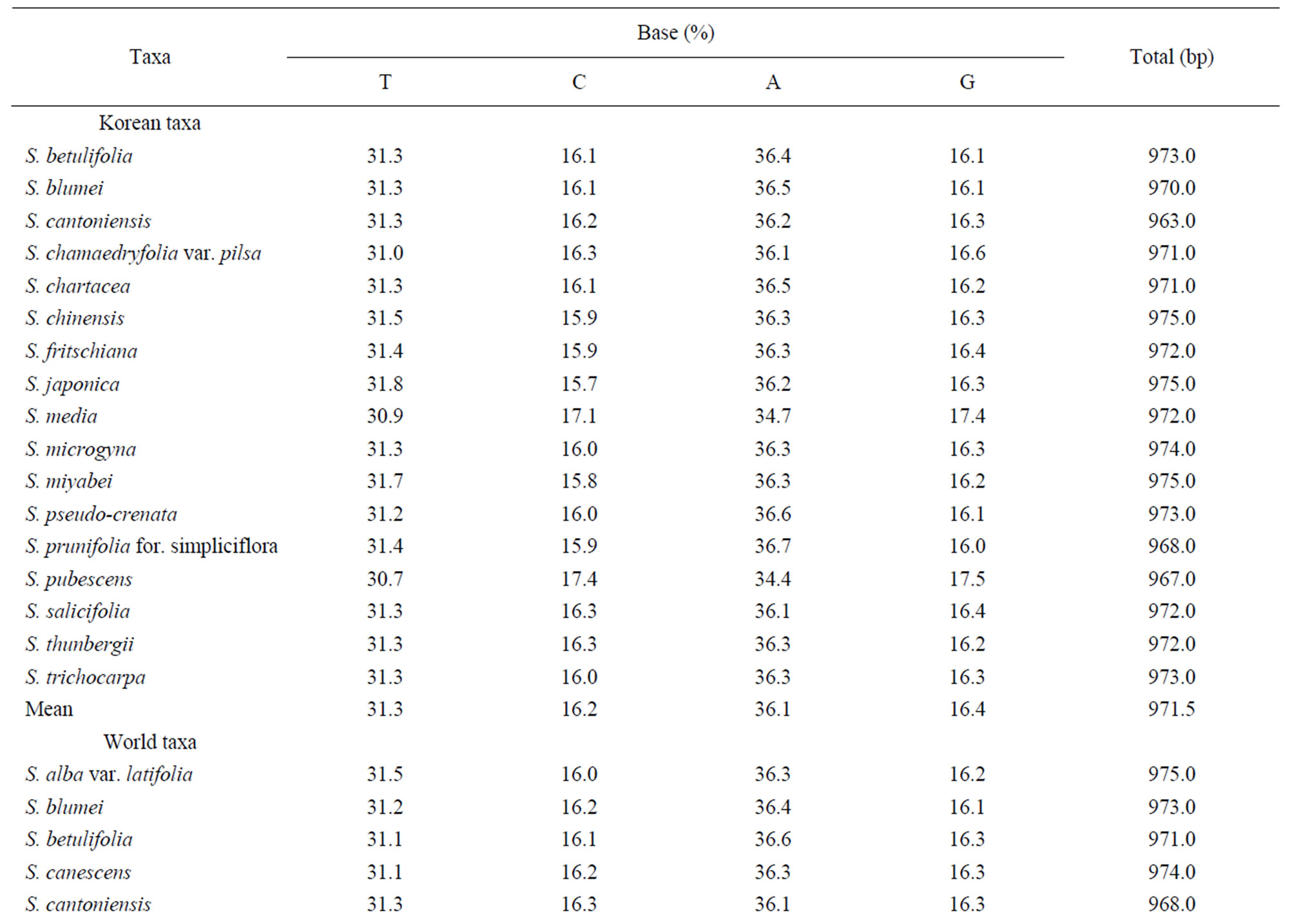

Table 3. Base frequencies and total length across fourteen taxa of genus Spiraea in Korea using trnL-trnF analysis.
