Crystal Structure Theory and Applications
Vol.06 No.01(2017), Article ID:74208,10 pages
10.4236/csta.2017.61001
Synthesis and Molecular Structures of Two Novel π-Conjugation Extended Dithia[3.3]metacyclophanes
Tetsuji Moriguchi1*, Daisuke Yakeya1, Venkataprasad Jalli1, Akihiko Tsuge1, Kenji Yoza2
1Department of Applied Chemistry, Faculty of Engineering, Kyushu Institute of Technology, Kitakyushu, Japan
2Bruker AXS, Yokohama, Japan

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: January 24, 2017; Accepted: February 14, 2017; Published: February 17, 2017
ABSTRACT
Two novel dithia[3.3]metacyclophanes substituted with ethynyl groups were synthesized as small-sized metacyclophanes from the corresponding brominated dithia-metacyclophanes via C-C bond formation reaction. These compounds were characterized by using 1H NMR, mass spectrometry and elemental analyses. The exact structures were determined by using X-ray diffraction analyses. X-ray diffraction analysis results revealed that these compounds have anti conformations because of existence of intermolecular π-π interaction and some short contacts in the crystal. The long wavelength fluorescence was observed for one of the compound having a pyrene moiety in solid state because of the existence of the intermolecular π-π interaction between two pyrene units.
Keywords:
Cyclophane, X-Ray Analysis, Suzuki Coupling, π-Conjugation

1. Introduction
Cyclophanes are cyclic compounds consisting of aromatic rings and bridge chains. Over the years, synthesis and molecular structures of short-bridged cyclophanes have been attracting many researcher in supramolecular chemistry due to the interesting conformations of these molecules [1] . The understanding of the preferred conformations of cyclophane is of importance in the design of various supramolecular systems. On the other hand, small-sized cyclophane molecules act as a model to explore the flexibility of such cyclophanes, due to the presence of various conformational processes including ring flipping, ring-tilting and syn- anti isomerization. Tsuge et al., have reported the small-sized cyclophane units as a platform to build cofacial bisporphyrins [2] .
Although thia-metacyclophanes considered as a member of the metacyclophane family, readily synthesized from base-induced condensation of bis-halomethyla- renes with bis-mercaptomethylarenes in the existence of alkaline catalyst using high-dilution technique [3] . Varieties of [3.3]metacyclophanes containing oxygen [4] and nitrogen [5] as a hetero atom have also been reported. In general, cyclic compounds such as small sized metacyclophanes have various conformations derived from their orientations of the aromatic parts [6] . As part of our ongoing research towards the synthesis and conformational analysis of various substituted [3.3]metacyclophanes [7] [8] , in this study, we report the synthesis and absolute structure determination of two novel small-sized metacyclophanes having thiaether bridges and ethynyl substituents.
2. Experimental
2.1. Materials and Methods
All reagents and solvents were obtained from commercial sources and are used without further purification. The 1H-NMR spectra were recorded on a Bruker ABANCE 400 spectrometer in CDCl3 with tetramethylsilane (Me4Si) as the internal reference. The electron impact (EI) mass spectra (MS) of the compounds were obtained on a JEOL JMS-SX102A spectrometer using dichloromethane (DCM) as the solvent. The instrument was operated in positive ion mode over an m/z range of 100 - 1200. Elemental analyses were performed on a YANAKO MT-5 CHN analyzer. Column chromatography was carried out on silica gel (5 - 50 g, Wako gel C-300).
2.2. Synthesis of 2a and 2b
Ethynylation of 2,11-Dithia[3.3]metacycloophanes 2a (Scheme 1).
