International Journal of Nonferrous Metallurgy
Vol.06 No.03(2017), Article ID:77489,20 pages
10.4236/ijnm.2017.63003
Reducing Greenhouse Gas Emission from the Neodymium Oxide Electrolysis. Part II: Basics of a Process Control Avoiding PFC Emission
Hanno Vogel, Bernd Friedrich
RWTH Aachen University, IME Institute of Process Metallurgy and Metal Recycling Intzestraße 3, Aachen, Germany

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: May 17, 2017; Accepted: July 4, 2017; Published: July 7, 2017
ABSTRACT
The neodymium electrolysis produces unnecessary high emission of CF4 and C2F6. These perfluorocarbons (PFCs) are potent greenhouse gases and are not filtered or destroyed in the off-gas. A process control in analogy to the aluminum electrolysis can reduce the PFC emission to a great extend and keep the process in a green process window. Therefore, a theoretical analysis is done of the cell voltage of the industrial neodymium electrolysis in dependence on the neodymium oxide concentration in the electrolyte. The analysis shows the different contributions to the cell voltage focusing on the impact of the anodic overvoltage on the cell voltage, by which the electrolysis process can be controlled. The model of the cell voltage is evaluated by laboratory neodymium electrolysis with a similar setup as the industrial cell. The relation of the oxide concentration, the anodic current density and the cell voltage with the cell resistance are measured. The continuous off-gas measurements show the gas concentration and PFC emissions. The effect of Nd2O3 feeding on the galvanostatic electrolysis is analyzed as well. Based on the results a process control strategy is proposed similar to the aluminum electrolysis strategy. The green process window is in a narrow oxide concentration range, making a continuous and precise oxide feeding essential.
Keywords:
Rare Earth, Neodymium, Electrolysis, Greenhouse Gas Emission, Process Control

1. Introduction
Neodymium as other rare earth elements is transformed to metal by oxide electrolysis in a fluoride electrolyte. The industrial rare earth electrolysis is not automated, is on low technological level and is still based on operator experience. Alternative processes are not likely to come into production in the next decade [1] . Because of its employment with about 30%*1 in permanent magnets, the production of neodymium metal increased rapidly from 1990s on [2] and reaches about 30,000 t in 2016 [3] . While conducting the electrolysis, the anode reaction produces mainly CO and CO2, but also CF4 and C2F6, which are PFCs with greenhouse gas potential of 7,390 and 12,200 CO2-equivalents respectively [4] . As shown in part I, the sum of CO2-eq. from a continuous PFC emitting electrolysis can be as high as 20 million tons per year [5] . Minimizing the PFC emission needs to be a primary goal of the rare earth electrolysis. The definition of a process window for a sustainable electrolysis is determined by the partial anode effect, where the first CF4 can be detected, as shown in Part I [5] . The current density should not be higher than the partial anode effect at a given oxide concentration in the electrolyte. The oxide feeding has to prevent an oxide depletion of the electrolyte. For the aluminum electrolysis, a decrease in greenhouse gas emission over time was achieved by improvements in its process control with an appropriate feeding strategy, lowering the number and time of anode effects. Because of the vertical electrode setup of the neodymium electrolysis cell, the behavior of the anode effect can be different as known from the horizontal anode setup of the aluminum electrolysis [1] . Nevertheless, the process control should manage the right amount of oxide in the electrolyte, avoiding the critical conditions of PFC formation. Therefore, a similar process control is needed for the rare earth electrolysis. Foremost the neodymium electrolysis should be equipped with an automated process control because of its production quantity. The control system can be applied to the other rare earth oxide electrolysis processes as well.
2. Process Control Strategy
Figure 1 shows the most important electrolysis parameters to control the anodic off-gas without PFC content.
The oxide concentration is the only value, which can be changed in the conventional rare earth electrolysis. The anode size and position is regarded as fixed
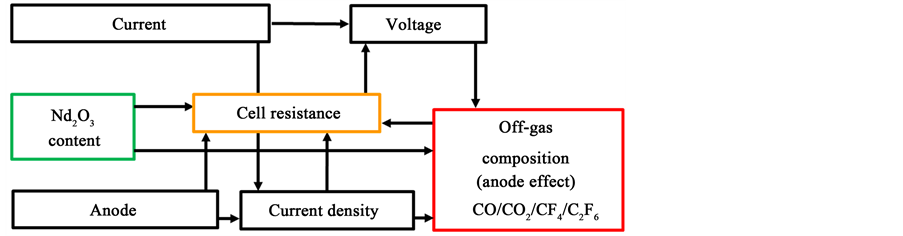
Figure 1. Relevant interrelations for a PFC controlling process control.
and the current is not changed in the galvanostatic process. The voltage, and by taking the current into account, also the electrical resistance, are the only directly visible values of the process. The aluminum electrolysis is controlled with a defined upper voltage, where the overfeeding of alumina is initiated before running into anode effect. The duration of this overfeeding period is calculated. Then the underfeeding starts to avoid a sludging of the cell due to undissolved oxides [6] [7] . The same principle of control also works for the rare earth electrolysis, because the relation of the oxide content of the electrolyte to the electrolysis voltage and critical current density is already shown [5] . The solubility of neodymium oxide in the NdF3-LiF electrolyte is smaller than for alumina in the cryolite electrolyte with only about 4% [8] . There are more differences in the electrolytic system and cell design making a theoretical analysis and an experimental investigation of the oxide related cell resistance and oxide feeding process in the galvanostatic electrolysis necessary.
In this approach, the control of the anodic product is the primary goal. The formation of the gaseous product is influenced by the current density, the voltage and the oxide concentration, while the anodic products can influence the electrical resistance, which is happening at the passivation phenomenon at the partial and full anode effect, clearly visible at the linear voltammograms in part I [5] . The dynamic cell voltage can be calculated theoretically by analyzing the oxygen concentration dependent overvoltages, in particular the anodic overvoltage according to the extensive work done by Vogt et al. for the aluminum electrolysis [9] [10] [11] .
The total cell voltage is the sum of the following voltage drops and overvoltage phenomena, some of which are dependent on the concentration of the oxygen containing ions. Each contribution will be discussed separately below:
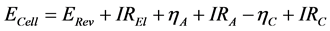 (1)
(1)
ECell: Cell voltage; ERev: theoretical decomposition voltage; I: Current; REl = Ohmic resistance of electrolyte; ηA: Anodic overvoltage; RA: Ohmic resistance of anode; ηC: cathodic overpotential; RC: Ohmic resistance of cathode
2.1. Theoretical Decomposition Voltage
The theoretical voltage of decomposition ERev of 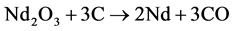 as the predominant electrolysis reaction can be expressed with its activity relation according to Nernst by [9] :
as the predominant electrolysis reaction can be expressed with its activity relation according to Nernst by [9] :
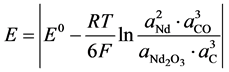 (2)
(2)
When 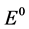 equals −1.297 V at 1050˚C [5] and Nd, CO und C are all unity in their standard state, the equation can be simplified by expressing the activity of Nd2O3 by the ratio of the concentration to the saturation concentration [12] .
equals −1.297 V at 1050˚C [5] and Nd, CO und C are all unity in their standard state, the equation can be simplified by expressing the activity of Nd2O3 by the ratio of the concentration to the saturation concentration [12] .