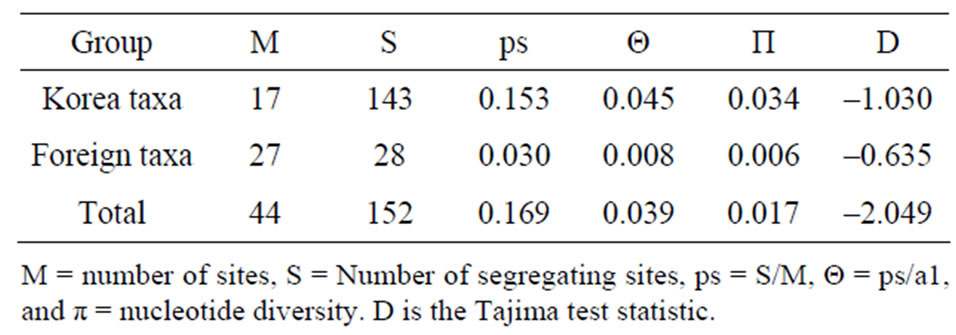
Table 4. Results from Tajima’s neutrality test for trnL-trnF sequences of genus Spiraea.
parsimony analysis with a CI of 0.897, a RI of 0.908, and a RC of 0.814. The most parsimonious tree was recovered from the parsimony analysis with a length of 358 steps and all taxa were most fully resolved (Figure 2). All species (including varieties) formed three or four distinct clades with moderate bootstrap support (for convenience, only bootstrap values under parsimony criterion are presented here). All species including varieties in the Korean did not formed one clade. Only S. japonica formed one clade. Three accessions (two worlds and one Korean) of S. japonica and three S. vergiana were divided into different clades. Outgroup was omitted in phylogenetic tree because of the very long branch between genus Spiraea and outgroup.
4. Discussion
The internal structure of the genus Spiraea is by and large in discordances with other recent and more densely sampled DNA phylogenies of the Spiraeea, such as those of Porter et al. [1]. The sparse sampling of Spiraea taxa in the present study, together with a relatively high DNA sequence mutation rate, made alignment of Spiraea sequences a difficult task, and because of this difficulty, an insertion/deletion complex unique to the one Spiraea taxa had to be removed from the data matrix before analysis.
Although all taxa in Korea exhibited well solved topology with high bootstrap, phylogenetic reconstructions revealed notable differences in worldwide taxa (Figure 2). This discordance in trees for Spiraea may be explained with several possible explanations. First, it is a possibility of species-misidentification. An accurate identification of Spiraea species has always been problematic even for expert botanists. This is because of the