6,15-Dibromo-2,11-dithia[3.3]metadibenzenophane 1a (1.0 g, 2.3 mmol) [9] and 15 ml of trimethylamine were dissolved in 100 ml of toluene solvent, and then, 0.20 g (0.29 mmol) of Pd(PPh3)2Cl2, catalytic amount of CuI, 0.27g (1.0
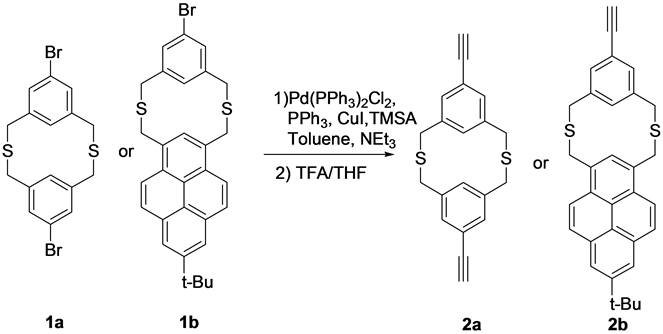
Scheme 1. Structures of the precursors (1a and 1b) and the products ethynyl substituted dithia[3.3]metacyclophanes (2a and 2b).
mmol) of PPh3, and 0.45 g (4.6 mmol) of trimethylsilyl acethylene were added the solution. The resulting reaction mixture was stirred for 20 hours at 90˚. The reaction mixture was washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was dissolved in 200 ml of THF solvent, and then, 75% trifluoroacetic acid 100 ml was added the solution and the mixed solution was vigorously stirred at room temperature for 30 min. The solvent was removed under reduced pressure, the resulting residue was chromatographed on silica gel (Wako C-300) using dichloromethane as an eluent to give the compound 6,15-diethynyl-2,11-dithia[3.3]metadibenzenophane 2a (0.49 g, 66%) as a white solid.
1H NMR (400 MHz, CDCl3) 2.99 (s, 2H, ethenyl H), 3.72 (s, 8H, thiaether bridge C-H), 6.91 (s, 2H, aryl H), 7.05 (s, 4H, aryl H). EI-MS (50 eV): m/z 320 (M+). Elemental analysis: C 75.11% (74.96%, calcd.), H 5.31% (5.03%, calcd.).
19-tert-Butyl-6-bromo-2,11-dithia[3.3]metabenzenopyrenophane 1b (1.0 g, 1.9 mmol) and 15 ml of trimethylamine were dissolved in 100 ml of toluene solvent, and then, 0.20 g (0.29 mmol) of Pd(PPh3)2Cl2, catalytic amount of CuI, 0.27 g (1.0 mmol) of PPh3, and 0.45 g (4.6 mmol) of trimethylsilyl acethylene were added the solution. The resulting reaction mixture was stirred for 20 hours at 90˚. The reaction mixture was washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. The resulting residue was dissolved in 200 ml of THF, and then, 75% trifluoroacetic acid 100 ml was added the solution and the mixed solution was vigorously stirred at room temperature for 30 min. The solvent was removed under reduced pressure, the resulting residue was chromatographed on silica gel (Wako C-300) using dichloromethane as an eluent to give the compound 19-tert-butyl-6-ethynyl-2,11-dithia[3.3]metabenzenopyrenophane 2b (0.51 g, 57%) as a yellow solid.
1H NMR (400 MHz, CDCl3) 1.53 (s, 9H, tert-butyl H), 3.65 (s, 4H, thiaether bridge C-H), 4.33 (s, 4H, thiaether bridge C-H), 6.66 (s, 1H aryl H), 6.71 (s, 1H aryl H), 7.15 (s, 2H, aryl H), 8.22 (s, 2H, pyrene H), 8.09 (d, 2H, pyrene H, J = 7.1 Hz), 8.11 (d, 2H, pyrene H, J = 7.1 Hz). EI-MS (50 eV): m/z 476 (M+). Elemental analysis: C 80.83% (80.63%, calcd.), H 5.81% (5.92%, calcd.).
Single crystals of these compounds (2a and 2b) were obtained from a solution of dichloromethane/hexane at room temperature using a vapor diffusion technique.
2.3. Single-Crystal X-Ray Analysis and Structure Determination
A colorless prismatic crystal of 6,15-diethynyl-2,11-dithia[3.3]metadibenzenophane (2a) with the approximate dimensions 0.50 × 0.15 × 0.10 mm was mounted on a glass fiber. The data collection was performed on a Bruker APEX II KY CCD diffractometer using graphite monochromatized Mo-Kα radiation (λ = 0.71073Å) and a nominal crystal-to-area detector distance of ca. 83 mm.