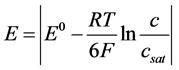 (3)
(3)
c = Concentration of Nd2O3, csat = Saturation concentration of Nd2O3 in electrolyte with 4%, 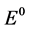 = Standard potential at 1050˚C, R = Gas constant, F Faraday constant, T = Temperature (1323 K).
= Standard potential at 1050˚C, R = Gas constant, F Faraday constant, T = Temperature (1323 K).
With a saturation concentration of 4% [8] at 1050˚C an increase in the decomposition voltage is found with decreasing oxide concentration. When the oxide concentration falls from 2% to 0.2% the voltage increases with 50 mV, as shown in Figure 2.
In addition, the temperature has a strong influence on the decomposition voltage. As Figure 3 shows, the decline of the enabling reaction of neodymium oxide with carbon to CO is stronger than to CO2. The theoretical decomposition voltage decreases from 1025˚C to 1100˚C with about 70 mV.
The temperature dependence plays a minor role because the system is regarded to stay at 1050˚C. The kinetic effects of the electrode reactions are much stronger, leading to various overvoltage phenomenon with a strong oxygen concentration dependency. Linear voltammograms of the system clearly show this behavior [5] .
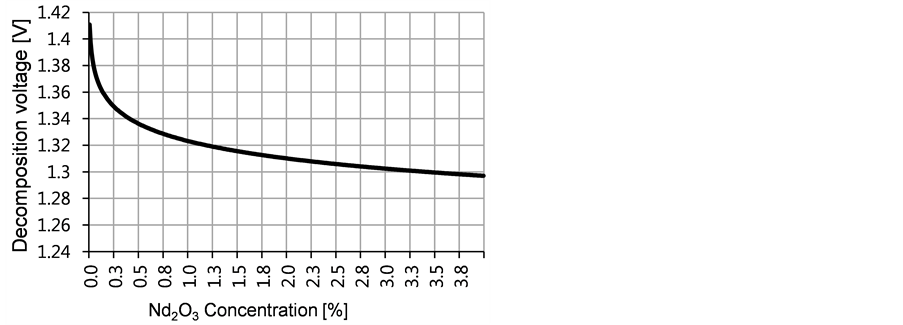
Figure 2. Theoretical decomposition voltage of Nd2O3 + 3C → 2Nd + 3CO in relation the neodymium oxide concentration in the electrolyte with saturation at 4% at 1050˚C.
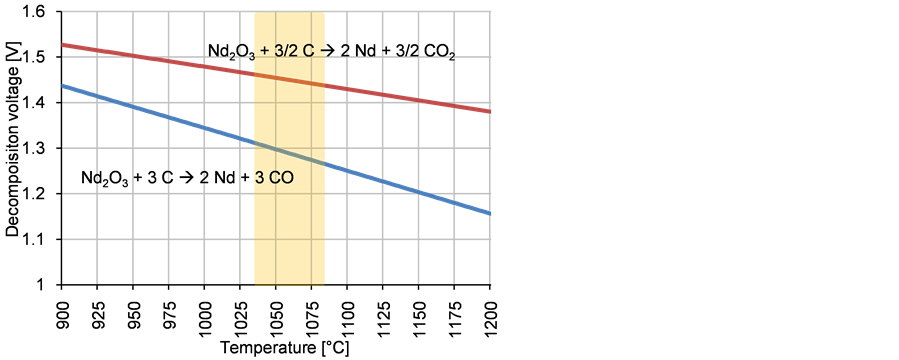
Figure 3. Theoretical decomposition voltage of both intended reactions in relation to the temperature with working temperature as shaded area, according to FactSageTM 6.4 (Data: Fact PS).
2.2. Anodic Overvoltage
The anodic overvoltage shows the strongest oxygen concentration dependence. Two major contributors to the overvoltage can be distinguished. This is the diffusion overvoltage as the kinetic effect of product transport from the anode to the electrolyte and the charge transfer overvoltage, including the diffusion of the reactant at the electrode interface [9] . Another study analyzes the anodic overvoltage of the Nd2O3-NdF3-LiF system by a slow scanning oscillogram at 2, 3 and 4% Nd2O3 concentration between a current density of 0.37 and 0.73 A/cm [13] . The minimum was found at high oxide concentration and low current density with 0.07 V and a maximum at 2% Nd2O3 and 0.73 A/cm2 with 0.34 V. However, the overvoltage should theoretically rise much more with lower oxide concentration, which is the important window for a process control similar to the aluminum electrolysis and needs further investigation.
2.2.1. Concentration Corrected Anodic Charge Transfer Overvoltage
The concentration of the oxygen containing ions at the anode has the strongest influence on the anodic overvoltage. Vogt and Thonstad [9] determined the charge transfer overpotential by considering the diffusion of this ion species in accordance to the concentration corrected Butler-Volmer equation. By this the concentration overvoltage for the reacting ion is included with the mass transfer coefficient. The formula fitted for the neodymium electrolysis system with the anodic reaction producing CO can be expressed as [9] [10] :
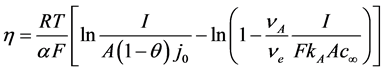 (4)
(4)
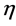 = Overvoltage,
= Overvoltage, 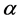 = Charge transfer number, F = Faraday constant, R = Universal gas constant, T = Temperature, I = Current, A = Anode surface,
= Charge transfer number, F = Faraday constant, R = Universal gas constant, T = Temperature, I = Current, A = Anode surface, 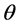 = Ratio of inactive anode surface,
= Ratio of inactive anode surface, 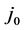 = exchange current density (ca. 100 mA/cm2);
= exchange current density (ca. 100 mA/cm2);
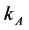 = Mass transfer coefficient,
= Mass transfer coefficient, 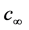 = bulk concentration of reagent,
= bulk concentration of reagent, 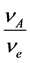 =
=
Number of reaction species per number of electrons.
The assumption of an exchange current density of 100 mA/cm2 seems to be valid, analyzing the linear voltammetry of the system [5] by a Tafel plot. With a mass transfer coefficient of 0.001 m/s, a charge transfer number of 0.5, a current of 2200 A and an anode surface of 1600 cm2 a fast rise in the anode potential below 0.4% Nd2O3 concentration is calculated. The total anodic overvoltage is the sum of the diffusion and charge transfer overvoltage, in which the diffusion of the oxygen containing reacting ions is included, changing with the neodymium oxide concentration as shown in Figure 4.
The anodic overvoltage increases rapidly below 0.4% Nd2O3, reaching infinity under 0.16%. The effect of an ongoing oxygen depletion at the anode leads to an increase in the anode potential, enabling the reaction of NdF3 with carbon resulting in PFC emission.
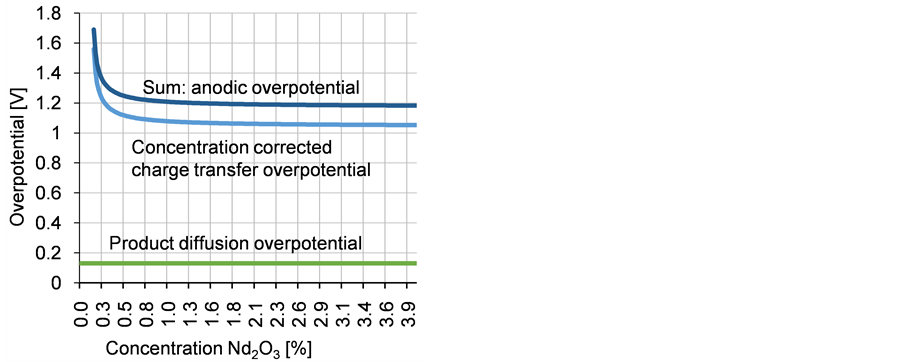
Figure 4. Anodic overvoltage in relation to the neodymium oxide concentration.
