Pharmacology & Pharmacy
Vol.10 No.04(2019), Article ID:91772,10 pages
10.4236/pp.2019.104014
The Effects of Rufinamide on in Vitro Spinal Muscular Atrophy Model
Shiori Ando1,2*, Arisu Sato1,2*, Michinori Funato2, Kazuki Ohuchi1,2, Satoshi Inagaki1,2, Shinsuke Nakamura1, Masamitsu Shimazawa1#, Hideo Kaneko2, Hideaki Hara1
1Molecular Pharmacology, Department of Biofunctional Evaluation, Gifu Pharmaceutical University, Gifu, Japan
2Department of Clinical Research, National Hospital Organization, Nagara Medical Center, Gifu, Japan

Copyright © 2019 by author(s) and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: March 1, 2019; Accepted: April 12, 2019; Published: April 15, 2019
ABSTRACT
Spinal muscular atrophy (SMA) is devastating genetic disease characterized by progressive loss of motor neuron and skeletal muscle weakness. SMA is the most common lethal genetic disease in infancy. SMA is caused by deletion or mutation of SMN1 gene and subsequent lack of SMN protein. Our purpose in this study was to evaluate the therapeutic potential of rufinamide, an antiepileptic drug. In this study, SMA patient-derived fibroblasts and differentiated spinal motor neurons (MNs) using SMA patient-derived iPSCs were used as in vitro SMA model. SMN mRNA was significantly increased by addition of rufinamide in type III SMA patient-derived fibroblasts. Furthermore, rufinamide stimulated neurite elongation in type III SMA patient derived-iPSCs-MNs. In contrast of the result using type III SMA patient-derived fibroblasts, the expression level of SMN mRNA was not changed after rufinamide treatment in type I SMA patient-derived fibroblasts, and rufinamide did not affect neurite outgrowth in type I SMA patients derived-iPSCs-MNs. These findings indicate that rufinamide may be one of the potential candidate drugs for mild type of SMA.
Keywords:
Spinal Muscular Atrophy, Rufinamide, iPSCs

1. Introduction
Spinal muscular atrophy (SMA) is autosomal recessive neurodegenerative disease characterized by progressive loss of motor neuron and skeletal muscle weakness [1] . SMA is caused by deletion or mutation of SMN1 (survival motor neuron 1) gene and subsequent lack of SMN protein [2] . SMN protein is encoded by SMN1 and SMN2 gene in human. SMN2 gene is highly homologous gene of SMN2 gene, but has a C to T transition compared to SMN1 gene [3] . This transition results in production of unstable form of SMN protein. Therefore, deletion and mutation of SMN1 gene cause a low expression level of SMN protein.
SMA patients are approximately 1 in 6000 - 10,000 births [4] . Clinically, SMA is classified into 4 types based on the onset and severity: type I (severe form; onset within the first six months), type II (intermediate form; onset before the age of three years), type III (milder form; onset after the age of three years), and type IV with adult onset [5] . SMA is one of the devastating neuromuscular disorders. Without invasive ventilation, many of type I SMA patients cannot survive in infancy. Type III SMA patients’ life-expectancy is comparable to normal lifespan. Recently, nusinersen, an antisense oligonucleotide which increases SMN protein level by modulating the splicing of the SMN2 mRNA transcript, has been approved as the first drug for SMA treatment [6] . However, nusinersen is available for intrathecal injection, which could be invasive and distressing for patients.
Rufinamide is used for treatment of Lennox-Gastaut syndrome, one of the most severe types of childhood-onset epilepsy, by oral administration. It modulates sodium-dependent neuronal refractory period [7] . Previous study demonstrated that rufinamide had neuroprotective effects on the kainic acid-induced excitotoxic neuronal death in the mouse hippocampus [8] . However, the effect of rufinamide for neurodegenerative disease is unknown.
