Modern Research in Catalysis
Vol.2 No.2A(2013), Article ID:32376,7 pages DOI:10.4236/mrc.2013.22A001
Catalytic Active Sites in Molybdenum Based Catalysts
1Chemistry Department, Kuwait University, Safat, Kuwait
2College of Health Sciences, Public Authority for Applied Education and Training (PAAET), Faiha, Kuwait
Email: *ali.katrib@ku.edu.kw
Copyright © 2013 S. Al-Kandari et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 27, 2013; revised April 25, 2013; accepted May 15, 2013
Keywords: Bifunctional Mo Catalyst; XPS-UPS; 2-Propanol; N-Hexane
ABSTRACT
In situ metal, acid and metal-acid (bifunctional) catalytic active functions were prepared following partial reduction by hydrogen of MoO3 deposited on TiO2 at temperatures between 623 K and 673 K. The bifunctional structure is obtained following the reduction of MoO3 to MoO2. The metallic properties of MoO2 are attributed to the delocalized p electrons above the Mo atoms place along the C-axis of the deformed rutile structure of this phase and observed as a density of states at the Fermi level. Hydrogen dissociation by this metallic function and bonding of the produced H atoms to surface oxygen atoms results in the formation of Brønsted acid Mo-OH function(s). Accordingly, a bifunctional (metal-acid) MoO2−x(OH)y structure is formed on the TiO2 support. The bifunctional properties enabled to perform isomerization reactions of light naphtha hydrocarbons into branched species of higher octane number. This catalyst is proposed as a possible replacement of the commercially used Pt deposited on chlorinated alumina catalysts in which toxic chlorine is employed and benzene is produced as a byproduct of n-hexane isomerization. The acid function in this bifunctional Mo system is quenched following the addition of controlled amount of sodium. The presence of only the metallic function in this modified NaMoTi system is monitored via the hydrogenation of olefins and enabled to define the bifunctional mechanism of the hydrocarbon isomerization process performed by MoO2−x(OH)y structure.
1. Introduction
Catalytic active species are classified as metallic and acidic. In some specific reactions, both metallic and acidic (bifunctional) functions are required. Preparation and identification of the active site(s), responsible for specific catalytic process(s), is a major challenge to research workers especially that the whole process takes place in situ following specific experimental protocol. Several characterization techniques can provide general information on the bulk structure and/or surface structure in a broad sense. However, none of these techniques alone can define exactly the real chemical state composition of the active chemical species presented on the outermost surface layer, responsible for the catalytic activity of a given system. This is due, in part, to the fact that the catalytic activity of a given surface. In this respect, in situ combination of XPS-UPS, ISS techniques measurements in parallel with catalytic measurements carried out at the same experimental conditions could provide valuable information concerning the electronic structure of the outermost surface layer responsible for observed catalytic reaction(s).
Hydroisomerization of n-hexane to monoand di-branched molecules is an important catalytic process in petroleum industry in order to increase the octane number and to improve the motor engine working conditions. These catalytic processes take place in several concerted catalytic steps, summarized in terms of a bifunctional (metal-acid) mechanism [1]. Platinum based catalysts are the most commonly used systems at the industrial and the fundamental research concerning the isomerization of light alkanes [2-4]. In these systems, the metallic function consists of finely dispersed platinum particles (few atoms), on acidic support such as chlorinated alumina. Platinum is rare and expensive. Also, there are several problems associated with the use of these catalysts such as sintering, the corrosion caused by chlorine, the poisoning of the platinum by sulfur and water, as well as the formation of toxic benzene as a by-product.
In view of the above problems associated with the use of Pt based catalysts in different applications in industry, extensive research is underway in order to find substitute(s) and efficient catalysts for reforming, hydrogenation and dehydrogenation reactions. In this respect, we were able to prepare bifunctional catalysts based on the partial reduction of molybdenum or tungsten trioxide MO3 (M = Mo, W) deposited on TiO2 [5-7]. In the case of MoO3 for example, exposure of the supported trioxide to hydrogen at temperatures between 623 K - 673 K enabled to produce molybdenum dioxide structure having metallic properties. The metallic function is attributed to the delocalized p electrons over the Mo-Mo atoms placed along the C-axis of the deformed rutile structure of MoO2. On the other hand, the acidic function is formed as a result of hydrogen dissociation by this metallic function followed by hydrogen atom(s) bonding to surface oxygen to produce BrØnsted Mo-OH acidic group(s). The bifunctional MoO2−x(OH)y/TiO2 structure is characterized by the Mo(3d) spin-orbit coupling binding energies, the oxygen 1s, FT-IR spectra of the BrØnsted acidic group(s), and the well-resolved p and s bands in the UPS spec- -trum [5-9]. One important feature of this bifunctional MoO2−x(OH)y/TiO2 structure resides in the fact that the metallic function consists of an atomic wire like of the transition element M in a stable MO2 state under experimental conditions. Consequently, different particle size(s) are not formed which might occur during the catalyst preparation and/or due to sintering problems as in the case in Pt based catalysts. Titanium dioxide is used in this case as a support in order to increase the surface area and add mechanical strength to the active MoO2−x(OH)y structure without any apparent catalytic activity. Addition of controlled amount of sodium or potassium to the Mo salt and calcination of the mixture at 773 K produced bronze system [10-12] with certain metallic properties. Moreover, exposure of this bronze structure to hydrogen at temperature up to 673 K results in neutralizing the BrØnsted Mo-OH acidic functions due to replacement of hydrogen by sodium atoms MoO2−x(OH)y/TiO2 having only metallic properties. This modified system will be used in view of the elucidation of the mechanism of hydroisomerization of n-hexane as well as dehydration and dehydrogenation of 2-propanol. Catalytic measurements will be conducted in parallel with in situ XPS-UPS and ISS spectroscopic techniques.
