Modern Research in Catalysis
Vol.2 No.1(2013), Article ID:27278,8 pages DOI:10.4236/mrc.2013.21001
Au/CuOx-TiO2 Catalysts for CO Oxidation at Low Temperature
1Department of Mechatronic Engineering, Ta Hwa University of Science and Technology, Hsin-Chu, Chinese Taipei
2Department of Chemical and Materials Engineering, National Central University, Chung-Li, Chinese Taipei
Email: *ywchen@ncu.edu.tw
Received December 3, 2012; revised January 5, 2013; accepted January 13, 2013
Keywords: CO Oxidation; Gold Catalysts; Copper; Nanometal
ABSTRACT
A series of Au/CuOx-TiO2 with various Cu/Ti ratios were prepared. CuOx/TiO2 was prepared by incipient-wetness impregnation with aqueous solution of copper nitrate. Au catalysts were prepared by deposition-precipitation method at pH 7 and 338 K. The catalysts were characterized by inductively-coupled plasma-mass spectrometry, temperature programming reduction, X-ray diffraction, transmission electron microscopy, high-resolution transmission electron microscopy and X-ray photoelectron spectroscopy. The reaction was carried out in a fixed bed reactor with a feed containing 1% CO in air at WHSV of 120,000 mL/h·g. High gold dispersion and narrow size distribution was obtained. The addition of CuOx in Au/TiO2 enhanced the activity on CO oxidation significantly. CuOx was in amorphous state which could stabilize the Au nanoparticles. Cu was in Cu1+ state. Cu donated partial electrons to Au. The interactions among Au, Cu1+ and TiO2 account for the high catalytic activity for CO oxidation. The significant promotional effect of CuOx on CO oxidation at low temperature was demonstrated.
1. Introduction
Carbon monoxide is a toxic, colorless and tasteless gas. It can cause human being to die in the short time. When gold is deposited as nanoparticles on metal oxides, it exhibits surprisingly high catalytic activity for CO oxidation at a temperature as low as −173˚C. The activity of gold catalysts also depends on support, preparation method and condition. Haruta and coworkers [1-3] found the high activity of supported gold catalysts for lowtemperature CO oxidation. It is believed to occur on the metal-support interface. To improve the metal-support interaction, one can add a second metal with gold on support which oxygen can be adsorbed and activated easily. AuCu/SiO2 and AuCu/SBA-15 were reported [4,5] to be active for CO oxidation. However, Au and Cu were alloy and Cu was in metallic state in these studies. Copper oxide and supported copper oxides are known to be highly active for CO oxidation, however, only at elevated temperature (>573 K) [6]. CuOx/TiO2 samples could be oxidized to Cu2O by annealing at 473 K [7]. CO oxidation on Au catalysts has been extensively studied [1-3, 8-11]. CuOx was reported to be active for CO oxidation, but not active at room temperature.
In this study, low Au metal loading (0.7 wt%) was used. CuOx was added in Au/TiO2 catalyst to improve the metal-support interaction and catalytic activity for CO oxidation reaction. The effects of CuOx loading on the catalytic properties of Au/TiO2 was elucidated.
2. Experimental
2.1. Catalyst Preparation
Reagents used here were analytical grade. P25 TiO2 was obtained from Evonik-Degussa Company. CuOx-TiO2 was prepared by incipient-wetness impregnation method. Various contents of Cu(NO3)2 aqueous solutions were added into TiO2 powder under stirring. It was calcined at 473 K for 4 h. the temperature was not too high to have crystalline phase of CuO. Au was then added by deposition-precipitation technique. Au catalysts were prepared by deposition-precipitation (DP) method. An aqueous solution of HAuCl4 was added into the solution containing suspended CuOx-TiO2 support at a rate of 10 mL/min. The temperature of the solution was maintained at 338 K. 1 M NH4OH solution was used to adjust the pH value to 7. After aging for 2 h, the precipitate was filtered and washed with hot water until no chloride ions were detected. Finally, the sample was dried overnight in air at 80˚C, and then calcined at 453 K for 4 h. This temperature was high enough to reduce cationic Au to metallic Au, but not too high to cause aggregation of Au.
2.2. Characterization
The catalysts were characterized by inductively-coupled plasma-mass spectrometry (ICP-MS), X-ray diffraction (XRD), transmission electron microscopy (TEM), highresolution transmission electron microscopy (HRTEM), temperature programming reduction (TPR), and X-ray photoelectron spectroscopy (XPS).
The exact gold content was analyzed by ICP-MS (PE-SCIEX ELAN 6100 DRC. The cross flow pneumatic nebulizer and double pass scott type spray chamber was used to nebulize the samples. The solution was transferred by peristaltic pump, and used the nebulizer to nebulize the samples into spray chamber detected by DRC-ICP-MS. A CEM MDS-2000 (CEM, Matthews, NC, USA) microwave apparatus equipped with Teflon vessels was used to digest the powder samples.
XRD (Burker KAPPA APEX II) analysis was performed using a Siemens D500 powder diffract meter using CuKα1 radiation (0.15405 nm) at a voltage and current of 40 kV and 40 mA, respectively. The sample was scanned over the range of 2θ = 20˚ - 70˚ at a rate of 0.05˚/min to identify the crystalline structure. Samples for XRD were prepared as thin layers on a sample holder.
The morphologies and particle sizes of the samples were determined by TEM on a JEM-2000 EX II operated at 160 kV and HRTEM on a JEOL JEM-2010 operated at 160 kV. Initially, a small amount of sample was placed into the sample tube filled with a 95% methanol solution and after agitating under ultrasonic environment for 10 min, one drop of the dispersed slurry was dipped onto a carbon-coated copper mesh (300#) (Ted Pella Inc., CA, USA), and dried in an oven for 1 h. Images were recorded digitally with a Gatan slow scan camera (GIF). Based on the several images of TEM or HRTEM, more than 100 particles were counted and the size distribution graph was obtained.