Several studies have shown that antiepileptic drug is effective for SMA. Some clinical trials demonstrated potential benefit of valproic acid for SMA treatment [9] [10] . Valproic acid increased SMN protein [11] . Another trial revealed gabapentin improved muscle weakness of type II and III SMA patients [12] . Therefore, we hypothesized that rufinamide is effective for SMA treatment.
The purpose of this study was to clarify the effect of rufinamide on SMA pathology. To investigate the effect of rufinamide, SMA patients-derived fibroblasts and spinal motor neuron derived from SMA-iPSCs were used. In this study, we evaluated the effect of rufinamide on SMN mRNA and neural elongation.
2. Materials and Methods
2.1. Ethics Statement
The pathological analysis and establishment of patient-derived cells were approved by the Ethics Review Committee of the National Hospital Organization, Nagara Medical Center (Approval number: 26-15), and informed consents were obtained from the parents of our pediatric patients. Informed consents were obtained from the subjects after explanation of the nature and possible consequences of the study.
2.2. Fibroblasts Culture and Drug Assays
Fibroblasts taken from two SMA type III patients (SMA01 and SMA05) and three SMA type I patients (SMA06, SMA08 and SMA12) were used in the present study. The control human fibroblast cell line TIG-119 was obtained from the National Institutes of Biomedical Innovation, Health and Nutrition (Osaka, Japan). TIG-119 and fibroblasts derived from SMA patients were maintained in Dulbecco’s modified Eagle medium (Life Technologies, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS, biowest, Nuaillé, France) and 500 U/mL penicillin/streptomycin (PS, Life Technologies) under a humidfied atmosphere of 5% CO2 at 37˚C. These cells were passaged by trypsinization every 2 - 3 days.
Fibroblasts were seeded at a density of 100,000 cells per well into 6 well plate, and incubated in 5% CO2 at 37˚C. After 24 h incubation, cells were treated by rufinamide (Tokyo chemical industry, Tokyo, Japan) for 24 h.
2.3. iPSCs Culture, Spinal Motor Neuron Differentiation and Drug Assays
The iPSCs derived from an SMA type III patient (SMA01) and an SMA type I patient (SMA06) were used in this study. The iPSCs were maintained in primate ES cell medium (ReproCELL, Kanagawa, Japan) supplemented with 4 ng/mL basic fibroblast growth factor (Wako, Osaka, Japan) and 500 U/mL PS (Life Technologies). The iPSC colonies were cultured in 5% CO2 at 37˚C and passaged every 7 days.
For spinal motor neuron differentiation from iPSCs, 9000 single iPSCs were seeded into a primary differentiation medium containing Dulbecco’s modified Eagle medium (DMEM)/F12 (Life Technologies), 5% Knockout Serum Replacement (KSR, Life Technologies), and 500 U/mL PS with 2 μM dorsomorphin (Sigma-Aldrich, St. Louis, MO, USA), 10 μM SB431542 (SB, Cayman, San Diego, CA, USA), and 10 μM Rho-associated coiled-coil forming kinase inhibitor, Y-27632 (Wako). The medium was changed the medium supplemented with 2 μM dorsomorphin and 10 μM SB431542 after 3 days. Spheres were then plated in Matrigel-coated 24-well plates (Becton, Dickinson and Company, NJ, USA) in medium supplemented with 2 μM dorsomorphin and 10 μM SB431542 for another 7 days. The neural precursor cells were then cultured in the second differentiation medium containing DMEM/F12, 1% N2 supplement (Life Technologies), and 500 U/mL PS with 0.1 μM retinoic acid (RA, Sigma Aldrich). The cells were then cultured in the third differentiation medium with 1 μM purmorphamine (PMN, MiltenyiBiotec, Bergisch, Gladbach, Germany) and 0.1 μM RA for 7 days. Finally, the generated spinal motor neurons were cultured in the supplemented medium containing 10 ng/mL brain-derived neurotrophic factor (BDNF, R & D Systems Inc., Minneapolis, MN, USA), 10 ng/mL glial cell line-derived neurotrophic factor (GDNF, R&D), 1 μM cyclic adenosine monophosphate (cAMP, Wako) and 200 ng/ml ascorbic acid (AA, Sigma-Aldrich). During all differentiation stages, medium was changed every 2 or 3 days. The differentiated spinal motor neurons were exposed to rufinamide for 42 to 56 days of our induction protocol by changing the medium every 3 days.
