Crystal Structure Theory and Applications
Vol.05 No.04(2016), Article ID:72044,11 pages
10.4236/csta.2016.54006
Syntheses, Characterization and DFT Analysis of Two Novel Thiaheterohelicene Derivatives
Tetsuji Moriguchi*, Keiichi Mitsumoto, Yuki Nishizawa, Daisuke Yakeya, Venkataprasad Jalli, Akihiko Tsuge
Department of Applied Chemistry, Faculty of Engineering, Kyushu Institute of Technology, Kitakyushu, Japan

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: October 27, 2016; Accepted: November 13, 2016; Published: November 16, 2016
ABSTRACT
Two novel thiaheterohelicene derivatives were synthesized from the corresponding 2,2'-(2,6-naphthalenediyl-di-2,1-ethenediyl) bis-thiophene and its dimethyl substituted analogue 2,2'-(2,6-naphthalenediyldi-2,1-ethenediyl) bis-2’’-methylthiophene using oxidative photo cyclization reaction. The compounds were characterized by 1H NMR, electron impact-mass spectrometry, elemental analyses, and the absolute molecular structures were determined by single crystal X-ray diffraction analysis. They crystallized under monoclinic system with space group P21/n for the unsubstituted compound and P21/c for the methyl substituted compound, respectively. The dihedral angle between the terminal thiophene ring and the molecular center was observed to be 20.82˚ for the unsubstituted compound and 14.27˚ for the methyl substituted compound, respectively. Furthermore, molecules oriented as herringbone structures by intermolecular π-π stacking in the crystals. The relative study of the actual arrangement of these molecules has been carried out using X-ray diffraction analysis. The two molecules have different crystal packing. The molecule 3b has herring bone like arrangement due to the substituent bulkiness and weak CH-π interaction. On the other hand, the molecular packing of molecule 3a is not herringbone probably due to the multiple weak intermolecular CH-S short contacts between columns consisting of stacked molecules.
Keywords:
Crystal Structure, Thiaheterohelicene Derivative, Strained Structure, Single Crystal X-Ray Study

1. Introduction
Helicenes are ortho fused polycyclic aromatic compounds. They have attracted great attention of researchers because of the unique properties associated with their structure. Helicenes have found many applications in various fields such as organic electronic materials [1] [2] , liquid crystals [3] [4] and molecular recognition [5] [6] [7] . There are two classes of helicenes, carbohelicene and heterohelicene. Although the carbohelicenes are more studied, recently heterohelicene molecules having one or more heteroatom have also gained great attention due to the extended π-electron conjugation compared with carbohelicene. They have been employed as organic light-emitting diodes [8] , organic field effect transistors [9] [10] and photovoltaics [11] . Over the past decade intense efforts have been made for the synthesis of novel heterohelicene molecules comprising oxygen [12] [13] [14] , sulfur [15] [16] [17] [18] and boron [19] as a heteroatom. Linear fused thiahelicene molecules exhibit good organic field effect transistor (OFET) mobility, easier functionalization and better chemical stability. As part of our continuous interest towards the development of novel polycyclic compounds as semiconductive materials, herein, we report the syntheses of two new thiahelicene derivatives having H (3a) and -CH3 (3b) substituents at α position to the S atom. The aim of this research is to study the effect of the -CH3 substitution on the actual arrangement of the molecules using X-ray structural determination of the two molecules. We have taken -CH3 substituent for the comparative study of the effect of substation on the actual arrangement of these molecules because making of this molecule (3b) is easy, economically cheaper compared to the other substituents.
2. Experimental
2.1. Materials and Measurements
All reagents and solvents were purchased from commercial sources and are used without further purification. The 1H-NMR spectrum was recorded on a JEOL JNM A-500 spectrometer in CDCl3 with tetramethylsilane (Me4Si) as the internal reference. The electron impact (EI) mass spectrum (MS) of the compound was obtained on a JEOL JMS-SX102A spectrometer using dichloromethane (DCM) as the solvent. The instrument was operated in positive ion mode over an m/z range of 100 - 1200. Elemental analysis was performed on a YANAKO MT-5 CHN analyzer.
2.2. Synthesis
Typical procedure for the synthesis of the compounds 2a and 2b.