The existence of interactions between copper and gold was proved by means of temperature-programmed reducetion TPR. 40 mg sample was put into U-shape tube, the gas flow rate was 50 ml/min, the composition of the reaction gas was 5 volume % H2 in Ar, the temperature ramp was 10 K/min, and analyzed by a gas chromatography equipped with TCD (China Chromatography 9800).
The XPS spectra were recorded with a Thermo VG Scientific Sigma Prob spectrometer. The XPS spectra were collected using AlKα radiation at a voltage and current of 20 kV and 30 mA, respectively. The base pressure in the analyzing chamber was maintained in the order of 10−7 Pa. The spectrometer was operated at 23.5 eV pass energy and the binding energy was corrected by contaminant carbon (C 1s = 284.5 eV) in order to facilitate the comparisons of the values among the catalysts and the standard compounds. Peak fitting was done using XPSPEAK 4.1 with Shirley background and 30:70 Lorentzian/Gaussian convolution product shapes. The fullwidth at half maximum (FWHM) in the entire spectra was 1.3 eV.
2.3. Catalytic Activity
The catalytic activities of CO oxidation in air were carried out in a downward, fixed-bed continuous-flow, pyrex glass-tubular reactor loaded with 0.05 g of catalyst. The reactant gas containing 1% CO in air was fed into reactor with a flow rate of 100 ml/min, (WHSV = 120,000 mL/h·g). The outlet gas was analyzed by a gas chromatograph (China Chromatography 8700T) equipped with a MS-5A column and a thermal conductivity detector. Calibration of the gases was done with a standard gas containing know concentration of the components. The CO conversion was calculated as follows: CO conversion,
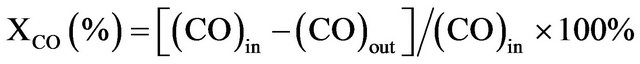 (1)
(1)
where (CO) is the concentration of CO.
3. Results and Discussion
3.1. ICP-MS
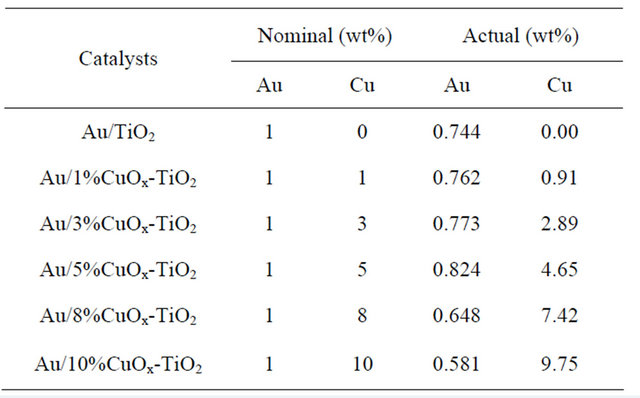
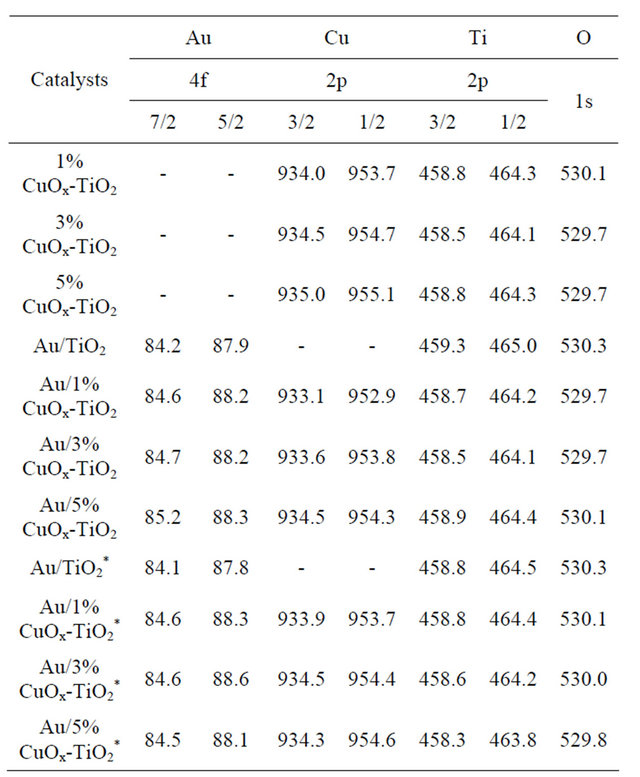
The ICP-MS results shown in Table 1 unfold the real amounts of gold and copper loadings in the catalysts. In this study, the nominal Au loading was 1 wt% and the Cu loadings were between 1 and 10 wt%. Only about 60% - 70% original Au in solution was deposited on the support by the DP method. The results are in agreement with literature data [1-3,8-13].
Most of Cu did not leach out during DP process. Au/5%CuOx-TiO2 catalyst had the highest Au loading among all catalysts, inferring that adding suitable amount of CuOx could change the surface properties of support and resulting in higher Au loading [12].
Table 1. Actual Au and Cu loadings in the catalysts.
3.2. XRD
Figure 1 shows the XRD patterns of Au/CuOx-TiO2. All catalysts containing TiO2 support showed intense XRD peaks for anatase phase 2θ = 25.23˚ (101), 37.72˚ (004), 47.89˚ (200), 53.77˚ (105) and 62.51˚ (204) and rutile phase 2θ = 27.45˚ (110), 36.10˚ (101) and 54.36˚ (211), as expected. The peaks at 2θ = 35.4˚, 38.7˚, 44.2˚ and 61.5˚ corresponding to CuO or Cu2O were not clearly observed in the XRD patterns. No distinct gold peaks at 2θ = 38.2˚ and 44.5˚ were observed, possibly because the particle size of gold particles was too small to be detected.