The data were collected at a temperature of 90 K to a maximum 2θ value of 25.02˚ (0.84 Å resolution). APEX2 software was used for preliminary determination of the unit cell [10] . Determination of the integrated intensities and unit cell refinement were performed using the SAINT program [11] . Out of the 3587 reflections that were collected, 1249 were unique (Rint = 0.0151); equivalent reflections were merged. The linear absorption coefficient, μ, for Mo-Kα radiation was 0.337 cm−1.
An empirical absorption correction was applied that resulted in transmission factors ranging from 0.941 to 0.967 [10] . The data were corrected for Lorentz and polarization effects.
The structure was solved using SHELXS-2014/7 direct method [12] , and subsequent structure refinements were performed using SHELXL-2014/7 [12] . All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms at carbon atoms were added geometrically and refined using a riding model (constrained). The final cycle of full-matrix least-squares refinement on F2 was based on 3587 observed reflections and 100 variable parameters and converged with un-weighted and weighted agreement factors of: 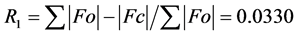 and
and
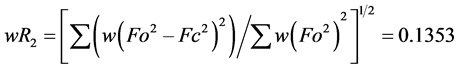 , respectively. The standard
, respectively. The standard
deviation for the observations of the unit weights was 1.227 [13] , and the sigma weights were used in the LS calculation [14] .
Similarly, a yellow prismatic crystal of 19-t-butyl-6-ethynyl-2,11-dithia[3.3]me- tabenzenopyrenophane (2b) with the approximate dimensions 0.20 × 0.20 × 0.15 mm was mounted on a glass fiber and the data collection was performed on a Bruker APEX II KY CCD diffractometer using graphite monochromatized Mo- Kα radiation (λ = 0.71073 Å) and a nominal crystal-to-area detector distance of ca.83 mm.
The data were collected at a temperature of 90 K to a maximum 2θ value of 25.04˚ (0.84 Å resolution). APEX2 software was used for preliminary determination of the unit cell [10] . Determination of the integrated intensities and unit cell refinement were performed using the SAINT program [11] . Out of the 10028 reflections that were collected, 4233 were unique (Rint = 0.0121); equivalent reflections were merged. The linear absorption coefficient, μ, for Mo-Kα radiation was 0.240 cm−1.
An empirical absorption correction was applied that resulted in transmission factors ranging from 0.900 to 0.960 [10] . The data were corrected for Lorentz and polarization effects.
The structure was solved using SHELXS-2014/7 direct method [12] , and subsequent structure refinements were performed using SHELXL-2014/7 [12] . All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms at carbon atoms were added geometrically and refined using a riding model (constrained). The final cycle of full-matrix least-squares refinement on F2 was based on 4233 observed reflections and 311 variable parameters and converged with un-weighted and weighted agreement factors of: 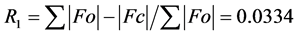 and
and
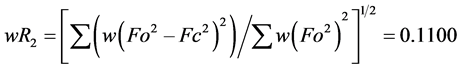 , respectively. The standard
, respectively. The standard
deviation for the observations of the unit weights was 1.215 [13] , and the sigma weights were used in the LS calculation [14] .
The results are summarized in Table 1.
3. Results and Discussion
Brominated dithia[3.3]metacyclophanes 1a and 1b precursors were synthesized using cesium ion template effect and high dilution principle technique according to previous reported methods. The cyclization reaction conditions are quite general for synthesis of cyclic compounds. The obtained brominated metacyclophanes 1a and 1b were reacted with trimethylsilyl acetylene using Suzuki Coupling C-C bond formation reaction conditions, followed by the deprotection of trimethylsilyl group by using trifluoroacetic acid in THF solvent yielded the final products 2a and 2b in moderate yields (Scheme 1).
The desired ethynyl compounds 2a and 2b were readily separated by simple silica-gel column chromatography and obtained as colorless solids in moderate

Table 1. Crystallographic data for ethynyl dithia[3.3]metacyclophanes 2a and 2b.
yield (66% for 2a, 57% for 2b, respectively).
In the EI mass spectra of the compounds 2a and 2b using 50 eV ionization voltage, only one intensive signal with characteristic isotopic patterns for [M]+ were detected.