2,6-Bis(triphenylphosphinnomethyl)-naphthalene dichloride Wittig Salt (0.75 g, 1.0 mmol) 1 was dissolved in 200 ml of dry THF in round bottom flask under argon stream. To this thiophene carboxaldehydes (2.0 mmol) was added dropwise, the mixture was stirred for 14 h, quenched with 1.0% HCl aq., and extracted twice with CH2Cl2. The organic layers were washed once with 50 mL water, twice with 50 mL of brine solution, dried over MgSO4, and the solvent was removed under reduced pressure. The precursor 2a and 2b were obtained using silica gel (Wako gel C-300) column chromatography (280 mg, 82% yield for 2a, 270 mg, 72% for 2b) with CH2Cl2 as an eluent.
2a1H NMR (400 MHz, CDCl3): 7.04 (d, 2H, J = 8.0 Hz, ethenyl H), 7.05 (d, 2H, J = 7.0 Hz, aryl H), 7.12 (s, 2H,aryl H), 7.22 (d, 2H, J = 7.0 Hz, aryl H), 7.36 (d, 2H, J = 8.0 Hz, ethenyl H), 7.67 (d, 4H, J = 6.6 Hz, aryl H), 7.78 (t, 2H, J = 6.6 Hz, aryl H); EI-MS: m/z 344 (M+).
2b1H NMR (400 MHz, CDCl3):2.50 (6H, s, CH3), 6.66 (d, 2H, J = 7.7 Hz, ethenyl H), 6.90 (d, 2 H, J = 7.7 Hz, ethenyl H), 6.95 (d, 2H, J = 7.4 Hz, aryl H), 7.25 (d, 2H, J = 7.4 Hz, aryl H), 7.63 (d, 2 H, J = 7.2 Hz, aryl H), 7.74 (s, 2H, aryl H), 7.75 (d, 2H, J = 7.2 Hz, aryl H); EI-MS: m/z 372 (M+).
Typical procedure for the Photo cyclization of the compounds 2a and 2b.
4,9-Bis{2’-(2”-substituted thiophenyl)ethenyl}-naphthalene (1.0 mmol) 2 was dissolved in 200 ml of benzene in round bottom flask. To this iodine(10 mmol) was added, the mixture was stirred and irradiated UV light using high-pressure Hg lump for 14 h, quenched with 1.0 mol/L Na2S2O3 solution, allowed to warm to room temperature, and extracted twice with AcOEt. The organic layers were washed once with 50 mL water, twice with 50 mL of brine solution, dried over MgSO4, and the solvent was removed under reduced pressure. The title compound was obtained using silica gel (Wako gel C-300) column chromatography (190 mg, 56% yield for 3a, 120 mg, 33% for 3b) with CH2Cl2 as an eluent.
3a M.p.: 226˚C - 232˚C; 1H NMR (400 MHz, CDCl3): 7.68 (d, 2H, J = 7.3 Hz, aryl H), 7.87 (d, 2 H, J = 7.5 Hz, aryl H), 8.02 (d, 2H, J = 7.2 Hz, aryl H), 8.05 (d, 2H, J = 7.3 Hz, aryl H), 8.54 (d, 2H, J = 7.2 Hz, aryl H), 9.04 (d, 2 H, J = 7.5 Hz, aryl H); EI-MS: m/z 340 (M+); Analysis: C22H12S2, Found: C: 77.74%, H: 3.61%, Calculated: C: 77.61%, H: 3.55%).
3b M.p.: 232˚C - 236˚C; 1H NMR (400 MHz, CDCl3): 2.80 (s, 6H, CH3), 7.85 (d, 2H, J = 7.3 Hz, aryl H), 8.00 (d, 2 H, J = 7.3 Hz, aryl H), 8.04 (d, 2H, J = 7.4 Hz, aryl H), 8.26 (s, 2H, aryl H), 9.06 (d, 2 H, J = 7.4 Hz, aryl H); EI-MS: m/z 368 (M+); Analysis: C24H16S2, Found: C: 78.34%, H: 4.49%, Calculated: C: 78.22%, H: 4.38%).
2.3. Single Crystal X-Ray Analysis and Structure Determination
Single crystals of two compounds 3a and 3b were obtained at room temperature from a solution of dichloromethane/n-hexane (v/v = 1/1).