2. Experimental
2.1. Catalyst Preparation
The supported MoO3/TiO2 (MoTi) catalyst system was prepared by calcination at 773 K for 12 h of wet impregnated titania pellets with the equivalent of 5 monolayers of MoO3 of ammonium heptamolybdate ((NH4)6Mo7O24∙ 4H2O, 99.9% pure product of STREM). The impregnation was carried out according to Pines et al. [13] and the titania support was Degussa P25 (75% anatase) with pore volume of 0.5 cm3/g and BET surface area of 50 ± 5 m2/g. It is worth mentioning that, the loaded ammonium heptamolybdate was quantitatively decomposed into MoO3 species during the calcination process as characterized by XPS. 2.5% concentration by mass of sodium in form of NaNO3 with respect to Mo were added following postimpregnation of the dried heptamolybdate-impregnated TiO2 pellets prior to the calcination process at 773 K (NaMoTi). In situ reduction of the MoO3/TiO2 system before and after Na addition in a flow of 40 cm3 H2/ min.as a function of temperature was carried out in different ways in order to elucidate the surface structure and its stability in relation to catalytic activity. The hydrogen gas was a 99.9% pure product of KOAC (Kuwait).
2.2. Catalyst Characterization
X-ray Photoelectron Spectroscopy (XPS) was conducted using a Thermo Scientific ESCALAB-250Xi spectrometer. The radiation source was monochromatic of AlK Operating at the power of 300 W (15 kV, 20 mA). Ultraviolet Photoelectron Spectroscopy (UPS) He(I) resonance 584 Å radiation of 21.217 eV was employed for the Valence Band (VB) energy region measurements. Vacuum in the analysis chamber was better than 7x10−9 mbar during all measurements. The in situ reduction was carried out in a high-pressure gas cell housed in the preparation chamber. Binding energies were based on the carbon contamination C1s at 284.8 eV within an experimental error of ±0.2 eV. Ion Scattering Spectroscopy (ISS) measurements were performed on the same test sample using He+ with a kinetic energy of 1 KeV.
2.3. Catalyst Tests
Catalytic reactions of 2-propanol, 1-hexene and n-hexane were studied on the test catalysts by introducing the reactants as pluses of 5 mL and time on stream modes over a fixed-bed quartz reactor under atmospheric hydrogen pressure. A continuous H2 flow of 40 cm3/min was allowed through 500 mg of the catalyst which contains 65 mg of Mo. The reaction mixture was separated and ana- - lyzed with an on-line gas chromatograph Chemito, India 1000 equipped with a Petrocol-DH column and a flame ionization detector.
3. Results and Discussion
3.1. Catalyst Characterization
3.1.1. XPS-UPS Spectra
Identification of the chemical composition of the outermost surface layer is essential in order to correlate catalytic activity and surface chemical structure. However, there is no one technique which enables to clearly define the exact chemical structure of the species responsible for a given catalytic activity. Combination of different XPS-UPS, ISS spectroscopic techniques could help in this respect.