contradictory classification systems proposed by various researchers, primarily based on morphological characters that could be highly variable depending on the environmental conditions. For example, S. chamaedryfolia var. pilsa has hairs in flowers. S. chamaedryfolia var. pilsa is separated from S. chamaedryfolia by this character. However ther are very similar to each other. S. chamaedryfolia var. pilsa are the varieties of S. chamaedryfolia. However, they are distinct from each other (Figure 2). Second, the evidence that hybridization events most
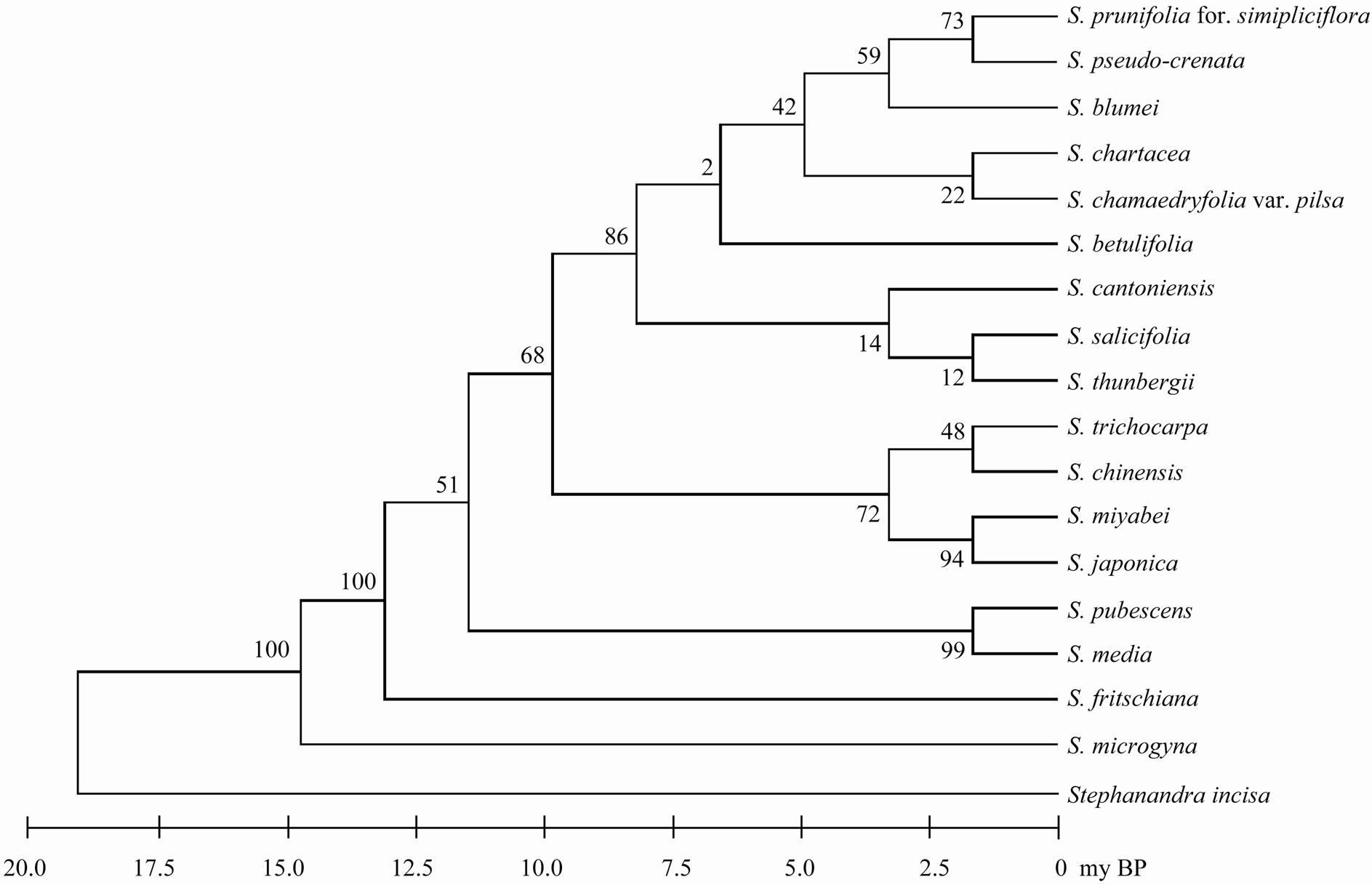
Figure 1. The maximum parsimonious tree for Korean taxa of genus Spiraea based on trnL-trnF using MEGA5. The values of bootstrap were shown in side of vertical lines.
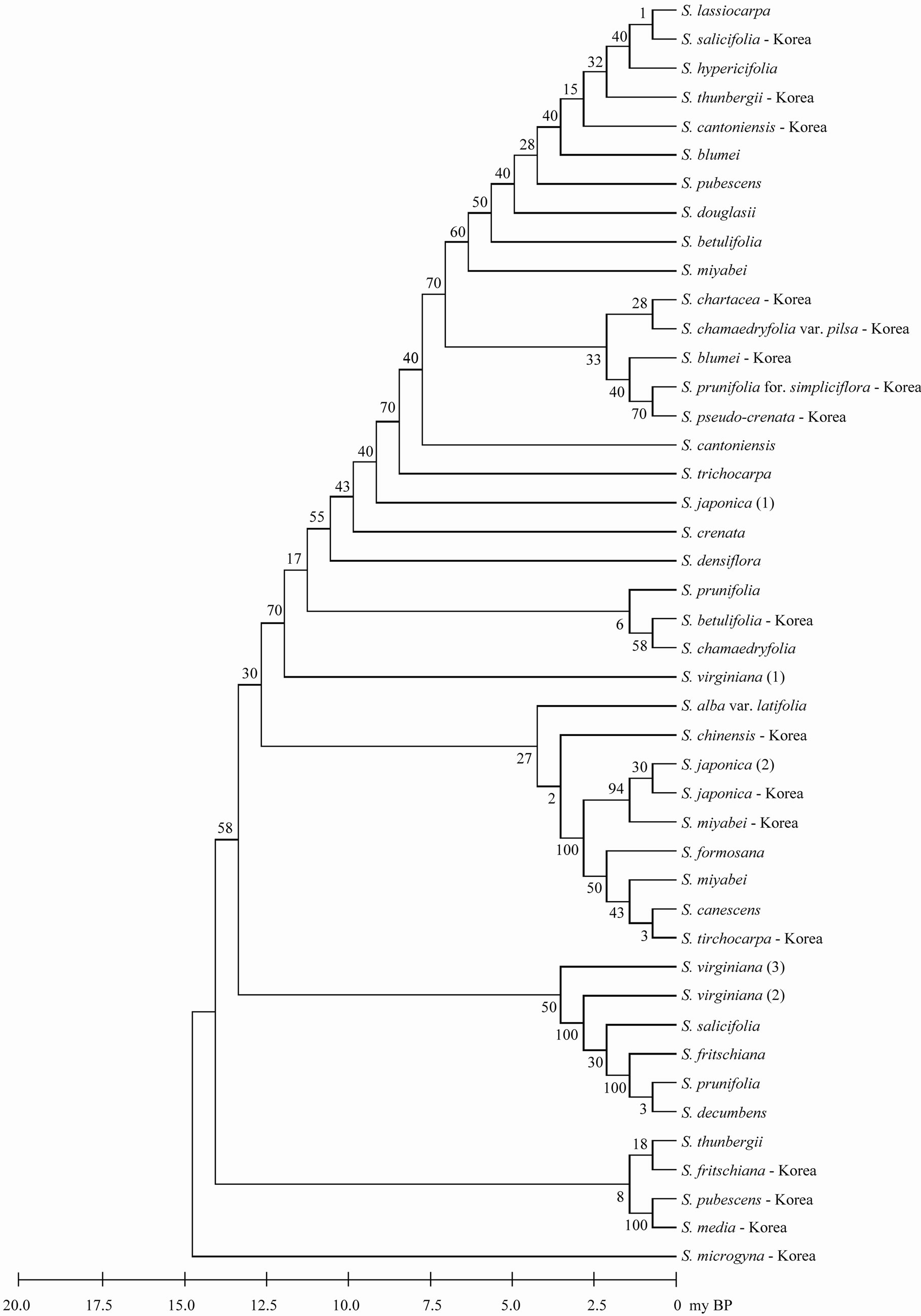
Figure 2. The maximum parsimonious tree for genus Spiraea in the worldwide based on trnL-trnF using MEGA5. The values of bootstrap were shown in side of vertical lines.
likely occurred in several places was provided by the presence in the introduced meadowsweet populations [17]. Although all varieties of S. japonica are not hybridized species, hybridization cannot be ruled out. Many varieties of S. japonica (var. acuta, var. acuminata, var. fortune, var. glabra, var. japonica, var. incise, var. ovalifolia, and var. stellaris) are occurred in same regions. Third, the methods and primers of trnL-trnF may be different ways according to authors. For example, the sequences of combined primer set (forward and reverse primers) are slightly different for each person or species. Some species were well amplified, but other species needed some modified primers for amplification. For one reason or another, fourteen overlapped taxa between Korea and other countries did not same clades for same species (Figure 2). In addition, several sequencies of S. japonica in GenBank were not accordance with each other.