2.2.2. Diffusion Overvoltage of Anode Product
The anodic product is transported off the anode as gas bubbles and dissolved gas in the electrolyte [14] [15] . The formation of bubbles depends on the wettability and fluid dynamics condition [11] . The bubble coverage ratio 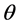 is reducing the active anode surface. Because of the vertical anode setup and fast electrolyte movement [16] , this ratio should be small for a current density below the partial anode effect. Still there is a substantial amount of gas dissolving in the electrolyte leading to a saturation dependent overvoltage. The anodic gaseous product is considered here for reasons of simplicity to be only CO. Even though the reaction kinetics should also lead to a CO2 formation, the major off-gas component was experimentally found to be CO. This diffusion overvoltage of the anodic CO formation can be expressed as [9] :
is reducing the active anode surface. Because of the vertical anode setup and fast electrolyte movement [16] , this ratio should be small for a current density below the partial anode effect. Still there is a substantial amount of gas dissolving in the electrolyte leading to a saturation dependent overvoltage. The anodic gaseous product is considered here for reasons of simplicity to be only CO. Even though the reaction kinetics should also lead to a CO2 formation, the major off-gas component was experimentally found to be CO. This diffusion overvoltage of the anodic CO formation can be expressed as [9] :
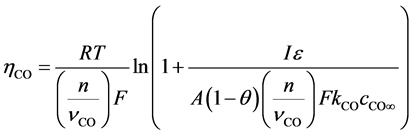 (5)
(5)
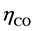 = Diffusion over voltage by dissolved CO, n =Charge number,
= Diffusion over voltage by dissolved CO, n =Charge number, 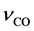 = Stoichiometric number CO,
= Stoichiometric number CO, 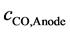 = Concentration of CO at the anode,
= Concentration of CO at the anode, 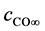 = Concentration of CO in bulk electrolyte,
= Concentration of CO in bulk electrolyte, 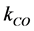 = Mass transfer coefficient CO, A = Anode surface,
= Mass transfer coefficient CO, A = Anode surface, 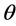 = Ratio of covered anode surface by bubbles,
= Ratio of covered anode surface by bubbles, 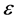 = current efficiency
= current efficiency
Assuming a current efficiency 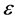 of 80%, a bubble coverage
of 80%, a bubble coverage 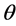 of 0.1, a mass transfer coefficient kCO of 0.001 m/s and a saturation concentration of CO of 100 mol/m3 a constant overvoltage of 0.13 V is obtained. This value can vary with its assumptions but seems to be an acceptable approximation, since the calculated value for the CO2 overvoltage within the aluminum electrolysis equals 0.18 V [9] .
of 0.1, a mass transfer coefficient kCO of 0.001 m/s and a saturation concentration of CO of 100 mol/m3 a constant overvoltage of 0.13 V is obtained. This value can vary with its assumptions but seems to be an acceptable approximation, since the calculated value for the CO2 overvoltage within the aluminum electrolysis equals 0.18 V [9] .
2.3. Ohmic Resistance Electrolyte
The ohmic resistance of the electrolyte is also dependent on the oxide concentration, but this time the voltage drop increases with increasing oxide content in the electrolyte. An empirical formula for calculating the ohmic resistance can be used, which attributes a strong influence of the oxide concentration on the decrease in electric conductivity [17] :
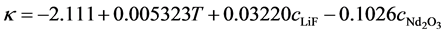 (6)
(6)
κ: Electrical conductivity (S・cm−1); T: Temperature (˚C), valid range: 1050˚C - 1150˚C; cLiF (%), valid range 5% - 15%; 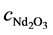 (%): valid range 1% - 3%.
(%): valid range 1% - 3%.
The voltage drop in the electrolyte depends on the specific resistance and cell constant, which can be approximated by the coaxial geometry [18] , giving a cell constant of the round 3 kA-cell of 0.010246 cm−1.
At 1 050˚C and a LiF content of 12.5% an electrical conductivity of 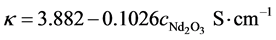 is obtained, resulting in the oxide dependent resistance of:
is obtained, resulting in the oxide dependent resistance of:
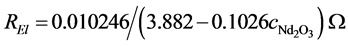 (7)
(7)
A linear increase in the electric resistance with rising oxide concentration is obtained. The exact behavior of the conductivity is an important value for the process control and should be investigated further.
2.4. Electric Resistances Independent of the Oxide Concentration
The constant ohmic resistances of the anode and cathode are dependent on the specific conductivity of the material, its cross section and the temperature and can be calculated individually. The graphite anode resistance of the 3 kA cell is approximated with 0.061 mΩ [19] and the tungsten cathode resistance with 0.07 mΩ [20] .
The cathode overvoltage is not investigated separately and is approximated to be about −0.1 V following an investigation on the cathodic overvoltage of the aluminum electrolysis [21] .
3. Model of the Cell Voltage
The theoretical model of the cell voltage of the conventional 3 kA technology of the Chinese industrial cell is according to equation 1 the sum of all over voltages and resistances. Figure 5 shows the dynamic oxide dependent behavior of the cell voltage with all contributions. The cell voltage is calculated to be between 8 and 9 V, which is in good accordance with literature data, stating a voltage of 9 to 11 V [22] . The peripheral voltage drops in the bus bar connection is neglected here, making the theoretical result a realistic value. The major contribution to the cell voltage of 8 to 9 V is the ohmic resistance of the electrolyte. The minimum cell voltage is reached at a concentration of 0.64% Nd2O3. To smaller oxide concentration, the voltage increases rapidly due to the charge transfer overvoltage and to higher oxide concentration, the voltage increases due to the increase in electric resistance of the electrolyte.
The validity of the model is tested in laboratory scale electrolysis by
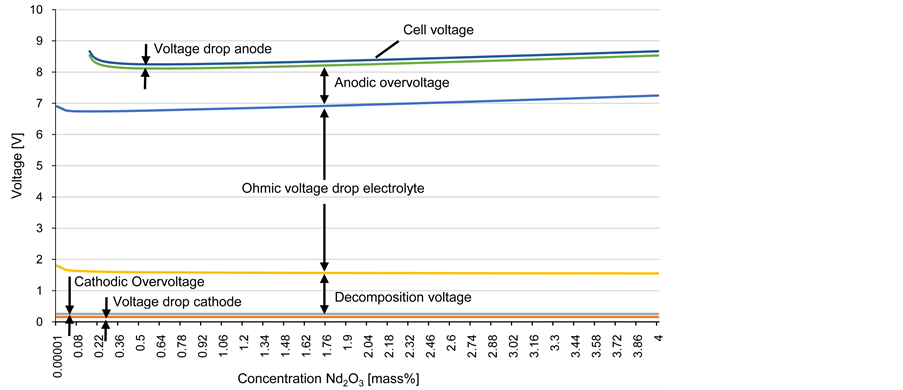
Figure 5. Cell voltage with individual contribution in relation to the Nd2O3 concentration according to the mathematical model.
determining the cell voltage with changing oxide concentration in dependence of the current density.