X-ray photoelectron spectroscopy is a valuable technique which provides information about the different chemical species presented in the upper 10 monolayers. Changes in the oxidation state(s) following, in situ, different treatments such as reduction by hydrogen will be closely monitored. Characterization of the Mo(3d) spin-orbit binding energies, the valence band energy region and the presence of a density of state structure at the Fermi level in parallel with ion scattering spectra ISS will provide important information in relation to the catalytic activity of the material under investigation. In this respect, it was observed that MoO3 is reduced by hydrogen to the MoO2 structure in a stable state at temperatures up to 673 K. At higher reduction temperature, lower valency metallic Mo(0) state is produced [14]. In this work, we present the XPS-UPS, ISS data following in situ samples reductions by hydrogen at 673 K for 12 h. Reference XPS binding energies for the Mo(3d5/2,3/2) are: 232.65 and 235.85 eV for MoO3, 231.7 and 234.9 eV for Mo2O5, 229.1 and 232.3 eV for MoO2 and 227.7 and 230.85 eV for Mo(0). On the other hand, Ti(2p3/2,1/2) binding energy values are observed at 458.5 and 464.2 eV for TiO2 [15]. XP spectra of the Mo(3d) energy region of the MoTi and NaMoTi samples following in situ hydrogen reduction at 673 K for 12 h in comparison with the unreduced MoTi are presented in Figure 1. In the case of untreated MoTi sample, two well-defined spectral lines at 232.65 and 235.85 eV characteristics of Mo(3d5/2,3/2) spin-orbit components of MoO3 [15]. Most of the Mo is reduced to the MoO2 state following sample exposure to hydrogen at 673 K for 12 h (Figure 1 (b)). This assignment is based on the Mo3d binding energies at 232.3 and 229.1 eV. The sample MoO3 conversion to MoO2 under these experimental conditions seems to be incomplete since lower concentration of MoO3 and the interphase Mo2O5 are still observed (Figure 1(b)). The efficiency of MoO3 reduction to MoO2 in NaMoTi at 673 K seems to be more pronounced as could be observed in Figure 1(c). Most probably, this is due to the fact that addition of sodium to the molybdenum oxide results in the formation of separated structural NaMoTi unit structure as compared to a polymeric type MoO3 structure. On the other hand, the Na1s binding energy measured at 1072.1 eV corresponds to Mo-ONa bond formation. Titanium 2p spin-orbit components at 458.5 and 464.2 eV remain unchanged in the two MoTi and NaMoTi before and after hydrogen reduction. This is a demonstration that TiO2 is not affected by the MoO3 reduction process.
The XPS of the valence band (VB) energy region of calcined MoTi shows a broad band which consists
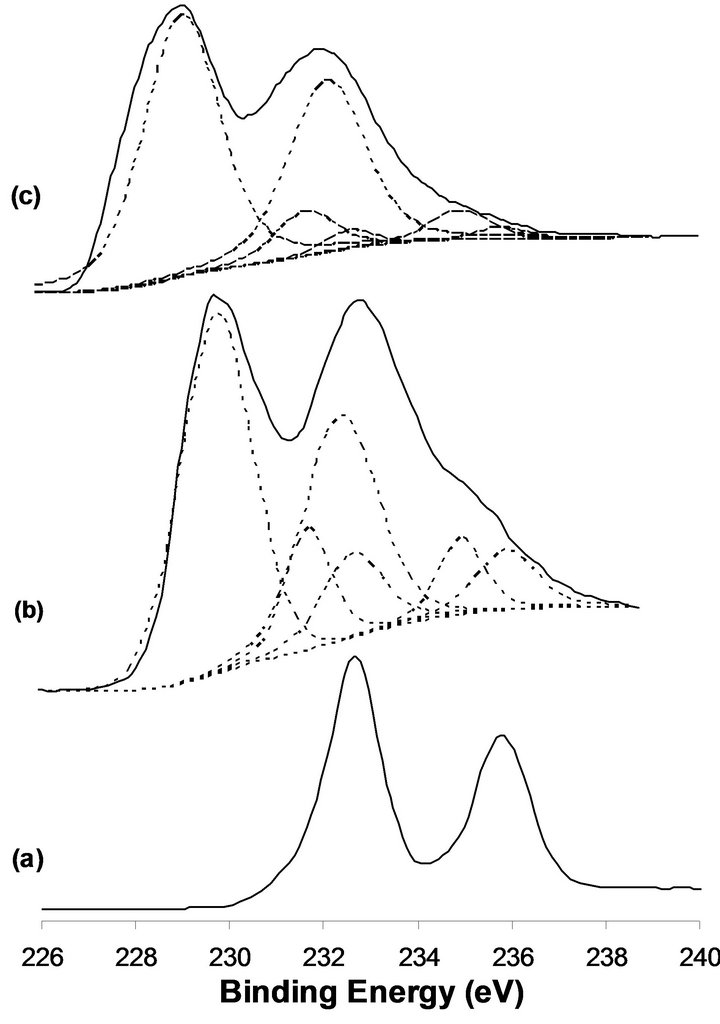
Figure 1. XPS of the Mo(3d) obtained for the calcined MoTi (a), reduced MoTi (b), 2.5%-NaMoTi (c).
mainly of O2p spectral line. No DOS structure is observed at the Fermi level which reflects the insulating properties of MoO3 structure (Figure 2(a)). Exposure of the sample to hydrogen at 673 K for 12 h produces beside the O2p band, a relatively high density structure at the Fermi level (Figure 2(b)), attributed to the metallic properties of MoO2. Higher resolution UPS of this energy region enables to clearly define the presence of s and p bands characteristics of the deformed rutile structure of MoO2 [5,6]. Similar structure is observed in the case of the Na promoted system after hydrogen reduction (Figure 2(c)).