3.3. TEM
Gold catalysts have high activity on CO oxidation when the particle size of Au is less than 3 nm [1-3]. If the particle is larger than 5 nm, gold catalysts will lose its activeity. Figure 2 shows the TEM micrographs and the corresponding gold particle size distributions of various Au/CuOx-TiO2 catalysts. The TEM images clearly show that the average particle sizes of Au in these catalysts are around 2.1 - 2.6 nm. The gold particles were observed as dark spots and dispersed very well on the support.
The electron diffraction pattern of Au/5%CuOx-TiO2 catalyst is shown in Figure 1. Many diffraction rings were observed, which are corresponding to the crystal lattices of TiO2, CuO and Au. It proves the existence of these species in Au/5%CuOx-TiO2. They were not amorphous in Au/5%CuOx-TiO2.
3.4. HRTEM
The HRTEM images of Au/5%CuOx-TiO2 catalyst are shown in Figure 3. The values in parentheses are the standard lattice distances, and the others are measured values. The particles of Au and CuOx were very close and they were deposited on TiO2. The lattice fringe of CuO (111) was not regular, which was caused by the
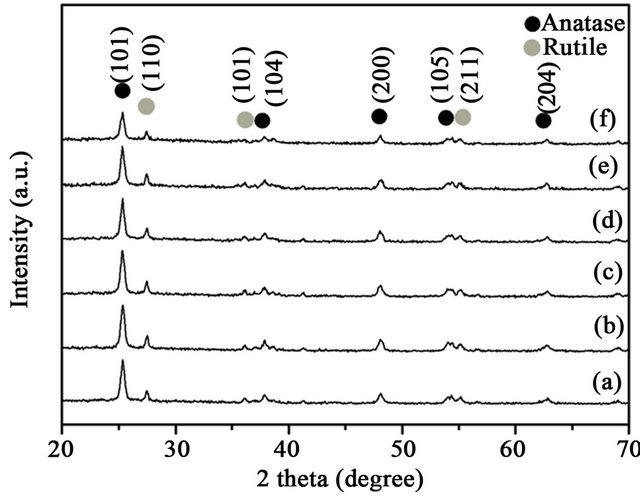
Figure 1. XRD of patterns of Au/CuOx-TiO2, (a) Au/TiO2; (b) Au/1%CuOx-TiO2; (c) Au/3%CuOx-TiO2; (d) Au/5% CuOx-TiO2; (e) Au/8%CuO-TiO2; (f) Au/10%CuOx-TiO2.
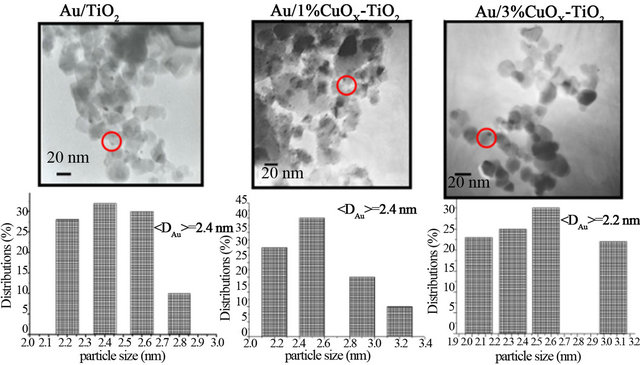
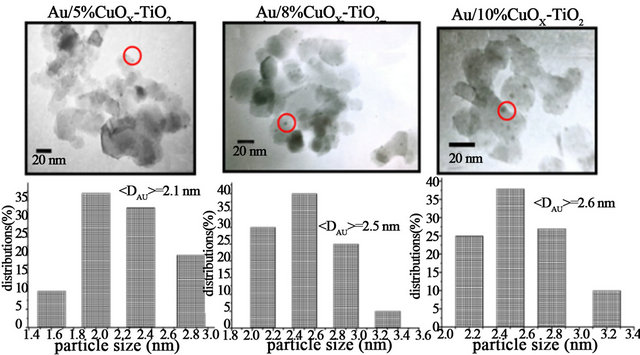
Figure 2. TEM images and particle size distributions of Au/CuOx-TiO2 catalysts.
strong interaction between CuO and TiO2 [6,14]. There was an interaction between Au, CuOx, and TiO2. It can be observe that the particle sizes of Au and CuO were very small. The Au particle size was about 2 nm, and CuO was about 4 nm. To compare TEM diffraction and HRTEM image, the diffraction rings were very close, and overlapped between Au (200) and TiO2 rutile (111). The distance of diffraction rings to central point was close to TiO2 rutile (111).
3.5. H2-TPR
The TPR profiles of the CuOx-TiO2 shown in Figure 4 are characterized by a single reduction peak for CuO at ~500 K [15]. The Tmax increased with the increase of copper content. After loading Au on CuOx-TiO2, the temperature of reduction for CuO decreased because the Au species could adsorb hydrogen and promote reduction of CuO. There was no reduction peak for AuO, because Au cation was reduced to Au by heating at 453 K. The peak corresponded to the reduction of CuO only. The peak area expressed the amount of H2 consumption. The peak areas were small when Au was deposited on CuOx-TiO2. The support of CuOx-TiO2 was calcined at 200˚C for 4 h and the Cu(NO3)2 on TiO2 was converted to CuO. CuO was converted to other copper oxide (CuOx), possibly Cu2O or Cu3O4 after depositing Au on the support, resulting in the less amount of H2 consumption on Au/CuOx-TiO2 than on CuO-TiO2.