4. Experimental Setup and Procedure
The electrolyte is prepared of 99.9% pure NdF3 (Treibacher Industrie AG) and 99.85% LiF (Alfa Aesar GmbH & Co KG). The powder mixture with 87.5% NdF3 and 12.5% LiF is dried over night at 300˚C and then pre-melted in a sealed furnace with 1.4 bar argon 5.0 atmosphere. The fluoride mixture is homogenized in a pure graphite crucible by superheating it shortly and then poured in a thin graphite mold for rapid solidification. The solid electrolyte is broken in pieces and about 2 kg is charged in the graphite crucible (SIGRAFINE® R8510; SGL Carbon) with an inner diameter of 10 cm. Similar to the industrial cell, the center cathode is a pure tungsten rod (99.95%; PLM GmbH) with 8 mm diameter surrounded by a hollow graphite cylinder anode (SIGRAFINE® R8510; SGL Carbon) of an inner diameter of 7.5 cm as depicted in Figure 6. A molybdenum crucible (99.95%; PLM GmbH) with 3 cm height and an inner diameter of 4 cm is placed underneath the cathode rod to collect the liquid neodymium. The steel cell is placed in a SiC resistance heated furnace. Through the lid, a Pt-PtRh- thermocouple with molybdenum sheath of 4 mm diameter (Omega Engineering GmbH) and an alumina insulation at the Swagelok connection is dipped in the liquid electrolyte to control the temperature at 1050˚C with a deviation due to electrolysis heat generation of about 10 K. Also the electrodes are insulated electrically at the water cooled lid by alumina sheaths. The sealed cell is flushed with pure argon 5.0 at 3 l/min. The voltammetry measurements are conducted by a potentiost at with a 40 A booster (Ivium Technologies).
Electrolyte samples are taken by pure glass pipettes (HSQ300; Quarzglas Hein- rich) filled with argon 5.0 with an inner diameter of 2.5 mm to determine the oxygen content. The pipette is dipped 3 cm in the electrolyte close to the anode, where the sample of about 1 g is taken out of the cell. The oxygen content in the
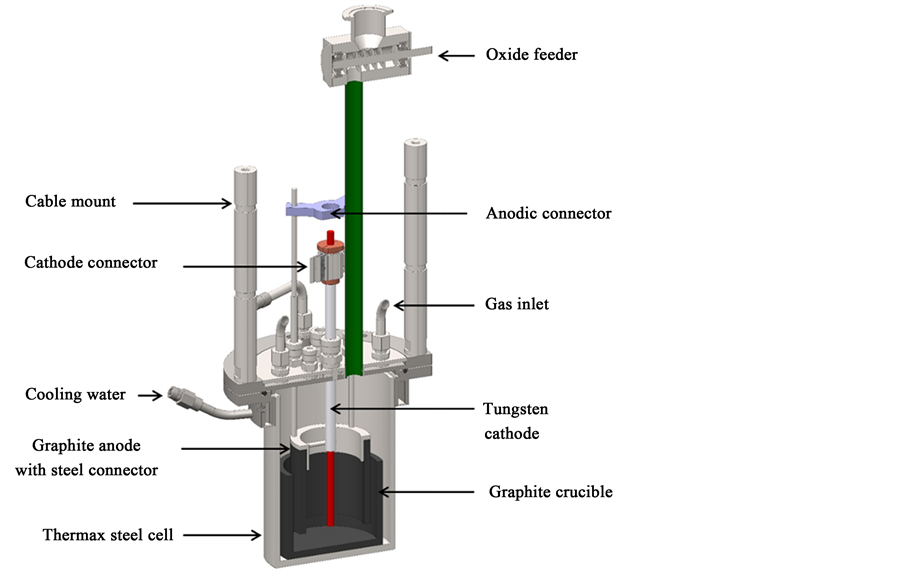
Figure 6. Cross section of the experimental setup of the electrolysis cell with an outer diameter of 15 cm.
solidified sample is analyzed 3 to 5 times by the carrier gas method (Leco) against an iron standard giving a deviation of about 4%.
The electrolyte is further purified by running a pre-electrolysis for 3 hours with CO emission. Thereafter the first electrolyte sample is taken. Then a staircase voltammetry is conducted with voltage steps of 0.2 V from 2.0 to 5.0 V. The off-gas is measured continuously to detect the first emission of CF4. Each voltage step is hold for 50 seconds to get a good average of the current response to calculate the electric resistance. The off-gas is measured continuously by taking 1 l/min from the cell atmosphere and pumping it through the 180˚C heated FT- IR-spectrometer (Gasmet, Ansyco), which is calibrated for all relevant gases. The measuring interval is 7 seconds. After each staircase voltammetry an oxide amount of 0.25% is fed in the electrolyte and dissolved for 20 minutes. Then the next identical voltammetry is conducted and an electrolyte sample is taken nearby the anode. This is done up to a concentration of 4% neodymium oxide addition. Furthermore the oxide feeding effect is tested with the galvanostatic electrolysis, where the power is delivered by a 200 A rectifier (Munk) and a digital controller (UT35A, Yokogawa) is programmed to automatically or manually feed 1% neodymium oxide in the cell if a certain voltage threshold is surpassed for more than one minute. The powder neodymium oxide with 99.9% purity (Alfa Aesar GmbH & Co KG) is dried over night at 120˚C and pressed into tablets, which are crushed and sieved. The fraction between 0.71 mm and 1.25 mm is used for the experiments. A digital recorder (GP10, Yokogawa) records the current, voltage and temperature.
5. Results and Discussion
The electrolyte contains up to 2% oxygen after drying and pre-melting the otherwise pure electrolyte. To lower the oxygen content a cleaning electrolysis is conducted for 3 hours at an average current of 6 A. The current density in the galvanostatic process is very low at about 0.08 A/cm2, to start the CO production anodically at a voltage of about 3.6 V as shown in Figure 7. The voltage increases slowly due to the decreasing oxide content and without a disturbance in the cell voltage, the CF4 emission starts. The production of PFC does not result in an observable passivation phenomenon. The concentration ratio CF4/CO rises slowly and reaches its maximum at 0.1. In the off-gas almost no CO2 and C2F6 are observed. Similar experiments show that the CF4 formation can vanish and return without voltage disturbances. These partial anode effect events [5] are not detectable in the voltage signal, making it almost impossible for the process controller to know about the state of emission coming from the cell.
After 3 hours running the electrolysis at 6 A the electrolyte sample analysis result in an oxygen concentration of 520 ppm, which equals a neodymium oxide concentration of 0.36%.