3.1.2. ISS Spectra
Ion scattering spectroscopy ISS is the appropriate technique in order to determine the elemental composition of the outermost surface area. As could be observed in Figure 3(a), the ISS of MoTi before hydrogen reduction reveals the presence of the constituent Mo, Ti and O beside the pollution C. Despite the fact that the equivalent of 5 monolayers of MoO3 deposited on titania pellets, yet Ti is still observed as part of the surface structure. This indicates that Mo deposition is in form of aggregates and not uniformly as expected. Most likely, Mo deposition on
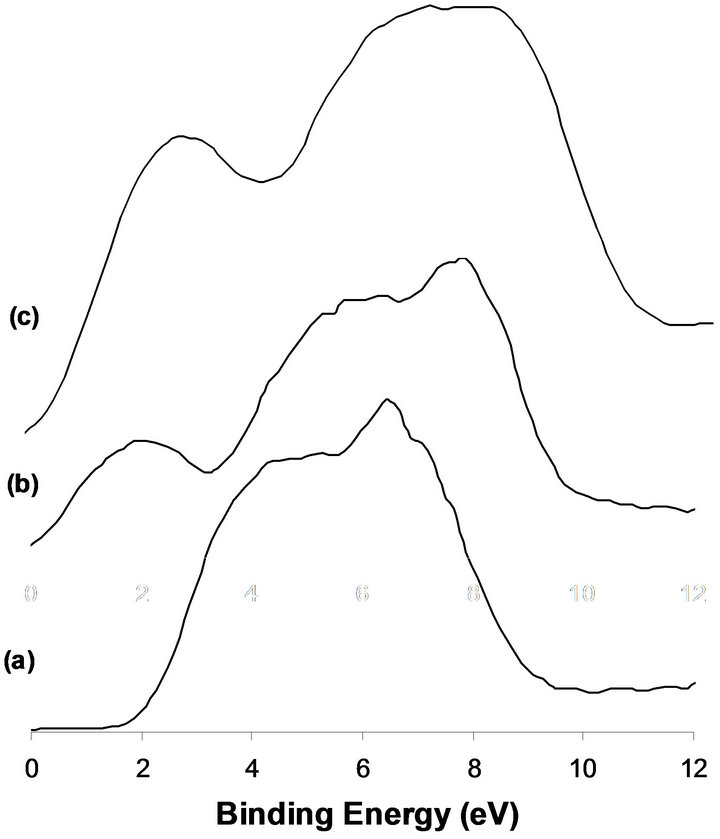
Figure 2. XPS of the VB obtained for the calcined MoTi (a), reduced MoTi (b), 2.5%-NaMoTi (c).
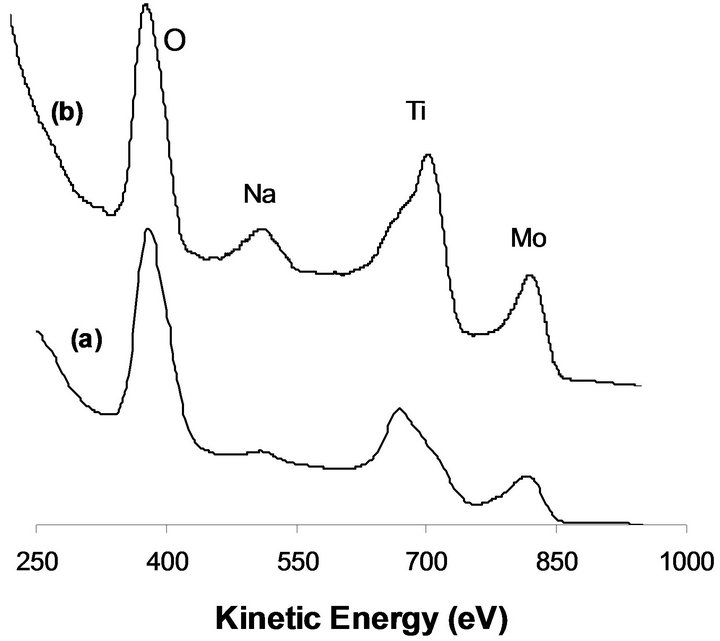
Figure 3. ISS spectra after sample exposure to hydrogen at 673 K for 12 h (a) MoTi, 2.5%-NaMoTi.
TiO2 surface takes place in form of “para-crystalline MoO3” phase that resembles bulk MoO3 and takes the form of needle-like crystallites growing away from the support surface as postulated by Bond et al. [16]. Similar behavior is observed in the case of NaMoTi (Figure 3(c)). The presence of Na on the sample surface is due to its bonding to the Mo via surface oxygen (Mo-ONa).