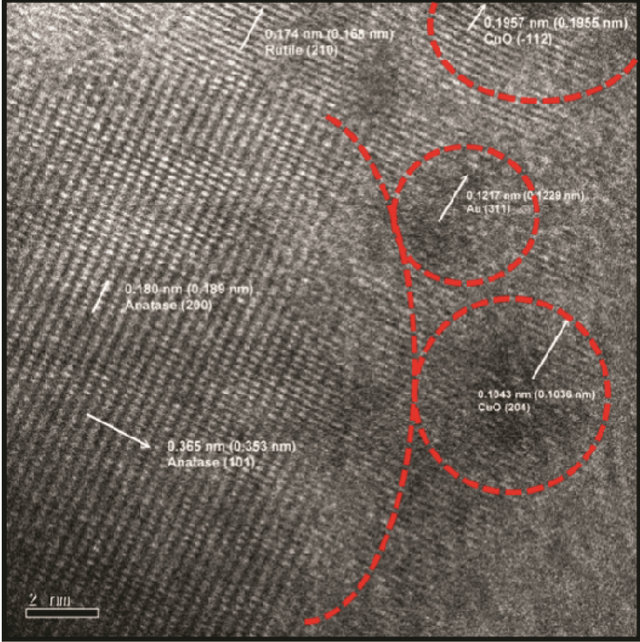
Figure 3. HRTEM image of Au/5% CuOx-TiO2.
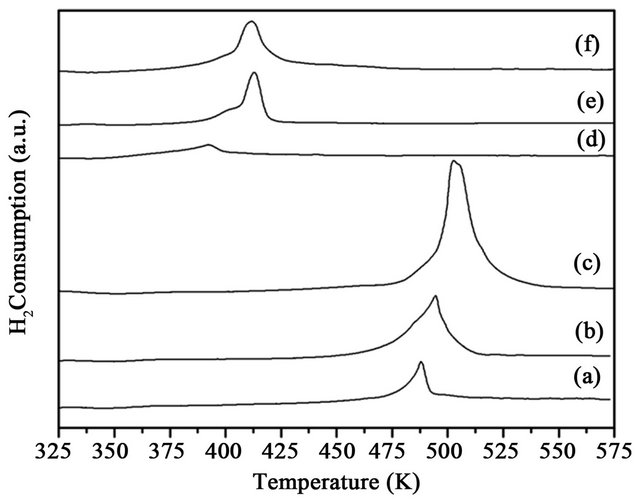
Figure 4. TPR profiles of the catalysts, (a) 1% CuOx-TiO2; (b) 3% CuOx-TiO2; (c) 5% CuOx-TiO2; (d) Au/1%CuOx- TiO2; (e) Au/3%CuOx-TiO2; (f) Au/5%CuOx-TiO2.
3.6. XPS
Electronic, structural and support effects have been considered, in turn, as the main requisites for an efficient gold catalyst. The XPS spectra of Au, Cu, Ti and O are presented in Figures 5-13, and the binding energies and the concentration of each species are tabulated in Tables 2 and 3. The Au particle size was considered to be the main factor. However, oxidized Au species has been suggested to be the active sites for CO oxidation [2,13]. Au 4f is characterized by the doublet of two spin orbit components, viz., Au 4f7/2 and Au 4f5/2. The binding energy of Au0 and Au+ in Au 4f7/2 was 84.0 and 85.5 eV [16]. The binding energy of Au 4f shifted to higher energy when CuOx was added in Au/TiO2 (Figure 5). The results indicate that Au supported on CuOx-TiO2 had strong metal-support interaction. The Au+ content increased with increasing the amount of CuOx in the catalyst. The presence of Au+ species has been reported to be effective in promoting the low temperature CO oxidation [13]. The XPS spectra for the samples after reaction
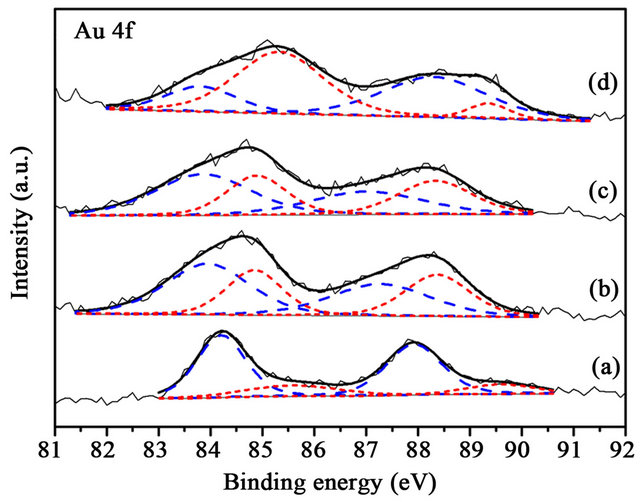
Figure 5. XPS Au 4f spectra of (a) Au/TiO2 (b) Au/1% CuOx-TiO2 (c) Au/3%CuOx-TiO2 (d) Au/5%CuOx-TiO2 before reaction. Au0 is the dash line, and Au+ is the shortdash line).
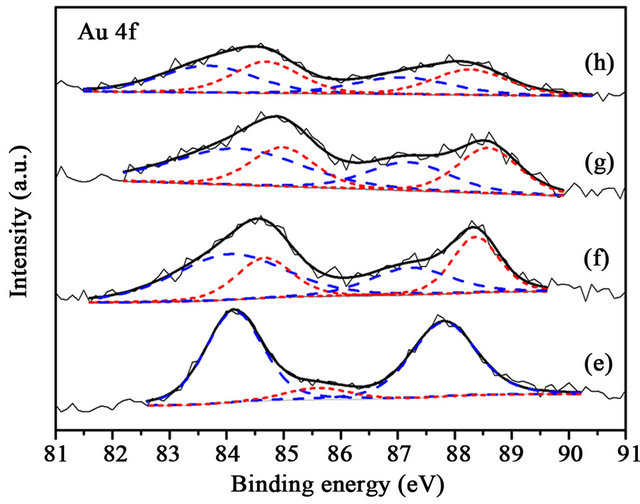
Figure 6. XPS Au 4f spectra of (a) Au/TiO2 (b) Au/1% CuOx-TiO2 (c) Au/3%CuOx-TiO2 (d) Au/5%CuOx-TiO2 after reaction. Au0 is the dash line, and Au+ is the shortdash line).