Feeding neodymium oxide in the electrolyte after the cleaning electrolysis does not result in the expected oxygen concentration. In Figure 8, the blue bars represent the actual measured oxygen concentrations from the upper part of the electrolyte, while the orange bars show the theoretical values, if the oxide is dissolved completely. An example of a solidified electrolyte is depicted in Figure 9 showing a clear separation of two phases, which happens in each experiment, where oxide is fed in the electrolyte. The dark phase at the bottom contains much more oxygen than the upper part, but the electrodes do not have contact to this oxygen rich dark bottom phase. Electron beam microanalysis images show the distribution and concentration of oxygen and fluorine as shown in Figure 10. The oxide is dissolved, because its concentration in the bright homogeneous phases, shown in Figure 10 on the right, reaches 6% in the line scan. In this phase the fluorine concentration drops to 17%. The oxygen seems to precipitate as a high melting NdOF, which does not dissolve again later on. No solid
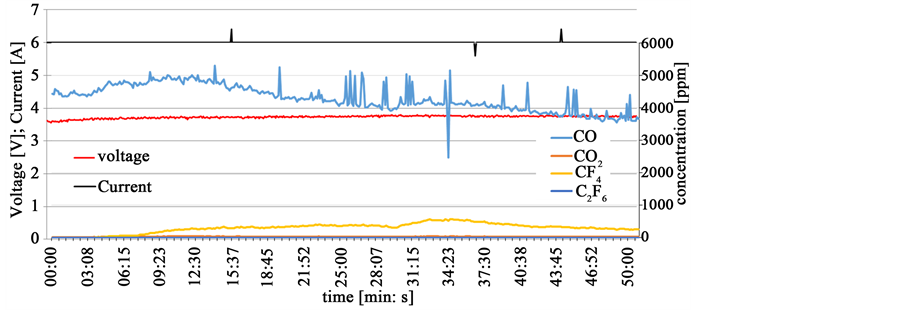
Figure 7. Galvanostatic neodymium electrolysis with slowly evolving CF4 emission without voltage disturbances.
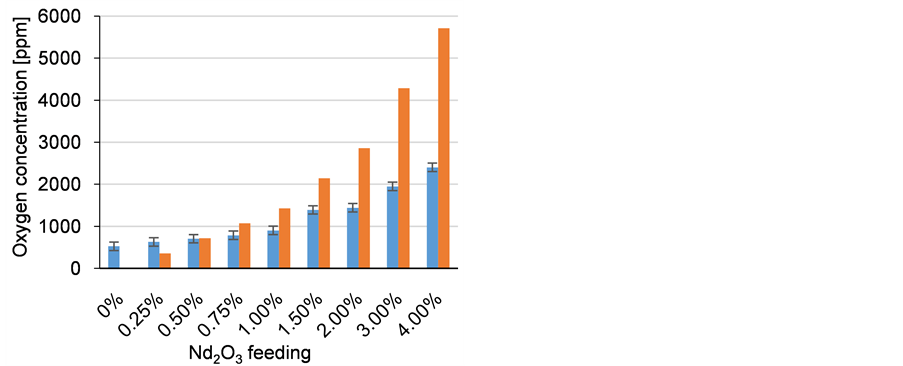
Figure 8. Real oxygen concentration after feeding of Nd2O3 in dark bars with theoretical values in light bars.

Figure 9. Solidified electrolyte with two distinguished phases after feeding of 6% neodymium oxide.
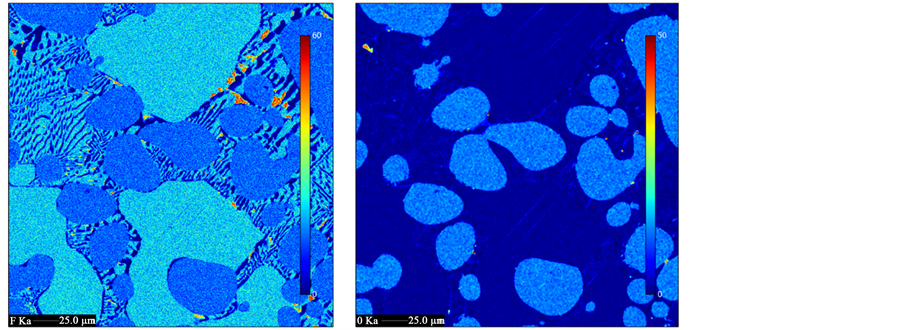
Figure 10. Electron beam microanalysis images of the bottom sludge phase after adding 6% Nd2O3 to the 87.5% NdF3, 12.5% LiF electrolyte. Left: intensity of fluorine, right: intensity of oxygen.
oxide particles are found in the bottom sludge phase, but the stoichiometry suggests a NdOF phase mixed with NdF3 fluorides, while LiF is the major constituent of the bright eutectic phase, as shown left in Figure 10.
There is an apparent difference in the amount of fed oxide and the true oxygen concentration in the electrolyte at the anode surface. To determine the behavior of the cell resistance in relation to the real oxygen content at the anode, a staircase voltammetry between 2 and 5 V is conducted for every 0.25% addition of Nd2O3 and an electrolyte sample near the anode is taken immediately after each measurement. The voltammetry after 1.0% addition of Nd2O3 and the real measured value of 785 ppm oxygen equaling an Nd2O3 concentration of 0.55% is shown in Figure 11. The continuous gas analysis shows the voltage at which the first CF4 is detected in accordance to the linear sweep voltammetry from part I of this work [5] . When CF4 is detected, the partial anode effect has happened. The process voltage should be kept below this measured voltage in dependence of the oxygen concentration and the current density. The electric resistance of the voltammetry measurements between 2.8 and 4.4 V are shown in Figure 12.
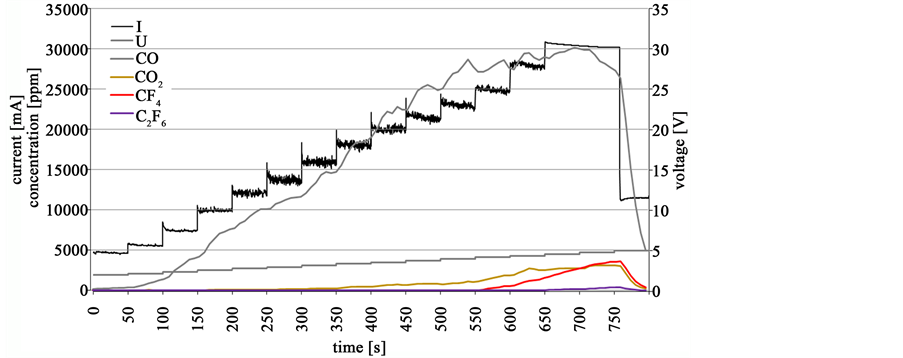
Figure 11. Staircase voltammetry with anodic gas emission between 2.0 and 5.0 V at 785 ppm oxygen concentration.
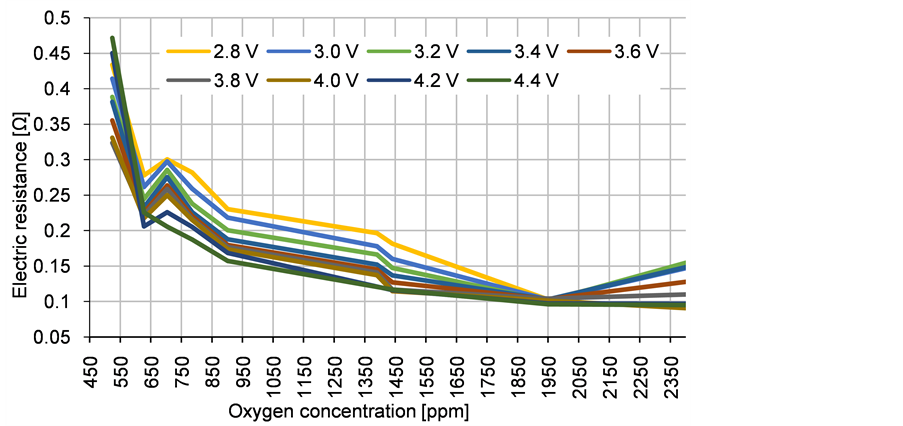
Figure 12. Electric resistance of the neodymium electrolysis cell over the measured oxygen concentration; 1430 ppm oxygen equals 1% Nd2O3.