3.2. Catalytic Measurements
Bifunctional (metal-acid) properties of MoO2−x(OH)y deposited on TiO2 obtained at 673 K reduction temperature and the expected neutralization of the Brønsted acid function following the addition of 2.5% by mass of Na will be evaluated in the case of catalytic reactions of 2-propanol, 1-hexene and n-hexane.
3.2.1. 2-Propanol
The catalytic activity of MoO2−x(OH)y/TiO2 (MoTi) is studied for 2-propanol as a function of reaction temperature (Figure 4). A conversion of 13.8% producing 72% propene and 28% propane. At higher reaction temperatures, all the reactant alcohol molecules were converted to propene and propane. However, the relative concentration of propene increases to a maximum of 76.7% at 473 K and decreases as a function of reaction temperature. This is in parallel increase in propane concentration to reach 92.5% at 673K (Figure 4). At 673 k, all propene molecules were hydrogenated to propane by the metallic function of the MoTi system. These results demonstrate the action of the acid and metal functions in the MoTisystem. The dehydration process of 2-propanol to pen-
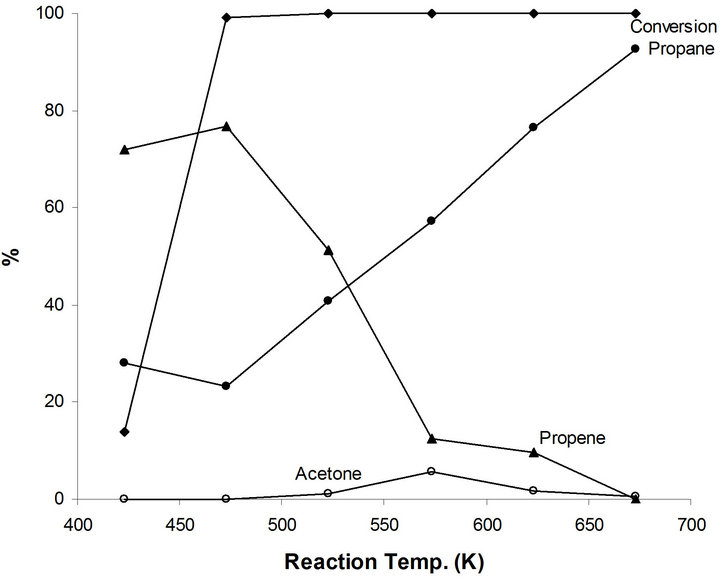
Figure 4. Products distribution of 2-propanol on MoTi at different traction temperatures.
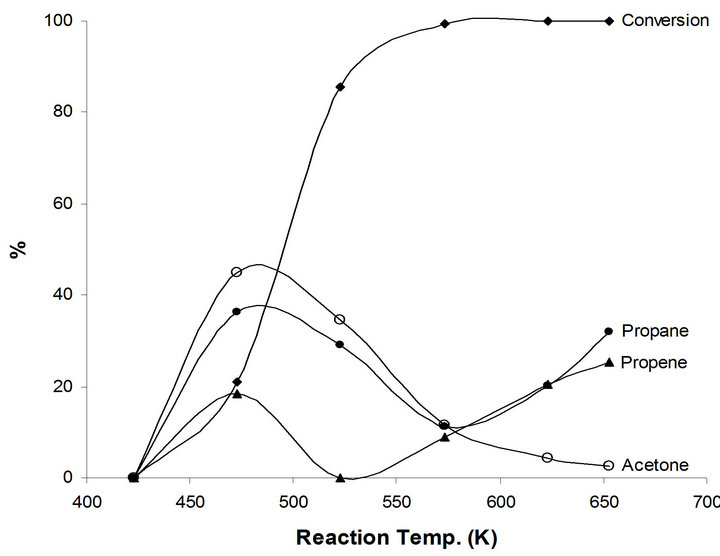
Figure 5. Products distribution of 2-propanol on 2.5%-Na MoTi at different traction temperatures.
tene, performed by the acid function of MoTi is the first and easier catalytic step. Hydrogenation of the produced propene to propane is catalyzed by the metallic function of MoTi. The efficiency of this second step catalytic process depends on the reaction temperature.
Different catalytic behavior takes place in the case of the modified system by sodium NaMoTi. No catalytic activity to very low of 21% at reaction temperatures below 500 K. Moreover, propene yield does not exceed 24 % although all reactant 2-propanol molecules were converted at 673 K reaction temperature (Figure 5). This is a clear indication of the neutralization of most, if not all, of the Brønsted Mo-OH acid by sodium Mo-ONa atoms. As a result, only the metallic function in MoO2−x(OH)y/ TiO2 remains active. Consequently, propane, acetone and polymeric compounds were produced. The effect of sodium addition on the metallic properties of MoTi could be observed in the case of the dehydrogenation of 2-propanol to acetone (Figure 5). This catalytic reaction did not take place in MoTi. Sodium bronze formation is most probably the reason for enhancing the metallic function in the modified system [10-12].