The crystal data were recorded on a Bruker APEX II KY CCD diffractometer equipped with graphite monochromatized (doubly curved silicon crystal) Mo-Kα-radiation (λ = 0.71073 Å) from a sealed micro focus tube, and a nominal crystal to area detector distance of 58 mm. X-ray generator settings were 50 kV and 30 mA. The data were collected at −183˚C (90 K). Data were acquired using four sets of Omega scans at different Phi settings and the frame width was 0.5˚. APEX2 software was used for the preliminary determination of the unit cell [20] . Integrated intensities and unit cell refinement were determined using the SAINT program [21] . Integration of the data yielded a total of 15,805 reflections to a maximum θ angle of 28.67˚ (0.74 Å resolution) for the compound 3a. And a total of 7585 reflections to a maximum θ angle of 25.55˚ (0.82 Å resolution) for the compound 3b.
The average residual for symmetry equivalent reflections were Rint = 4.13% with Rσ = 3.62% for 3a and Rint = 4.81% with Rσ = 3.85% for 3b, respectively. XPREP [22] determined the space groups to be P21/n (No. 14) with Z = 4 for the formula moiety of 3a, C22H12S2, and P21/c (No. 14) with Z = 2 for the formula moiety of 3b, C24H16S2.
Several scans in the ω direction were made to increase the number of redundant reflections, which were averaged in the refinement cycles. This procedure replaces an empirical absorption correction [23] . The structures were solved with direct methods (SHELXS-2014) and refined against F2 (SHELXL-2014) [24] .
Hydrogen atoms at carbon atoms were added geometrically and refined using a riding model (constrained), whereas the hydrogen atoms at carbon atoms were exact localized and refined isotropically with bond restraints of 89 pm for C-H. All non-hydrogen atoms were refined with anisotropic displacement parameters.
3. Results and Discussion
Synthetically, the two desired compounds 3a and 3b were obtained in 70 % isolated yield from the corresponding precursors bis(substituted-thienylethenyl) naphthalenes 2a and 2b by oxidative photo cyclization reaction in the presence of iodine as an oxidative reagent (Scheme 1). Structural properties in solution are in line with expectations, as shown by NMR spectroscopy. For the compounds 3a and 3b, the four protons (Ha and Hb), which located in Fjord regions were observed at low magnetic field region. The NMR shift values are 8.54 ppm and 9.04 ppm for 3a, 8.26 ppm and 9.06 ppm for 3b, respectively. The shifts were well explained by the strong ring current effects of the π-systems on the molecules.
The exact molecular structures of the compounds 3a and 3b are determined by using X-ray diffraction analysis (Figure 1, Figure 2). Suitable single crystals for X-ray structure analyses were obtained for the products 3a and 3b in dichloromethane/n-hexane (v/v = 1/1). The crystallographic details are summarized in Table 1. The compound 3a crystallizes in the centrosymmetric space group P21/n (No.14) with two non-planar
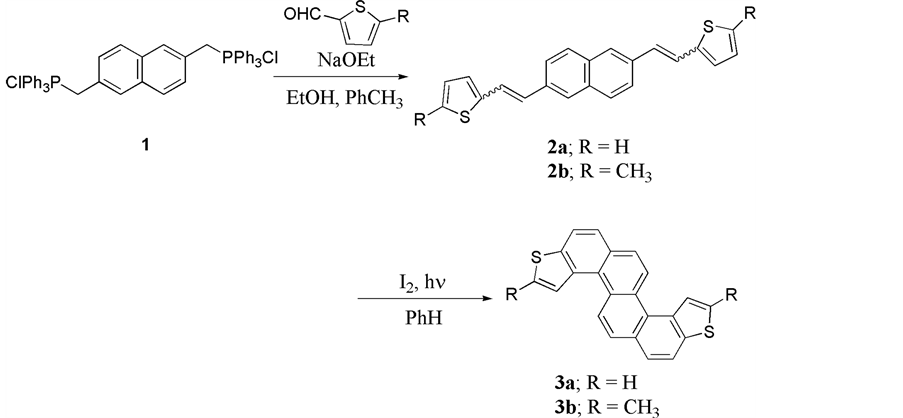
Scheme 1. Synthesis of the title compounds 3a and 3b.

Figure 1. Molecular structures of compounds 3a (above) and 3b (below).

Figure 2. Side view of structures of the compounds 3a (above) below (3b).
molecules in the unit cell (Figure 3). As the molecular shape was strained, the dihedral angle between two terminal thiophene rings was quite large (37.83˚). On the other hand, the compound 3b also crystallizes in the centrosymmetric space group P21/c (No.14) with four non-planar molecules in the unit cell (Figure 3). The molecular shape was also strained, the two terminal thiophene rings are parallel (the dihedral angle between them is 0˚).