In the 1H NMR spectra, quite simple aromatic proton signals corresponding to the phenyl groups of the compounds 2a and 2b were observed. Therefore, it was quite easy to determine the exact structure of the molecule via simple 1H NMR analysis. In addition, the bridge ether protons appeared as simple singlet(s), germinal coupling pattern(s) are not observed. Therefore, the molecular structures are readily moved in the chloroform-d solution at 297 K, in other words, the molecules are not rigid completely in solution.
A single-crystal X-ray diffraction study was performed to determine the structures of the compounds 2a and 2b in the solid state. To the best of our knowledge, the exact structures and crystal packing of these cyclophane compounds have not been previously characterized by X-ray analysis.
The compound 2a crystallizes in the space group P21/c (Z = 2, Figure 2).
Molecules 2a exist in anti conformation structure in the crystal (Figure 1). The orientation of the two ethynyl substituents are effectively stabilized by intermolecular short contacts between the hydrogen atoms of the ethynyl groups and the lone pairs of the bridge sulfur atoms (Figure 2).
On the other hand, the compound 2b crystallizes in the space group P − 1 (No.2) with the anti conformer in the unit cell (Z = 2, Figure 3). In the crystal packing of molecule 2b, there existed intermolecular π-π interaction between pyrene units resulted in complex packing (Figure 4).
We have also evaluated the photochemical properties of the two compounds 2a and 2b. UV-visible and fluorescence spectral measurements were performed, and the results are summarized in Table 2.
In general, the compounds, such as pyrene derivatives exhibit quite strong emission both in solutions and in the solid state. The emission peak maxima in solutions were observed at 334 nm for 2a and at 410 nm for 2b, respectively.
In the solid state, however, the emission maxima shifted to a much longer wavelength region. This phenomenon can be explained by the intermolecular interactions existing between the molecules when the compound 2b is in the solid form. This explanation is strongly supported by the results of the single crystal X-ray single crystal analysis.
Furthermore, HOMO and LUMO energy level calculations of the π systems of the compounds were carried out using density functional theory (DFT) B3-LYP 6-31G** level on SPARTAN14 Suite program [15] . The atomic coordinate data (x, y, z) of the X-Ray analyses were used in calculations. The calculation results were HOMO −6.05 eV LUMO −0.95 eV for 2a and HOMO −5.15 eV LUMO −1.54 eV for 2b, respectively (Figure 5).
4. Conclusion
In conclusion, two novel π-conjugation extended dithia[3.3]metacyclophanes 2a

Figure 1. Molecular views of 6,15-diethynyl-2,11-dithia[3.3]metabenzenophane 2a. Displacement ellipsoids are drawn at the 50% probability level. The yellow ellipsoids represent S atoms.

Figure 2. Crystal packing of 6,15-diethynyl-2,11-dithia[3.3]metabenzenophane 2a. The yellow ellipsoids represent S atoms.

Figure 3. Molecular views of 19-tert-butyl-6-ethynyl-2,11-dithia[3.3]metaben-zenopy- renophane 2b. Displacement ellipsoids are drawn at the 50% probability level. The yellow ellipsoids represent S atoms.

Figure 4. Crystal packing of 19-tert-butyl-6-ethynyl-2,11-dithia[3.3]metabenzenopyre- nophane 2b. The yellow ellipsoids represent S atoms.
Table 2. UV-Vis and Fluorescence spectral data for the compounds 2a and 2b.
aIn dichloromethane at 10−5 M. bIn chloroform at 10−5 M.
 (a)
(a) (b)
(b)
Figure 5. HOMO orbitals in the crystals of the compounds 2a (above) and 2b (below).
and 2b were synthesized using Suzuki coupling reaction and their absolute structures were determined via X-ray diffraction analysis. Further, we have also evaluated the UV-Visible and Fluorescence spectral properties of these compounds. The long wavelength fluorescence was observed for the compound 2b in solid state because of the existence of the intermolecular π-π interaction between two pyrene units.
Acknowledgements
We are grateful to the Center for Instrumental Analysis, Kyushu Institute of Technology (KITCIA) for the electron impact mass, 1HNMR spectra and X-ray analysis. This research was financially supported by JSPS KAKENH Grant Num- ber 15K05611.