3.2.2. N-Hexane
Hydroisomerization of n-hexane to monobranched 2, 3-methylpentanes and 2,2 DMB, 2,3 DMB of relatively high octane numbers in order to boost gasoline and improve motor engine efficiency is an important catalytic process in oil industry. These catalytic reactions were rationalized in terms of bifunctional mechanism [1]. The first catalytic step in this process is the dehydrogenation of n-hexane to the hexene olefin, performed by the metallic function, followed by the isomerization of the ole fin via carbenium ion mechanism performed by Brønsted acid function, then hydrogenation of the olefin isomer by the metallic function. Highly dispersed Pt particles deposited on chlorinated alumina or zeolites are the most commonly used catalysts to achieve such objective.
Very low conversion of 4.0% to only 2,3 MP takes place at 523 K. This is due to relatively difficult first step dehydrogenation process of n-hexane to hexene which requires relatively strong metallic function. The conversion increases as the reaction temperature increases to reach 88.6% at 673 K (Figure 6). On the other hand, the isomerization selectivity decreases from 100 % at 523 K to 30.4% at 673 K in favor of cracking products. It is interesting to note the isomerization products 2MP/3MP distribution remains almost constant at 1.4 regardless of the reaction temperature, which corresponds to the thermodynamic equilibrium ration.
No catalytic activity of n-hexane on the NaMoTi modified system at reaction temperatures up to 600 K. Very low conversion of 1.9% is observed at 623 K and reaches 7.8% at 673 K. The main products are cracking which reflects the metallic character of this modified system. In fact, it is expected that dehydrogenation of nhexane to hexene will take place even at low 523 K reaction temperature, but the reverse hydrogenation will take place in the absence of an acid function on the catalyst surface. This is a clear demonstration that indeed the isomerization of n-hexane to branched species takes place in a concerted dehydrogenation to olefin/isomerization of the olefin/hydrogenation of the olefin, using metal-acidmetal functions in what is known as the bifunctional mechanism.
The isomerization activity and efficiency of the MoO2−x(OH)y/TiO2 bifunctional are comparable to those of the industrial Pt deposited on chlorinated alumina. In fact, some advantages could be attributed to the Mo based catalyst which could summarized as follows: No toxic and corrosive chlorine is used, no toxic benzene compound is formed as a by-product, no sintering problems take place, molybdenum is much cheaper than platinum and the Mo catalyst is easy to regenerate in case of poisoning.
3.2.3. 1-Hexene
Additional hydrogenation catalytic reaction is presented in 1-hexene as compared to n-hexane in relation to the hydroisomerization reactions to produce high octane number branched species. This will enable to obtain further information concerning the exact catalytic active site(s) in MoTi and NaMoYi. Complete conversion of 1-hexene takes place on MoTi at all reaction temperature up to 673 K (Figure 7). Hydrogenation reaction to n-hexane (95.5%) and 4% 2,3-dimethyl pentanes were obtained at 423 K. In fact, isomerization reactions to different branched iC6 species increase at higher reaction temperature to reach a maximum 60% at temperatures between 573 K - 623 K (Figure 7). Beyond this temperature, the relative concentrations of cracking products
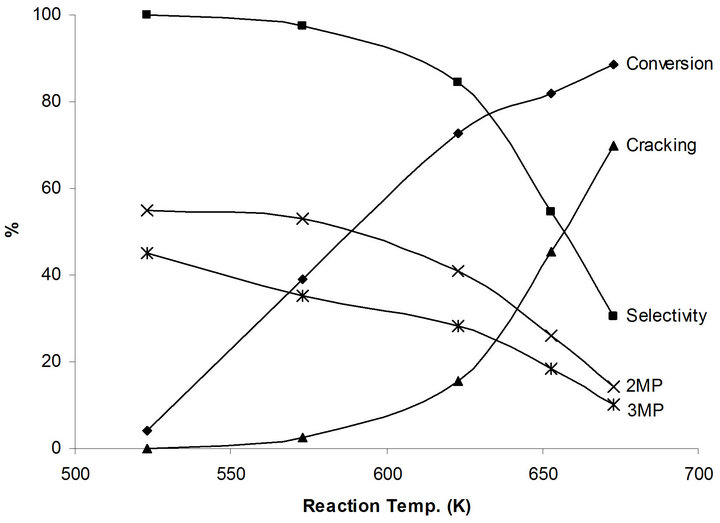
Figure 6. Products distribution of n-hexane on MoTi at different traction temperatures.
increase. This increase is in relation to thermodynamic effects. The 1-hexene isomerization to branched saturated compounds is interpreted as follows: Hydrogenation of 1-hexene to n-hexane takes place first as the easier catalytic process in this case. Consequently, the produced n-hexane undergoes the isomerization reactions to different branched compounds via the bifunctional mechanism as discussed previously.