Inflorescence type, the basis for the recognition of three sections within Spiraea [18] does not appear to be a reliable indicator of relationship within the genus (Figure 2), but some correlations were nonetheless observed. While compound corymbs, characteristic of section Calospira, are found in S. fritschiana var. obtusifolia, S. fritschiana, and S. miyabei. In this study, Calospira was sistered to section Chamaedryon (strongly supported by Bayesian analysis) in some of the most parsimonious trees.
The plants of genus Spiraea grow in forest-steppe, steppe, semi-dessert zones, and in subalpine belt of the mountains of the North Hemisphere [20]. This environmental heterogeneity could have resulted in more geographical barriers, higher selection pressures, greater isolation, and less gene flow between populations [18]. All these factors might explain the higher divergence and diversification among S. japonica varieties. It is a general principle that the higher the genetic divergence among phylogenetic lines, the earlier the many taxa diverged. This has been widely applied in the interpretation of biogeographical history using sequence data [19]. A possible explanation might be that different areas with different environments will vary in mutation accumulation or divergence maintenance and consequently differ in detectable evolutionary rate.
REFERENCES
- D. Potter, S. M. Still, D. Ballian and H. Kraigher, “Phylogenetic Relationships in Tribe Spiraeeae (Rosaceae) Inferred from Nucleotide Sequence Data,” Plant Systematics and Evolution, Vol. 266, No. 2, 2007, pp. 105-118. doi:10.1007/s00606-007-0544-z
- Committee of Chinese Academy of Sciences for Physical Geography of China, “Physical Geography of China: Pandect,” Science Press, Beijing, 1985.