The electric resistance of the cell is changing significantly with the oxygen concentration. At 1430 ppm oxygen equalling 1% Nd2O3 a medium cell resistance of about 0.15 Ω is measured. The cell resistance increases with lower oxygen concentration. A fast increase is measured below 600 ppm oxygen, equalling 0.42% Nd2O3. The theoretical investigation finds the beginning of a steep increase at 0.3% Nd2O3, probably underestimating the impact of the oxygen ion concentration on the anodic overvoltage. There seems to be a minimum in the cell resistance at 1950 ppm oxygen equalling 1.36% Nd2O3. Because of the low number of data points and uneven oxygen concentration distribution, it is probably not the precise minimum resistance value. Nevertheless, the theoretical analysis gives a minimum at 0.64%, quite lower than the experimental result. This leads to the conclusion, that the influence of the anodic overvoltage is stronger than the electric conductivity change of the electrolyte. This relation of the total cell resistance makes a voltage value based process control easier to handle than the theoretical approximation suggests. According to this measurement, the oxygen concentration should be controlled between 700 ppm and 2000 ppm oxygen equalling a range between 0.5% and 1.4% Nd2O3. This is a very narrow process window but the cell resistance due to the anodic overvoltage changes between 0.25 Ω and 0.1 Ω, which is a broad window, leading to easily detectable voltage changes.
If the current densities of all staircase voltammograms between 2 and 5 V are plotted against the true oxygen concentration and the cell voltage, the heat map of Figure 13 is obtained. In the galvanostatic industrial electrolysis a constant current density is applied. The iso current lines are visible, but cannot be regarded as precise and show the increase in cell voltage with decreasing oxygen concentration. At a current density of 0.3 A/cm2 the voltage increases from 2.9 V
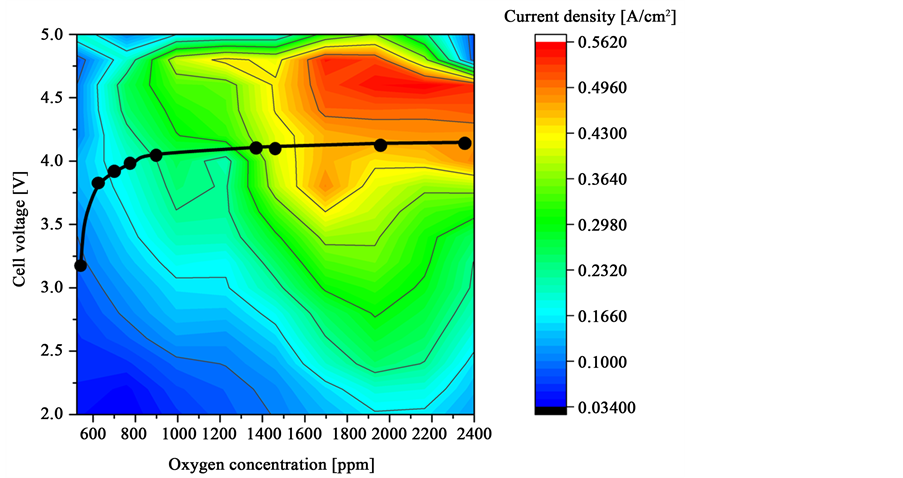
Figure 13. Heat map of the current density in relation to the cell voltage and oxygen concentration with first CF4 emission voltage as black points.
at 1800 ppm oxygen, equaling 1.26% Nd2O3, to a voltage of 4.5 V at 900 ppm oxygen, equaling 0.63% Nd2O3. The black dots in the diagram show the approximated voltages of first CF4 detection of the regarding voltammetry at one oxygen concentration. The line shows the approximate boundary between the safe process window and the risk of PFC formation. At a low oxygen concentration of 520 ppm, the first CF4 is detected at a cell voltage of 2.8 V equaling a critical current density below 0.1 A/cm2.
The oxide feeding effect on the galvanostatic electrolysis is tested at 30 A, equaling a current density of 0.4 A/cm2. Figure 14 shows the voltage behavior and the gas emission from the cell. The voltage is at 4.2 V and increases slightly while a few ppm CF4 can be detected in the off-gas at 4.3 V. With rising voltage at 13:34 also the CF4 concentration increases fast. When the cell runs into full anode effect, the voltage increases to the defined upper limit of the rectifier at 10 V. The computer controller initiates the oxide feeding, if a voltage threshold here at 5 V is surpassed for 1 minute. At the end of this minute, the CF4 emission reaches its maximum with about 1% of the former CO emission. C2F6 is detected in its maximum just in traces with 2% of the CF4 emission. At the moment of oxide feeding the cell voltage drops immediately to 3.6 V. Simultaneously the CF4 emission drops back to zero and the electrolysis returns to a PFC free process. With feeding of neodymium oxide, a sharp peak in water and CO2 is detected because of remaining humid air entrapment in the dried and pressed powder. The water content in the cell atmosphere leads to a HF formation shortly after feeding, but returning back to normal soon after.
Figure 15 shows the oxide feeding with 1% in the galvanostatic electrolysis at the very first detection of about 2 ppm CF4 in the off-gas, where the cell voltage stays at 4.5 V without rising. At the moment of feeding, the cell voltage falls rapidly to about 3.5 V. This means the anodic overvoltage decreases immediately with about 1 V and the CF4 evolution stops instantly bringing the electrolysis back to the green process window without PFC emission. An advanced process control with a higher level of security to prevent PFC emission could include a
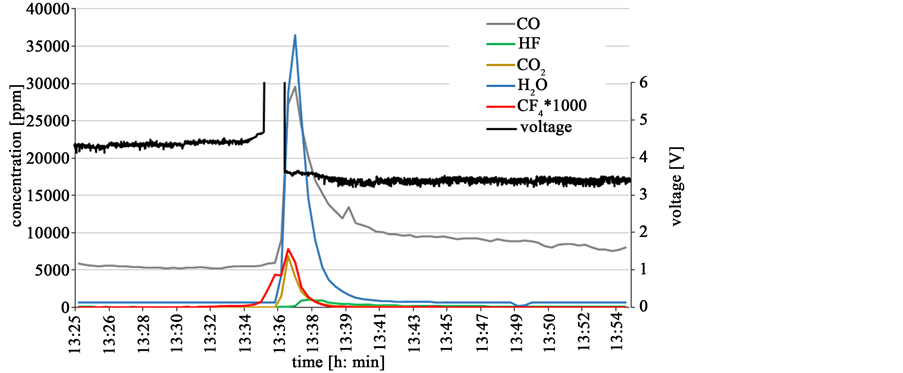
Figure 14. Automatic feeding of 1% neodymium oxide after 1 minute in full anode effect.