The only major catalytic reaction of 1-hexene on the modified NaMoTi is the hydrogenation to n-hexane. In fact, all reactant 1-hexene were converted at all reaction temperatures between 423 K and 673 K (Figure 8). n-hexane is the only product up to 600 K and decreases to 79% at 673 K in favor of cracking products as expected due to thermodynamic effect a clear demonstration of the neutralization of the BrØnsted Mo-OH acidic group(s) in the bifunctional MoO2−x(OH)y/TiO2 by the addition of sodium and producing mono-functional metallic MoO2−x(OH)y/TiO2 system. Also, a demonstration that the hydroisomerization of n-hexane to different branched iC6 species is performed by a concerted bifunctional mechanism.
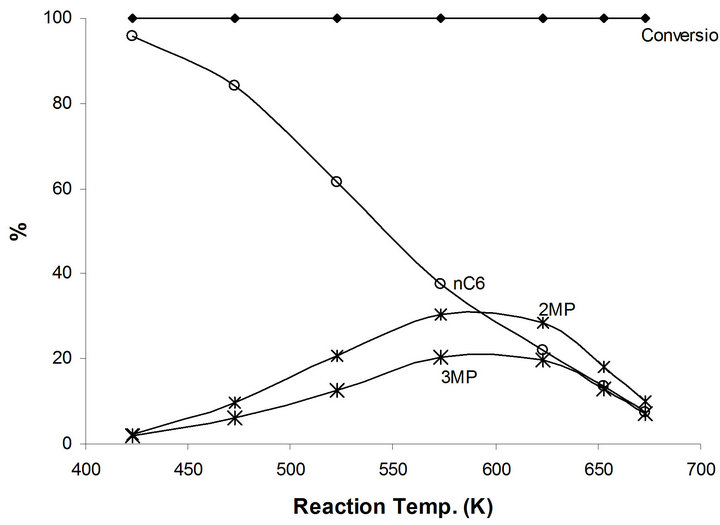
Figure 7. Products distribution of 1-hexene on MoTi at different traction temperatures.
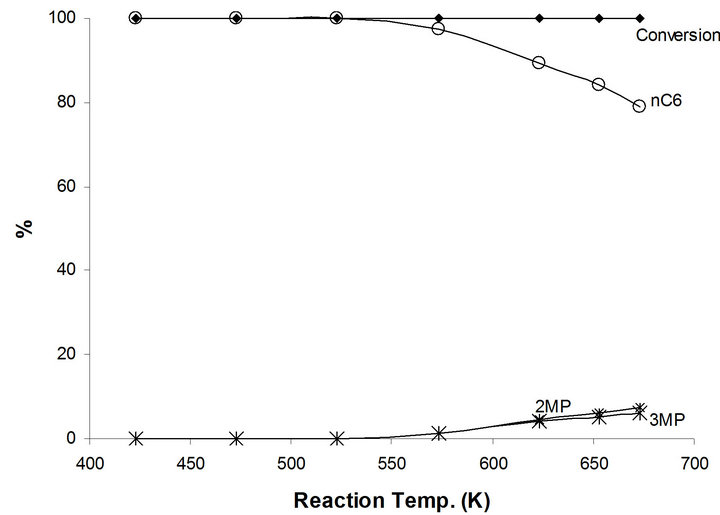
Figure 8. Products distribution of 1-hexene on 2.5%-Na MoTi at different traction temperatures.
4. Conclusion
In situ preparation of a bifunctional (metal-acid) catalytic system following controlled reduction of MoO3 to MoO2 structure has been characterized by the most appropriate surface XPS-UPS, ISS in parallel with model catalytic reactions using probe 2-propanol, n-hexane and 1-hexene molecules. Neutralization of the surface Brønsted MoOH acid function is realized following the addition of 2.5% by mass of sodium. Titanium dioxide is employed as a support in order to increase the surface area of the active Mo structure and add mechanical strength to the catalyst. The stability and efficiency of the bifunctional MoO2−x(OH)y/TiO2 catalytic system could be envisaged as possible replacement of the industrial Pt deposited on chlorinated catalysts for the hydroisomerization of light naphtha hydrocarbons. The main toxic and corrosive chlorine is not used, benzene as a by-product is not formed, low cost and easy to regenerate in case of loss of activity.
5. Acknowledgements
The support by Kuwait University through research grant # SC03/09 and GFS project # GS02/08 is gratefully acknowledged.