- G. M. Hewitt, “Speciation, Hybrid Zones and Phylogeography or Seeing Genes in Space and Time,” Molecular Ecology, Vol. 10, No. 3, 2001, pp. 537-549. doi:10.1046/j.1365-294x.2001.01202.x
- R. G. Olmstead and J. D. Palmer, “Chloroplast DNA Systematics: A Review of Methods and Data Analysis,” American Journal of Botany, Vol. 81, No. 9, 1994, pp. 1205-1224. doi:10.2307/2445483
- A. M. Dally and G. Second, “Chloroplast DNA Diversity in Wild Rice and Cultivated Species of Rice (Genus Oryza, Section Oryza). Cladistic-Mutation and GeneticDistance Analysis,” Theoretical and Applied Genetics, Vol. 80, No. 2, 1990, pp. 209-222. doi:10.1007/BF00224389
- D. R. Govindaraju, B. P. Dancik and D. B. Wagner, “Novel Chloroplast DNA Polymorphisms in a Sympatric Region of Two Pines,” Journal of Evolutionary Biology, Vol. 2, No. 2, 1989, pp. 49-59. doi:10.1046/j.1420-9101.1989.2010049.x
- L. Martin and R. G. Herrmann, “Gene Transfer from Organelles to the Nucleus: How Much, What Happens, and Why?” Plant Physiology, Vol. 118, No. 1, 1998, pp. 9-17. doi:10.1104/pp.118.1.9
- V. V. Goremykin, K. I. Hirsch-Ernst, S. Wölfl and F. H. Hellwig, “Analysis of the Amborella Trichopoda Chloroplast Genome Sequence Suggests That Amborella Is Not a Basal Angiosperm,” Molecular Biology and Evolution, Vol. 20, No. 9, 2003, pp. 1499-1505. doi:10.1093/molbev/msg159
- P. Taberlet, L. Gielly, G. Patou and J. Bouvet, “Universal Primers for Amplification of Three Non-Coding Regions of Chloroplast DNA,” Plant Molecular Biology, Vol. 17, No. 5, 1991, pp. 1105-1109. doi:10.1007/BF00037152
- D. Potter, F. Gao, P. Esteban Bortiri, S. H. Oh and S. Baggett, “Phylogenetic Relationships in Rosaceae Inferred from Chloroplast matK and trnL-trnF Nucleotide Sequence Data,” Plant Systematics and Evolution, Vol. 231, No. 1, 2002, pp. 77-89. doi:10.1007/s006060200012
- Y. M. Lee, 1992, “Taxonomic Study on the Genus Spiraea in Korea: Specially Referred to Morphology and Flavonoid Characteristics,” Ph.D. Thesis, Seoul National University, Seoul.
- T. J. Kim and B. Y. Sun, “Taxonomy of the Genus Spiraea in Korea,” Korean Journal of Plant Taxonomy, Vol. 26, No. 3, 1996, pp. 191-212.
- K. Tamura, D. Peterson, N. Peterson, G. Stecher, M. Nei and S. Kumar, “MEGA5 Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods,” Molecular Biology and Evolution, Vol. 28, No. 5, 2011, pp. 2731- 2739. doi:10.1093/molbev/msr121
- A. G. Kluge and J. S. Farris, “Quantitative Phyletics and the Evolution of Anurans,” Systematic Zoology, Vol. 18, No. 1, 1969, pp. 1-32. doi:10.2307/2412407
- M. J. Sanderson and M. J. Donoghue, “Patterns of Variation in Levels of Homoplasy,” Evolution, Vol. 43, No. 8, 1989, pp. 1781-1795. doi:10.2307/2409392
- J. S. Farris, “The Retention Index and Homoplasy Excess,” Systematic Zoology, Vol. 38, No. 4, 1989, pp. 406- 407. doi:10.2307/2992406
- Z. Zhang, L. Fan, J. Yang, X. Hao and Z. Gu, “Alkaloid Polymorphism and ITS Sequence Variation in the Spiraea Japonica Complex (Rosaceae) in China: Traces of the Biological Effects of the Himalaya-Tibet Plateau Uplift,” American Journal of Botany, Vol. 93, No. 5, 2006, pp. 762-769.
- A. Rehder, “Manual of Cultivated Trees and Shrubs,” 2nd Edition, Collier Macmillan, New York, 1940.
- S. H. Oh, L. Chen, S. H. Kim, Y. D. Kim and H. Shin, “Phylogenetic Relationship of Physocarpus Insularis (Rosaceae) Endemic on Ulleung Island: Implications for Conservation Biology,” Journal of Plant Biology, Vol. 53, No. 1, 2010, pp. 95-105. doi:10.1007/s12374-009-9093-z
- J. E. Richardson, R. T. Pennington, T. D. Pennington and P. M. Hollingsworth, “Rapid Diversification of a SpeciesRich Genus of Neotropical Rain Forest Trees,” Science, Vol. 293, No. 5538, 2001, pp. 2242-2245.