Furthermore, the molecular packing of the compound 3a and 3b in the crystals were quite different. The packing of 3b is called “Herring-Bone” structure. In general, almost organic semiconductive compounds consisting of polynuclear aromatic hydrocarbons and polynuclear heteroaromatics usually form two-dimensional herringbone molecular orientation in crystal. On the contrary, packing style of 3a is not “Herring-Bone” structure. Difference between crystal packing of 3a and 3b, is probably due to the substituent bulkiness and weak CH-π interaction. In the crystal of 3a, multiple weak intermolecular CH-S short contacts are existed between columns consisting of stacked molecules (Figure 4). In detail sulfur atoms on one molecule and aromatic protons on neighbor molecule, and the distance is 2.971 Å. On the other hand, such weak interaction could not be observed in the crystal of 3b. Only van der Waals forces are not existed between columns consisting of stacked molecules.
HOMO and LUMO energy level calculations of the π systems of the compounds were carried out using density functional theory (DFT) B3-LYP 6-31G(d) level on SPARTAN14 Suite program [18] . Atomic coordinate data (x, y, z) of the X-Ray analyses were used in calculations. The calculation results were HOMO −5.93 eV LUMO −1.93 eV for 3a and HOMO −6.95 eV LUMO 2.13 eV for 3b, respectively. The energy gaps were significantly large (4.00 eV for 3a and 9.00 eV for 3b). The large difference between the two energy levels is probably due to the molecular structures in the crystal packing.

Table 1. Crystal data and structure refinement details for compounds 3a and 3b.

Figure 3. Molecular packing of compounds 3a (above) below (3b).

Figure 4. Intermolecular short contact diagram of molecule 3a.
On the other hand, we also performed molecular calculations of these compounds 3a and 3b to compare with X-ray results (Figure 5). In molecular calculations, atomic coordinate data (x, y, z) were calculated using MMFF94 (Merck Molecular Force Field 94) program [25] . The resulting molecular structures are shown in Figure 6. The obtained molecular shapes are slightly different compared with those of the molecules in crystals. This implies that the molecules in the crystals are almost not affected by crystal packing force and many weak interactions such as π-π interaction, CH-π interaction, and so on.
From the calculated results, we also obtained HOMO and LUMO energy levels, HOMO −5.44 eV LUMO −1.36 eV for 3a and HOMO −5.20 eV LUMO 1.33 eV for 3b, respectively. The values are slightly different from those of the molecules in crystals, and these differences are probably due to the molecular structures. From the results the compounds 3a and 3b are seems to be good p-type semiconducting materials.


Figure 5. HOMO orbitals in the crystals of the compounds 3a (above) and 3b (below).

Figure 6. Calculated molecular structures of the compounds 3a (above) and 3b (below).
Furthermore, UV visible and fluorescence spectra were acquired to determine the photochemical properties of the two compounds 3a and 3b. The strong emissions were observed for the compounds 3a and 3b in chloroform and the peak maxima are detected at 405 nm for 3a and 403 nm for 3b, respectively. The strong emissions can be explained by the molecular shapes of the compounds 3a and 3b. In another words, the compounds have no flexible parts, therefore, the excited energy cannot be relaxed through molecular vibrations.
4. Conclusion
The novel air stable thiaheterohelicene derivatives were prepared in high yield and characterized by 1H NMR, elemental analyses and electron impact-mass spectrometry. The exact molecular structures and the molecular packing of the compounds were confirmed by single crystal X-ray analyses. The crystal packing was the same styles as other semiconductor compounds. Therefore, the semiconducting properties of these compounds are under investigation now.
Acknowledgements
We are grateful to the Center for Instrumental Analysis, Kyushu Institute of Technology (KITCIA), for elemental analyses, mass spectra and 1NMR spectra, and X-ray analyses. We also thank Dr. Kenji Yoza (Bruker AXS JAPAN) for experimental assistance during the refinements of the X-ray analyses. This research was financially supported by Kitakyushu Foundation for the Advancement of Industry Science and Technology (FAIS) and JSPS KAKENHI Grant Number 15K05611.