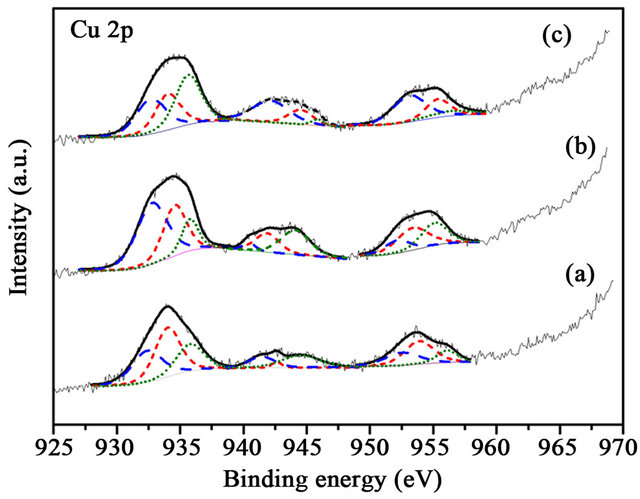
Figure 7. XPS Cu 2p spectra of (a) 1% CuOx-TiO2 (c) 3% CuOx-TiO2 (d) 5% CuOx-TiO2 (Cu0 is the dash line, and Cu+ is the short-dash line).
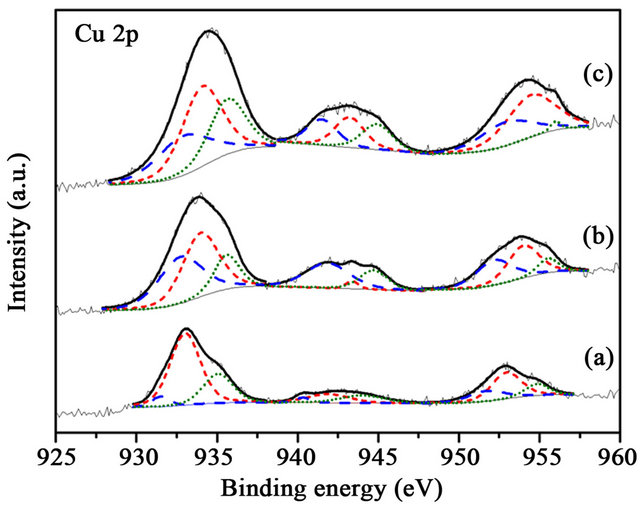
Figure 8. XPS Cu 2p spectra of the samples before reaction (a) Au/1%CuOx-TiO2; (c) Au/3%CuOx-TiO2; (d) Au/5% CuOx-TiO2 (Cu0 is the dash line, and Cu+ is the short-dash line.
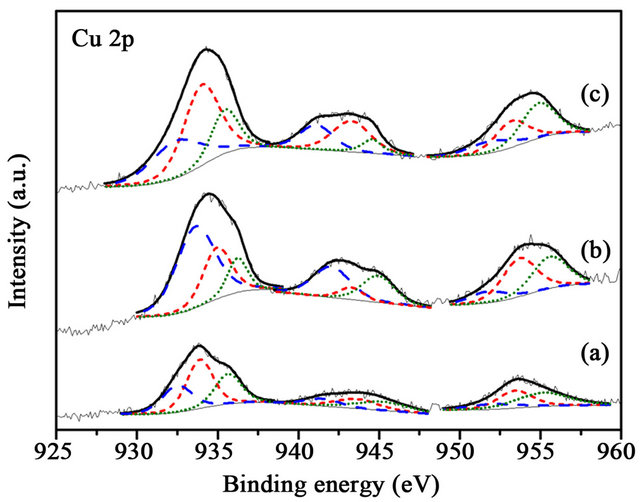
Figure 9. XPS Cu 2p spectra of the samples after reaction (a) Au/1%CuOx-TiO2; (c) Au/3%CuOx-TiO2; (d) Au/5% CuOx-TiO2 (Cu0 is the dash line, and Cu+ is the short-dash line.
(Figure 6) were similar with those before reaction. Only the content of Au+ in Au/TiO2 decreased after reaction, the content of Au+ in Au/CuOx-TiO2 samples did not change significantly after reaction. The results indicated that CuOx could stabilize the active sites in reaction.
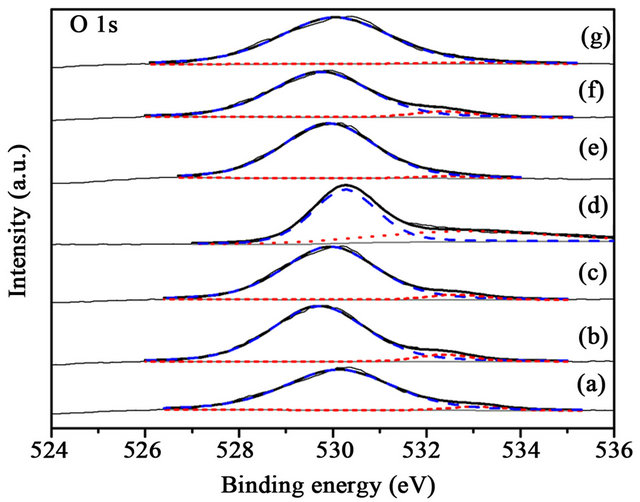
Figure 10. XPS O 1s spectra of the samples before reaction (a) 1% CuOx-TiO2; (b) 3% CuOx-TiO2; (c) 5% CuOx-TiO2; (d) Au/TiO2; (e) Au/1%CuOx-TiO2; (f) Au/3%CuOx-TiO2; (g) Au/5%CuOx-TiO2 (O2−is the dash line, and OH− is the short-dash line).