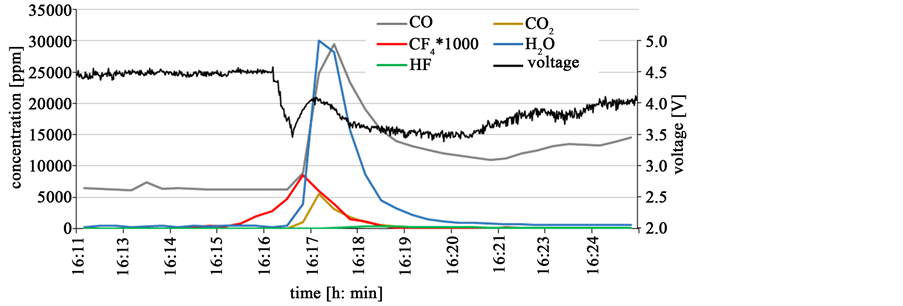
Figure 15. Feeding of 1% neodymium oxide directly after first CF4-detection.
continuous off-gas measurement with an alarm, as soon as 2 ppm CF4 is surpassed triggering the oxide feeding.
A continuously high PFC emitting neodymium electrolysis is shown in Figure 16, where the galvanostatic process runs without Nd2O3 feeding at 50 A without disturbances in current. The voltage is agitated but remains at a level of about 4.8 V. While the CO concentration declines continuously, the CF4 concentration rises continuously to 65% of the CO concentration in its maximum. Here the C2F6 concentration reaches 10% of the CF4 concentration. The electrolysis runs just above the partial anode effect, without jumping into full anode effect as shown in Figure 14. The reason for this different behavior is not apparent, but might be due to different amounts or structure of the bottom sludge phase. The sludge could have an impact by redissolving some oxide ions and depleting the electrolyte more slowly than at the first feeding cycles, where the bottom sludge is just starting to build up.
6. Comparison of the Model of the Laboratory and Industrial Cell with the Experimental Investigation
The process control needs to be based on the cell voltage behavior in relation to the depleting oxide concentration. The already presented calculated voltage drops of the industrial 3 kA cell are compared to the theoretical values of the laboratory cell, which are calculated by the same model and evaluated with the experimental results of the voltammetry study, all shown in Table 1. The references of the basic or derivate parameters are given. The ohmic resistances and the subsequent voltage drops are approximated by material specific data at 1050˚C.
The direct comparison of the modelled resistances and voltage drops of the laboratory cell at 30 A with the measured values at 30 A and a real oxide concentration of about 1% shows some deviations. The total cell voltage is experimentally quite higher with 3.4 V in comparison to a calculated 2.7 V. The ohmic resistances of the anode and cathode as well as the approximately constant cathode overvoltage were not determined experimentally. By measuring the cell constant by impedance spectroscopy between 500 to 10,000 Hz a more precise
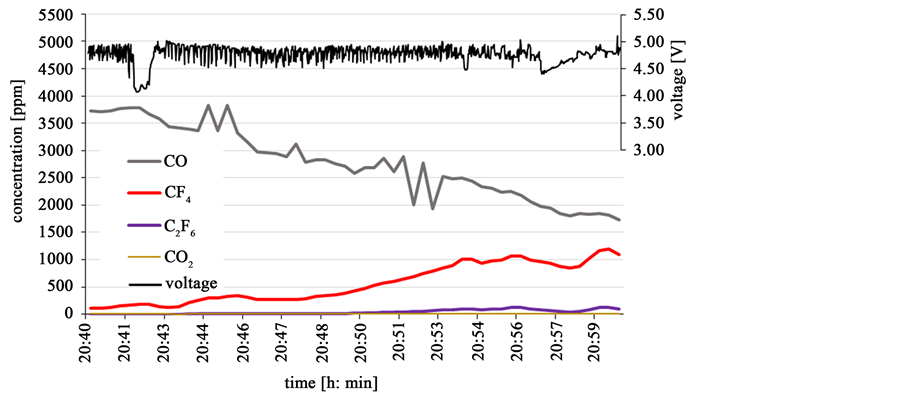
Figure 16. Galvanostatic electrolysis at 50 A without feeding and strong PFC emission.

Table 1. Comparison of the Chinese modelled 3 kA-Cell with the modeling of the laboratory cell and its experimental results all at 1% Nd2O3 and 1050˚C.
voltage drop of the electrolyte could be determined, but the uncertainty of the empirical formula [17] of the conductivity remains. The resistance of the electrolyte was found to be slightly smaller than calculated. While the theoretical decomposition voltage as the thermodynamic value at the given temperature is not changed, the anodic overvoltage has a much stronger influence than calculated. Adding 1% of Nd2O3 to the galvanostatic electrolysis, a voltage drop of about 1 V can be detected, which is not in good accordance to the theoretical model of the charge transfer overvoltage, which predicts just about 0.1 V. This result can be transferred to the industrial neodymium cell. This means, the voltage drop of the electrolyte becomes smaller, but remains the major contributor. The anodic overvoltage will have a greater influence on the cell voltage, with a minimum at higher oxide concentration. The theory predicts the minimum at 0.6% Nd2O3 while the experiments show a minimum value at 1.4% Nd2O3. The industrial process is not directly comparable to the laboratory electrolysis, because the current density as a crucial value is quite different. The laboratory cell runs at 0.4 A/cm2, and the industrial is supposed to run at 1.4 A/cm2, even though the optimal current density is about 1.0 to 1.25 A/cm2 according to an experimental study on the industrial cell between 2 and 3 kA [25] . The anodic current density of the industrial process is very high and needs to be always very close or already over the partial anode effect, depending of the oxygen concentration at the anode. Therefore, the emission of high amounts of PFC gases is very likely.
To run a sustainable rare earth electrolysis without frequent or permanent PFC emission, the cell voltage in dependence of the oxygen concentration and the CF4 emission should be measured simultaneously. Below 0.7% Nd2O3 the anodic overvoltage increases rapidly. Here the exact voltage at a given current density of the first CF4 emission should be determined and a lower voltage with a safety gap defined. This safety voltage should not be surpassed. In best case, a continuous feeding of rare earth oxide is maintained to keep the electrolyte rich in oxygen but not overfeeding, which leads to a fast sludging of the cell bottom. The mass flow of oxide is calculated by the faradaic oxygen consumption, taking the current efficiency into account, equalling a feeding rate of 100%. An underfeeding with e.g. 90% leads to a slow increase in cell voltage, until the defined safety voltage is reached. Then the overfeeding cycle starts for example with 110% for a defined time of e.g. 5 min, before the underfeeding starts again. By this measure, it should be possible to maintain the electrolysis in a narrow oxide concentration range, running the process PFC free. Only in the case of a process instability, the cell runs into partial or full anode effect emitting PFCs, which still is by far less than a continuous PFC emission. If the cell off-gas is measured continuously, the anode effect program of the pot control unit could feed at the first detection of CF4 shortly a high amount of oxide e.g. 150% to immediately stop the CF4 formation. In the case that the oxide needs more time to dissolve than the direct response of the laboratory cell suggests (Figure 14 and Figure 15), the cell current could be decreased for a short time, e.g. 5 min, to decrease the anode current density to get the cell out of its critical state.
If the industrial neodymium electrolysis always runs above the partial anode effect with a continuous PFC emission, more fundamental changes have to be made. If the oxide feeding is already appropriate, the current has to be decreased to lower the anode current density and switch to a PFC free anode process, because the anode position cannot be changed in this cell. By lowering the current, also the voltage decreases, leading to a lower power input and lower heating of the cell. Hence, the heat generation is out of balance and a better thermal insulation or a bigger cell becomes necessary.