Cite this paper
Moriguchi, T., Mitsumoto, K., Nishizawa, Y., Yakeya, D., Jalli, V. and Tsuge, A. (2016) Syntheses, Characterization and DFT Analysis of Two Novel Thiaheterohelicene Derivatives. Crystal Structure Theory and Applications, 5, 63-73. http://dx.doi.org/10.4236/csta.2016.54006
References
- 1. Storch, J., Zadny, J., Strasak, T., Kubala, M., Sykora, J., Dusek, M., Cirkva, V., Matejka, P., Krbal, M. and Vacek, J. (2015) Synthesis and Characterization of a Helicene-Based Imidazolium Salt and Its Application in Organic Molecular Electronics. Chemistry—A European Journal, 21, 2343-2347.
https://doi.org/10.1002/chem.201405239 - 2. Field, J.E., Muller, G., Riehl, J.P. and Venkataraman, D. (2003) Circularly Polarized Luminescence from Bridged Triarylamine Helicenes. Journal of the American Chemical Society, 125, 11808-11809.
https://doi.org/10.1021/ja035626e - 3. Nuckolls, C., Shao, R., Jang, W.G., Clark, N.A., Walba, D.M. and Katz, T.J. (2002) Electro-Optic Switching by Helicene Liquid Crystals. Chemistry of Materials, 14, 773-776.
https://doi.org/10.1021/cm010628o - 4. Nuckolls, C. and Katz, T.J. (1998) Synthesis, Structure, and Properties of a Helical Columnar Liquid Crystal. Journal of the American Chemical Society, 120, 9541-9544.
https://doi.org/10.1021/ja982025s - 5. Wang, D.W. and Katz, T.J. (2005) A HELOL Analogue That Senses Remote Chirality in Alcohols, Phenols, Amines and Carboxylic Acids. The Journal of Organic Chemistry, 70, 8497-8502.
https://doi.org/10.1021/jo0512913 - 6. Nuckolls, C., Katz, T.J., Verbiest, T., Elshocht, S.V., Kuball, H.G., Kiesewalter, S., Lovinger, A.J. and Persoons, A. (1998) Circular Dichroism and UV-Visible Absorption Spectra of the Langmuir-Blodgett Films of an Aggregating Helicene. Journal of the American Chemical Society, 120, 8656-8660.
https://doi.org/10.1021/ja981757h - 7. Yamamoto, K., Ikeda, T., Kitsuki, T., Okamoto, Y., Chikamatsu, H. and Nakazaki, M. (1990) Synthesis and Chiral Recognition of Optically Active Crown Ethers Incorporating a Helicene Moiety as the Chiral Centre. Journal of the Chemical Society, Perkin Transactions., 1, 271-276.
https://doi.org/10.1039/p19900000271 - 8. Tsuji, H., Mitsui, C., Ilies, L., Sato, Y. and Nakamura, E. (2007) Synthesis and Properties of 2,3,6,7-Tetraarylbenzo[1,2-b:4,5-b’]difurans as Hole-Transporting Material. Journal of the American Chemical Society, 129, 11902-11903.
https://doi.org/10.1021/ja074365w - 9. Mitsui, C., Soeda, J., Miwa, K., Tsuji, H., Takeya, J. and Nakamura, E. (2012) Naphtho[2,1-b:6,5-b’]difuran: A Versatile Motif Available for Solution-Processed Single-Crystal Organic Field-Effect Transistors with High Hole Mobility. Journal of the American Chemical Society, 134, 5448-5451.
https://doi.org/10.1021/ja2120635 - 10. Nakano, M., Niimi, K., Miyazaki, E., Osaka, I. and Takimiya, K. (2012) Isomerically Pure Anthra[2,3-b:6,7-b’]-difuran (anti-ADF), -dithiophene (anti-ADT), and -diselenophene (anti-ADS): Selective Synthesis, Electronic Structures, and Application to Organic Field-Effect Transistors. The Journal of Organic Chemistry, 77, 8099-8111.
https://doi.org/10.1021/jo301438t - 11. Wu, J.-S., Lin, C.-T., Wang, C.-L., Cheng, Y.-J. and Hsu, C.-S. (2012) New Angular-Shaped and Isomerically Pure Anthradithiophene with Lateral Aliphatic Side Chains for Conjugated Polymers: Synthesis, Characterization, and Implications for Solution-Prossessed Organic Field-Effect Transistors and Photovoltaics. Chemistry of Materials, 24, 2391-2399.