Additional Information
CCDC No. 812371 and 1515091 contain the supplementary crystallographic data for the compounds 2a and 2b, respectively. The data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif by e-mailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallography Data Centre, 12 Union Road, Cambridge, CB2 IEZ, UK. Fax: +44(0) 1223-336033.
Cite this paper
Moriguchi, T., Yakeya, D., Jalli, V., Tsuge, A. and Yoza, K. (2017) Synthesis and Molecular Structures of Two Novel π-Conjugation Extended Di- thia[3.3]metacyclophanes. Crystal Structure Theory and Applications, 6, 1-10. https://doi.org/10.4236/csta.2017.61001
References
- 1. Ramaiah, D., Prakash, P.N., Akhil, K.N. and Rekha, R.A. (2010) Functional Cyclophanes: Promising Hosts for Optical Biomolecular Recognition. Chemical Society Reviews, 39, 4158-4168.
https://doi.org/10.1039/b920032k - 2. Tsuge, A., Ikeda, Y., Moriguchi, T. and Araki, K. (2012) Preparation of Cyclophanes Having Cofacial Bisporphyrins and Their Binding Properties. Journal of Porphyrins and Phthalocyanines, 16, 250-254.
https://doi.org/10.1142/S1088424612004562 - 3. McMahon, G., O’Malley, S., Nolan, K. and Diamond, D. (2003) Important Calixarene Derivatives—Their Synthesis and Applications. Arkivoc, 7, 23-31.
http://www.arkat-usa.org/get-file/19101/ - 4. Bodwell, G.J. (1996) The New Inductees in the “Hall of Phane”—No Phane, No Gain. Angewandte Chemie, 35, 2085-2088.
https://doi.org/10.1002/anie.199620851 - 5. Diederich, F., Stang, P.J. and Tykwinski, R.R. (2008) Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis. Wiley, New York.
https://doi.org/10.1002/9783527621484 - 6. Boekelheide, V. (1983) Synthesis and Properties of the [2n]Cyclophanes. In: Vögtle, F., Ed., Topics in Current Chemistry, Springer, Berlin, 87-143.
https://doi.org/10.1007/3-540-12397-0_2 - 7. Moriguchi, T., Egami, T., Yakeya, D., Jalli, V., Yoza, K. and Tsuge, A. (2016) Synthesis and Crystal Structure Determination of 4’,9’,4”,9”-Tetra-t-Butyl-1’, 6’,1”,6”-Tetramethoxy-2,5-Dithia[3.3] Metabiphenylophane. Crystal Structure Theory and Applications, 5, 56-62.
https://doi.org/10.4236/csta.2016.53005 - 8. Moriguchi, T., Yakeya, D., Jalli, V., Yoza, K. and Tsuge, A. (2016) Syn-6,15-Dihy-droxy-2,11-Dithia[3.3]Metacyclophane Ethyl Acetate Monosolvate. IUCrData, 1, x160082.
https://doi.org/10.1107/S2414314616000821 - 9. Morisaki, Y., Ishida, T. and Chujo, Y. (2003) Synthesis and Optical Properties of Novel Through-Space π-Conjugated Polymers Having a Dithia[3,3]Metacyclophane Skeleton in the Main Chain. Polymer Journal, 35, 501-506.
https://doi.org/10.1295/polymj.35.501 - 10. APEX2 Version 2009.9. Bruker AXS Inc., Madison.
- 11. SAINT Version 7.68A (2009) Bruker AXS Inc., Madison.
- 12. Sheldrick, G.M. (2008) SHELXS Version 2014/6. Acta Crystallographica, A64, 112-122.
https://doi.org/10.1107/S0108767307043930 - 13. Standard Deviation of an Observation of Unit Weight: [Σw(Fo2-Fc2)2/(No-Nv)]1/2 Where: No = Number of Observations, Nv = Number of Variables.
- 14. Least Squares Function Minimized: (SHELXL-2013): Σw(Fo2-Fc2)2 Where w = Least Squares Weights.
- 15. Halgren, T.A. (1996) Merck Molecular Force Field: I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry, 17, 490-519.
https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P