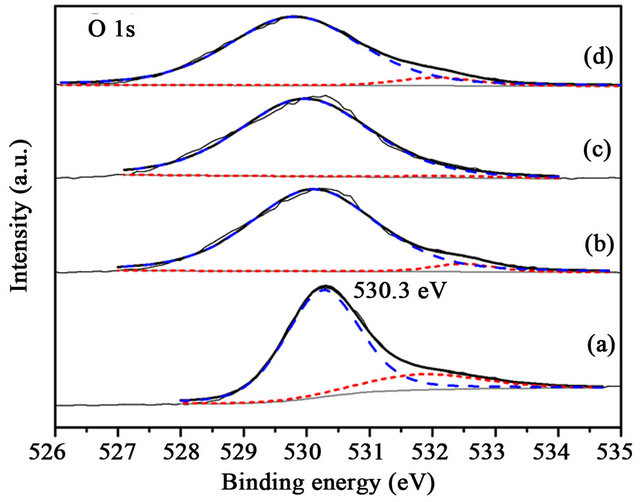
Figure 11. XPS O 1s spectra of the samples after reaction (a) Au/TiO2; (b) Au/1%CuOx-TiO2; (c) Au/3%CuOx-TiO2; (d) Au/5%CuOx-TiO2 (O2− is the dash line, and OH− is the short-dash line).
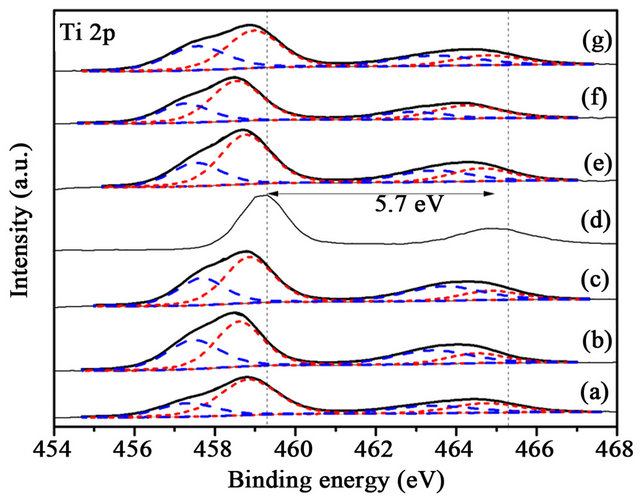
Figure 12. XPS Ti 2p spectra of the samples before reaction, (a) 1% CuOx-TiO2; (b) 3% CuOx-TiO2; (c) 5% CuOx-TiO2; (d) Au/TiO2; (e) Au/1%CuOx-TiO2; (f) Au/3%CuOx-TiO2; (g) Au/5%CuOx-TiO2 (Ti3+ is the dash line, and Ti4+ is the short-dash line).
Cu is an easily oxidized element, and the oxides of Cu are Cu2O and CuO. The binding energy of Cu, Cu2O, and
Table 2. The binding energies of various species on Au catalysts.
*after reaction.
Table 3. The concentrations of various species of the catalysts.
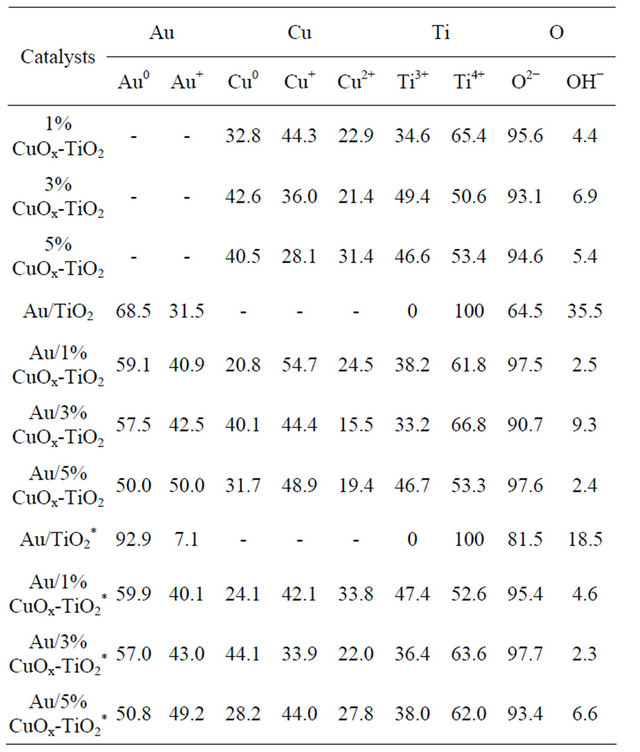
*after reaction.
CuO in Cu 2p3/2 was 932.2, 932.6, and 933.2 eV [13,16]. The Cu 2p was characterized by the doublet of two spin orbit components, viz., Cu 2p3/2 and Cu 2p1/2 (Figure 7). The content of Cu oxides did not have any correlation in these supports. Au was deposited on various supports. It can be observed (Figure 7) that the content of Cu+ was more than those of Cu0 and CuO2+ in these catalysts. CO-Cu+ interaction is much stronger than those of CO-Cu2+ and CO-Cu0, as the result, CO adsorbed on Cu+ are the main species.15 The Cu+ was reinforced the CO adsorbed on catalysts. The binding energy shifted to lower energy after depositing Au on supports. There was an interaction between Au and CuOx-TiO2. It has been reported [15] that much of the CO oxidation react with copper uses an inert gas, such N2, 1% - 2% CO, and 19% O2, which readily oxidizes the copper to CuO. Concentrations of O2 which result in a CO-O2 ratio lower than 2:1 will reduce the copper to Cu0. The change between before and after reaction (Figures 8 and 9) was not significant.