7. Conclusion
The neodymium electrolysis can run continuously with high CF4 and C2F6 emission. A process control in example of the aluminum electrolysis can be applied to the rare earth electrolysis. The theoretical analysis of the neodymium cell voltage in analogy to the aluminum electrolysis shows the impact of the different contributions but deviates from laboratory measurements. According to the experimental results, the oxide concentration should be managed to stay between 0.5 and 1.5%. The upper critical voltage of the laboratory cell with first CF4 emission at 1% Nd2O3 concentration and a current density of 0.4 A/cm2 were found to be 4.1 V. The minimum cell voltage in dependence on the oxide concentration at 0.4 A/cm2 was found to be 2.8 V at about 1.4% Nd2O3, which is higher than expected. This minimum voltage points at the galvanostatic process and the electrical conductivity of the electrolyte in dependence on the oxide content needs further investigation. The oxide concentration of the electrolyte has a stronger influence on the anodic overvoltage as predicted by the theoretical analysis, making a process control by a safety voltage definition possible. The feeding of 1% Nd2O3 has an immediate effect on the cell voltage and gas composition. The voltage drops about 1 V and the PFC emission vanishes. With the feeding of Nd2O3 particles, a high H2O and consequently also a HF content is detected in the off-gas. The oxide should be dried thoroughly before feeding it into the electrolyte. The sludging of the cell by inactive oxyfluorides is problematic, making a careful process control necessary to not overfeed and supersaturate locally the electrolyte. If the industrial process often emits PFCs when running above the partial anode effect or running into full anode effect, the implementation of the suggested process control could lead to a substantial decrease in PFC emission. However, if the industrial process runs continuously on PFC emission, which would be an unacceptable situation for the world atmosphere pollution, the implementation of the suggested process control would have a tremendous effect on the overall PFC reduction.
Cite this paper
Vogel, H. and Friedrich, B. (2017) Reducing Greenhouse Gas Emission from the Neodymium Oxide Electrolysis. Part II: Basics of a Process Control Avoiding PFC Emission. International Journal of Nonferrous Metallurgy, 6, 27-46. https://doi.org/10.4236/ijnm.2017.63003
References
- 1. Vogel, H. and Friedrich, B. (2015) Development and Research Trends of the Neodymium Electrolysis—A Literature Review. Proceedings of the 8th European Metallurgical Conference, Duesseldorf, 14-17 June 2015.
- 2. Zhang, Z., et al. (2001) Present Situation and Latest Progress of Process for Producing Metallic Neodymium by Electrolysis of Neodymium Oxide with Fluoride Salts: Translated from Chinese by Pan Q. Non-Ferrous Smelting, 30, 23-25.
- 3. Kingsnorth, D. (2015) The Global Rare Earth Industry Today—Plagued by Illegal Production in China. 11th International Rare Earth Conference, Singapore.
- 4. Hall, C. (2009) Key Greenhouse Gases and Global Warming Potentials.
http://www.ipcc.ch/publications_and_data/publications_ipcc_fourth_assessment_report_wg1_report_the_physical_science_basis.htm - 5. Vogel, H., Flerus, B., Stoffner, F. and Friedrich, B. (2017) Reducing Greenhouse Gas Emission from the Neodymium Oxide Electrolysis: Part I: Analysis of Anodic Gas Formation. Journal of Sustainable Metallurgy, 3, 99-107.
https://doi.org/10.1007/s40831-016-0086-0 - 6. Schneller, M. (2010) In Situ Cell Control. Light Metals, 2010, 563-568.
- 7. Iffert, M. (2007) Aluminium Smelting Cell Control and Optimisation. Doctoral Thesis, The University of New South Wales.
- 8. Guo, X., Sietsma, J. and Yang, Y. (2014) Solubility of Rare Earth Oxides in Molten Fluorides. ERES2014, 149-155.
- 9. Vogt, H. and Thonstad, J. (2002) The Voltage of Alumina Reduction Cells Prior to the Anode Effect. Journal of Applied Electrochemistry, 32, 241-249.
https://doi.org/10.1023/A:1015533928104 - 10. Vogt, H. (2013) On the Various Types of Uncontrolled Potential Increase in Electrochemical Reactors—The Anode Effect. Electrochimica Acta, 87, 611-618.
- 11. Vogt, H. and Thonstad, J. (2002) The Complex Mechanisms Inducing Anode Effects in Aluminium Electrolysis. Light Metals, 2002, 495-501.
- 12. Liu, K.R., et al. (2001) Theoretical Decomposition Voltage for Some Related Substance on Neodymium Electrolysis: Translated from Chinese by Pan Q. Chinese Rare Earths, 22, 30-33.
- 13. Liu, K.R., et al. (2003) Study of Anodic Overvoltage in Neodymium Electrolysis. Acta Metallurgica Sinica, 16, 355-359.
- 14. Vogt, H. (1980) On the Supersaturation of Gas in the Concentration Boundary Layer of Gas Evolving Electrodes. Electrochimica Acta, 25, 527-531.
- 15. Vogt, H. (1984) The Rate of Gas Evolution of Electrodes—I. An Estimate of the Efficiency of Gas Evolution from the Supersaturation of Electrolyte Adjacent to a Gas-Evolving Electrode. Electrochimica Acta, 29, 167-173.
- 16. Liu, Z. and Qi, S. (2007) Numerical Simulation of Flow Field for 3 kA Neodymium Electrobath. Rare Metal Materials and Engineering, 36, 194-196.
- 17. Hu, X.W., et al. (2007) Electrical Conductivity and Nd Solubility of NdF3-LiF-Nd2O3 Melts. Characterization of Minerals, Metals and Materials, TMS, 79-82.
- 18. Fu, S. (2007) An Experimental Study on Constant of Electric Resistance in Rare Earth Electrolysis Cell: Translated from Chinese by Wang J. Chinese Rare Earth, 28, 45-47.
- 19. Mal’tseva, L.F. and Marmer, é.N. (1962) Determination of the Electrical Properties of Graphite at High Temperatures. Soviet Powder Metallurgy and Metal Ceramics, 1, 34-38.
https://doi.org/10.1007/BF01135029 - 20. Haynes, W.M. (2012) CRC Handbook of Chemistry and Physics.
- 21. Thonstad, J. and Rolseth, S. (1978) On the Cathodic Overvoltage on Aluminium in Cryolite-Alumina Melts—I. Electrochimica Acta, 23, 223-231.
- 22. Zhang, X., Deng, Z., Wang, J. and You, H. (2003) Continuous Electrolysis of RE Metals with Block-Like Multi-Anodes: Translated by Wang J. Jiangxi Nonferrous Metals, 17, 28-30.
- 23. Zhang, Z., Liang, X., Ju, J., et al. (2000) The Current Situation and Latest Progress of the Preparation of Nd in Fluoride System by Nd Oxide Electrolysis: Translated from Chinese by Wang J. Chinese Society of Rare Earth, 207-211.
- 24. Ren, Y., Wang, J., He, Y. and Liu, Z. (2003) Calculation of Electric Field and Analysis on Body Voltage in Rare Earth Electrolysis Cell: Translated from Chinese by Wang J. Journal of Baotou University of Iron and Steel Technology, 22, 313-317.
- 25. Lai, H. and Wang, L. (2002) Study of the Effect of Current on RE in Molten-Salt Electrolysis: Translated by Wang J. Jiangxi Nonferrous Metals, 16, 23-25.
NOTES

1Percentage in this work is by mass unless otherwise marked.