https://doi.org/10.1021/cm301176s - 12. Areephong, J., Ruangsupapichart, N. and Thongpanchang, T. (2004) Enantioselective Bioreduction of Ethyl 4,4,4-Trihalide-3-oxobutanoate by Kluyveromyces marxianus. Tetrahedron Letters, 45, 3067-3070.
https://doi.org/10.1016/j.tetlet.2004.02.105 - 13. Salim, M., Akutsu, A., Kimura, T., Minabe, M. and Karikomi, M. (2011) Novel Synthesis of Oxahelicenes by Lawesson’s Reagent-Mediated Cyclization of Helical Quinone Derivatives. Tetrahedron Letters, 52, 4518-4520.
https://doi.org/10.1016/j.tetlet.2011.06.033 - 14. Irie, R., Tanoue, A., Urakawa, S., Imahori, T., Igawa, K., Matsumoto, T., Tomooka, K., Kikuta, S., Uchida, T. and Katsuki, T. (2011) Synthesis and Stereochemical Behavior of a New Chiral Oxaheterohelicene. Chemistry Letters, 40, 1343-1345.
https://doi.org/10.1246/cl.2011.1343 - 15. Roncali, J. (1992) Conjugated Poly(thiophenes): Synthesis, Functionalization, and Applications. Chemical Reviews, 92, 711-738.
https://doi.org/10.1021/cr00012a009 - 16. Roncali, J. (1997) Synthetic Principles for Bandgap Control in Linear π-Conjugated Systems. Chemical Reviews, 97, 173-206.
https://doi.org/10.1021/cr950257t - 17. Yamamoto, T., Arai, M., Kokubo, H. and Sasaki, S. (2003) Copolymers of Thiophene and Thiazole. Regioregulation in Synthesis, Stacking Structure, and Optical Properties. Macromolecules, 36, 7986-7993.
https://doi.org/10.1021/ma030167n - 18. Yamamoto, T., Komarudin, D., Arai, M., Lee, B.-L., Suganuma, H., Asakawa, N., Inoue, Y., Kubota, K., Sasaki, S., Fukuda, T. and Matsuda, H. (1998) Extensive Studies on π-Stacking of Poly(3-alkylthiophene-2,5-diyl)s and Poly(4-alkylthiazole-2,5-diyl)s by Optical Spectroscopy, NMR Analysis, Light Scattering Analysis, and X-Ray Crystallography. Journal of the American Chemical Society, 120, 2047-2058.
https://doi.org/10.1021/ja973873a - 19. Katayama, T., Nakatsuka, S., Hirai, H., Yasuda, N., Kumar, J., Kawai, T. and Hatakeyama, T. (2016) Two-Step Synthesis of Boron-Fused Double Helicenes. Journal of the American Chemical Society, 138, 5210-5213.
https://doi.org/10.1021/jacs.6b01674 - 20. Bruker AXS Inc. (2009) APEX2 Version 2009.9.
- 21. Bruker AXS Inc. (2009) SAINT Version 7.68A.
- 22. Sheldrick, G.M. (2008) XPREP Version 2008/2. Bruker AXS Inc., Madison.
- 23. Sheldrick, G.M. (2008) SADABS, Version 2008/1. Bruker AXS Inc., Madison.
- 24. Sheldrick, G.M. (2008) SHELX Version 2014/6. Acta Crystallographica, A64, 112-122.
https://doi.org/10.1107/S0108767307043930 - 25. Halgren, T.A. (1996) Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. Journal of Computational Chemistry, 17, 490-519.
https://doi.org/10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P
Appendix A. Supplementary Material
CCDC no. 1010570 for the compound 3a and 1010571 for the compound 3b contain the supplementary crystallographic data. The data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif by e-mailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallography Data Centre, 12 Union Road, Cambridge, CB2 IEZ, UK. Fax: +44(0) 1223-336033.

Submit or recommend next manuscript to SCIRP and we will provide best service for you:
Accepting pre-submission inquiries through Email, Facebook, LinkedIn, Twitter, etc.
A wide selection of journals (inclusive of 9 subjects, more than 200 journals)
Providing 24-hour high-quality service
User-friendly online submission system
Fair and swift peer-review system
Efficient typesetting and proofreading procedure
Display of the result of downloads and visits, as well as the number of cited articles
Maximum dissemination of your research work
Submit your manuscript at: http://papersubmission.scirp.org/
Or contact csta@scirp.org