The binding energy of lattice oxygen in TiO2 was 529 eV [16], and the binding energy of OH- group in TiO2 was 531.8 eV [16], as shown in Figure 10. The TiO2 from Evonic-Degussa Company contained less OH- group. The amount of OH- decreased when adding CuOx. The binding energy in Au/CuOx-TiO2 catalysts was shifted to lower energy than that of Au/TiO2. There is only a slightly difference between before and after reaction (Figures 10 and 11).
Ti 2p is characterized by the doublet of two spin orbit components, viz., Ti 2p3/2 and Ti 2p1/2, as shown in Figure 12. The gap between Ti 2p3/2 and Ti 2p1/2 is 5 - 6 eV, it is ascribed to Ti4+ (458.9 eV) [17]. The Ti3+ (456.8 eV) [17] only exists in Au/CuOx-TiO2. The intrinsic oxygen vacancy existed in TiO2, and Cu added in TiO2 caused production of extrinsic oxygen vacancy. The binding energy and the content of Ti3+ and Ti4+ in catalysts was
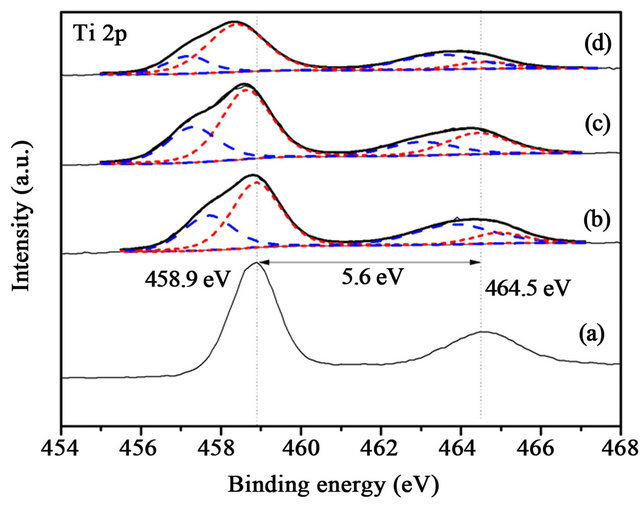
Figure 13. XPS Ti 2p spectra of the samples after reaction, (a) Au/TiO2; (b) Au/1%CuOx-TiO2; (c) Au/3%CuOx-TiO2; (d) Au/5%CuOx-TiO2 (Ti3+ is blue dash line, and Ti4+ is red dash line).
non-regular between the samples before and after reaction, as shown in Figures 12 and 13.
3.7. Catalytic Activity in CO Oxidation
Figure 14 shows the CO conversion on various catalysts and various reaction temperatures. The space velocity was very high WHSV = 120,000 mL/h·g. If the space velocity was 90,000 mL/h·g, the conversions were all 100%. It shows that all of the catalysts were very active even only 0.7 wt% Au was used. Figure 3 shows that adding small amount of Cu in Au/TiO2 enhanced the activity of the Au catalyst. The activity decreased if the amount of CuO was greater than 5 wt%. In this study, the best catalyst was Au/5% CuOx-TiO2, somewhat different from that obtained by Haruta [2] for Au/TiO2, because different amount of gold were used; i.e., 1 wt% Au/TiO2 in this study and 10 wt% Au/TiO2 in their study [2]. Copper has various oxidation states, such as Cu2O and CuO, but Cu2O was found to be more active than CuO for CO oxidation [18].
4. Conclusion
Au catalyst is well-known to be an active catalyst for CO oxidation. However, most of researchers used high Au loadings for this reaction. In this study, low Au metal loading was used. A series of CuOx-TiO2 supports were prepared by incipient-wetness impregnation method. Au/CuOx-TiO2 were prepared by DP method. The results showed small Au particles size (2.1 - 2.6 nm), narrow Au particle size distribution, and well Au dispersions on all catalysts. The Au/5%CuOx-TiO2 had the highest gold loading due to the change of isoelectric point of support by adding CuOx. Au, CuO, and TiO2 had interactions between each other. Au/CuOx-TiO2 had high activity because CuOx could stabilize the Au nanoparticles and Au+ species during reaction. Cu+ had strong ability to adsorb CO and Ti4+ was reduced to Ti3+ and created the oxygen vacancies. Both are beneficial for CO oxidation.
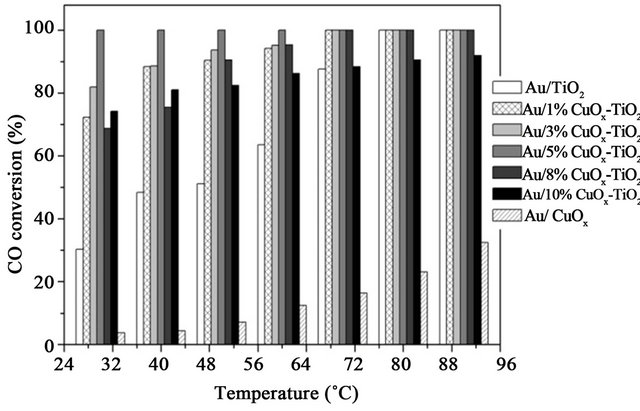
Figure 14. The CO conversion on Au/CuOx-TiO2 catalysts. (Reactant gas: 1% CO in air; WHSV=120,000 ml/h·g).