REFERENCES
- F. G. Gault, “Mechanisms of Skeletal Isomerization of Hydrocarbons on Metals,” Advances in Catalysis, Vol. 30, 1980, pp. 1-95. doi:10.1016/S0360-0564(08)60325-9
- T. Loften and E. A. Blekkan, “Isomerization of n-Hexane on Sulphated Zirconia Modified by Noble Metals,” Applied Catalysis A: General, Vol. 299, 2006, pp. 250-257. doi:10.1016/j.apcata.2005.10.045
- T. Kumra, “Development of pt/SO42−/ZrO2 Catalyst for Isomerization of Light Naphtha,” Catalysis Today, Vol. 81, No. 1, 2003, pp. 57-63. doi:10.1016/S0920-5861(03)00102-0
- C. Travers and P. Leprince, “Petroleum Refining 3: Conversion Processes,” Editions Technip, Paris, 2001, pp. 229-256.
- H. Al-Kandari, A. M. Mohamed, F. Al-Kharafi, M. I. Zaki and A. Katrib, “Modification of the Catalytic Properties of MoO2−x(OH)y Dispersed on TiO2 by Pt and Cs Additives,” Applied Catalysis A: General, Vol. 417-418, 2012, pp. 298-305. doi:10.1016/j.apcata.2012.01.006
- H. Al-Kandari, F. Al-Kharafi and A. Katrib, “Surface Electronic Structure-Catalytic Activity of Different Mo Oxidation States for Olefins and Saturated Hydrocarbon Molecules,” Catalysis Letters, Vol. 139, No. 3-4, 2010, pp. 134-140. doi:10.1007/s10562-010-0414-0
- H. Al-Kandari, S. Al-Kandari, F. Al-Kharafi and A. Katrib, “Molybdenum-Based Catalysts for Upgrading Light Naphtha Linear Hydrocarbon Compounds,” Energy & Fuel, Vol. 23, No .12, 2009, pp. 5737-5742. doi:10.1021/ef900617d
- A. Guilino, S. Parker, F. H. Jones and R. G. Egdell, “Influence of Metal-Metal Bonds on Electron Spectra of MoO2 and WO2,” Journal of the Chemical Society, Faraday Transactions, Vol. 92, No. 12, 1996, pp. 2137-2141. doi:10.1039/ft9969202137
- F. H. Jones, R. G. Egdell, A. Brown and F. R. Wondre, “Surface Structure and Spectroscopy of WO2(012),” Surface Science, Vol. 374, No. 1-3, 1997, pp. 80-94.
- M. Yang, B. Han and H. Cheng, “First-Principles Study of Hydrogenation of Ethylene on a HxMoO3(010) Surface,” The Journal of Physical Chemistry C, Vol. 116, No. 46, 2012, pp. 24630-24638. doi:10.1021/jp308255a
- A. Siokou, G. Leftheriotis, S. Papaefthimiou and P. Yianoulis, “Effect of the Tungsten and Molybdenum Oxidation States on the Thermal Coloration of Amorphous WO3 and MoO3 Films,” Surface Science, Vol. 482-485, No. 1, 2001, pp. 294-299. doi:10.1016/S0039-6028(01)00714-2
- C. Martin, I. Martin, V. Rives, B. Grzybowska and I. Gressel, “A FTIR Spectroscopy Study of Isopropanol Reactivity on Alkali-Metal-Doped MoO3/tio2 Catalysts,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, Vol. 52, No. 7, 1996, pp. 733-740. doi:10.1016/0584-8539(96)01665-0
- H. Pines, R. C. Olberg and V. N. Ipatieff, “Studies in the Terpene Series. VIII. Effect of Catalyst, Solvent and Temperature on the Dehydrogenation of Pinane and pMenthane,” Journal of the American Chemical Society, Vol. 70, No. 2, 1948, pp. 533-537. doi:10.1021/ja01182a031
- H. Al-Kandari, F. Al-Kharafi and A. Katrib, “Isomerization Reactions of n-Hexane on Partially Reduced MoO3/ TiO2,” Journal of Molecular Catalysis A: Chemical, Vol. 287, No.1-2, 2008, pp. 128-134. doi:10.1016/j.molcata.2008.03.007
- C. D. Wagner, W. M. Riggs, L. E. Davis and J. F. Moulder and G. E. Muilenberg, “Handbook of XPS,” PerkinElmer, Waltham, 1979.
- G. C. Bond, S. Flamerz and L. van Wijk, “Structure and Reactivity of Titania-Supported Molybdenum and Tungsten Oxides,” Catalysis Today, Vol. 1, No.1-2, 1987, pp. 229-243. doi:10.1016/0920-5861(87)80042-1
NOTES
*Corresponding author.