In summary, adding CuOx in Au/TiO2 significantly increased the activity on CO oxidation. Au/5%CuOx-TiO2 catalyst had the highest CO conversion at low temperature.
5. Acknowledgements
The authors thank the financial supports from Ministry of Economic Affairs, Chinese Taipei.
REFERENCES
- M. Haruta, “Sizeand Support-Dependency in the Catalysis of Gold,” Catalysis Today, Vol. 36, No. 1, 1997, pp. 153-166. doi:10.1016/S0920-5861(96)00208-8
- M. Haruta, “Gold as a Low-Temperature Oxidation Catalyst: Factors Controlling Activity and Selectivity,” Studies in Surface Science and Catalysis, Vol. 110, 1997, pp. 123-134. doi:10.1016/S0167-2991(97)80974-3
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, “Novel Gold Catalysts for the Oxidation of Carbon Monoxide at a Temperature Far Below 0 ˚C,” Chemistry Letters, Vol. 16, No. 2, 1987, pp. 405-408. doi:10.1246/cl.1987.405
- X. Liu, A. Wang, T. Zhang, D. S. Su and C. Y. Mou, “Au-Cu Alloy Nanoparticles Supported on Silica Gel as Catalyst for CO Oxidation: Effects of Au/Cu Ratios,” Catalysis Today, Vol. 160, No. 1, 2012, pp. 103-108. doi:10.1016/j.cattod.2010.05.019
- X. Liu, A. Wang, X. Wang, C. Y. Mou and T. Zhang, “Au-Cu Alloy Nanoparticles Confined in SBA-15 as a Highly Efficient Catalyst for CO Oxidation,” Chemical Communications, No. 27, 2008, pp. 3187-3189.
- B. Skarman, D. Grandjean, R. E. Benfield and A. Hinz, “Carbon Monoxide Oxidation on Nanostuctured CuOx/CeO2 Composite Particles Characterized by HREM, XPS, XAS and High Energy Diffraction,” Journal of Catalysis, Vol. 211, No. 1, 2002, pp. 119-133. doi:10.1006/jcat.2002.3735
- K. Y. Song, Y. T. Kwon, G. J. Choi and W. I. Lee, “Photocatalytic Activity of Cu/TiO2 with Oxidation State of Surface Loaded Copper,” Bulletin of the Korean Chemical Society, Vol. 20, No. 8, 1999, pp. 957-960.
- M. Haruta, “Nanoparticulate Gold Catalysts for LowTemperature CO Oxidation,” Journal of New Materials for Electrochemical Systems, Vol. 7, No. 3, 2004, pp. 163-172.
- K. Y. Ho and K. L. Yeung, “Effects of Ozone Pretreatment on the Performance of Au/TiO2 Catalyst for CO Oxidation Reaction,” Journal of Catalysis, Vol. 242, No. 1, 2006, pp. 131-141. doi:10.1016/j.jcat.2006.06.005
- M. M. Schubert, S. Hackenberg, A. C. Veen, M. Muhler, V. Plzak and R. J. Behm, “CO Oxidation Over Supported Gold Catalysts—‘Inert’ and ‘Active’ Support Materials and Their Role for the Oxygen Supply During Reaction,” Journal of Catalysis, Vol. 197, No. 1, 2001, pp. 113-122. doi:10.1006/jcat.2000.3069
- B. Schumacher, Y. Denkwitz, V. Plazk, M. Kinneand and R. J. Behm, “Kinetics, Mechanism, and the Influence of H2 on the CO Oxidation Reaction on a Au/TiO2 Catalyst,” Journal of Catalysis, Vol. 224, No. 2, 2004, pp. 449-462. doi:10.1016/j.jcat.2004.02.036
- L. H. Chang, Y. W. Chen and N. Sasirekha, “Preferential Oxidation of Carbon Monoxide in Hydrogen Stream Over Au/MgOx-TiO2 Catalysts,” Industrial & Engineering Chemistry Research, Vol. 47, No. 12, 2008, pp. 4098-4105. doi:10.1021/ie071590d
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, “XPS Study of Supported Gold Catalysts: The Role of Au0 and Au+δ Species as Active Sites,” Surface and Interface Analysis, Vol. 38, No. 4, 2006, pp. 215-218. doi:10.1002/sia.2180
- K. Yan, Z. Yanhua, W. Xiaoshu, W. Jun, W. Haiqin, D. Lin and Y. Qijie, “Catalytic Performance of Cu-MCM-41 with High Copper for NO Reduction by CO,” Studies in Surface Science and Catalysis, Vol. 165, 2007, pp. 749- 753. doi:10.1016/S0167-2991(07)80429-0
- G. G. Jernigan and G. A. Somorjai, “Carbon Monoxide Oxidation over Three Different Oxidation States of Copper: Metallic Copper, Copper (I) Oxide, and Copper (II) oxide—A Surface Science and Kinetic Study,” Journal of Catalysis, Vol. 147, No. 2, 1994, pp. 567-577. doi:10.1006/jcat.1994.1173
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. E. Bomben, “Handbook of X-Ray Photoelectron Spectroscopy,” Physical Electronics, 1995.
- D. Gonbeau, C. Guimon, G. Pfister-Guillouzo, A. Levasseur, G. Meunier and R. Dormoy, “XPS Study of Thin Films of Titanium Oxysulfides,” Surface Science, Vol. 254, No. 1-3, 1991, pp. 81-89. doi:10.1016/0039-6028(91)90640-E
- T. J. Huang, D. H. Tsai, “CO Oxidation Behavior of Copper and Copper Oxides,” Catalysis Letters, Vol. 87, No. 3-4, 2003, pp. 173-178. doi:10.1023/A:1023495223738
NOTES
*Corresponding author.