2.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
RNA purification was performed using RNeasy Mini Kit (QIAGEN, Hilden, Germany). The concentration of RNA extracted was determined spectrophotometrically in a NanoView Plus (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). First-strand cDNA was synthesized from 1 µg of total RNA using ReverTra Ace (Toyobo, Osaka, Japan). The cDNA samples were subjected to PCR amplification using a thermal cycler 2720 (Applied Biosystems, Carlsbad, CA, USA).
The PCR assay was performed using power SYBR green PCR master mix (Applied Biosystems) containing 250 nM of SMN primer. The SMN primer sequences were described in the previous report [13] . Amplification of SMN mRNA was performed as follows; 50˚C for 2 min, 95˚C for 10 min, followed by 45 cycles of 95˚C for 15 s and 60˚C for 1 min. The amplification of HB9 was performed as follows; 95˚C for 10 min, followed by 40 cycles of 95˚C for 15 s, and 60˚C for 1 min. The samples were run using the comparative CT (ΔΔCT) method. The GAPDH transcript levels were determined as reference genes for qPCR.
2.5. Immunocytochemistry
Plated cells were washed with PBS three times. Cells were fixed in 4% paraformaldehyde (NacalaiTesque, Kyoto, Japan) for 20 min at 4˚C and then washed again with PBS three times. Cells were blocked with 5% donkey serum in PBS for 30 min at room temperature, and incubated with primary antibodies for 24 h at 4˚C. After washing two times with PBS, the cells were labelled with the appropriate secondary antibody-tagged fluorescent dye for 1 h. Nuclear staining was performed using Hoechst 33342 (diluted 1:1000; Life Technologies). The following primary antibodies were used: rabbit anti-TUJ1 (βIII-tubulin) antibody (diluted 1:1000; BioLegend, San Diego, CA, USA), the secondary Alexa Fluor-labeled antibodies were 488 donkey anti-rabbit IgG (diluted 1:1000; Life Technologies). All images were taken by BIOREVO BZ-9000 (Keyence, Osaka, Japan).
2.6. Statistical Analysis
Data are presented as mean ± S.E.M. Statistical comparisons were performed using Student’s t-test. P values less than 0.05 were considered statistically significant. All statistical analyses were performed using SPSS Statistics (IBM, Armonk, NY, USA).
3. Results
3.1. The Effect of Rufinamide on Type III SMA Patient-Derived Fibroblasts
In SMA pathology, systemic deletion of SMN protein is observed. Fibroblasts derived from SMA patients have often been used for drug discovery research for SMA. To investigate the effect of rufinamide on SMN mRNA expression, we used type III SMA patient-derived fibroblasts (SMA01 and SMA05). Each patient has 3 copies of SMN2 gene. TIG-119 was used as the control human fibroblasts. Type III SMA patients-derived fibroblasts were treated by 5 µM rufinamide for 24 h. SMN mRNA was significantly increased by addition of rufinamide in type III SMA patient-derived fibroblasts (Figure 1).
3.2. The Effect of Rufinamide on Type I SMA Patient-Derived Fibroblasts
We then determined the effect of rufinamide on SMN mRNA in more severe disease class, type I SMA patient-derived fibroblasts (SMA06, SMA08 and SMA12). Each patient has two copies of SMN2 gene. In contrast of the result using type III SMA patient-derived fibroblasts, the expression level of SMN mRNA was not changed after rufinamide treatment in type I SMA patient-derived fibroblasts (Figure 2).
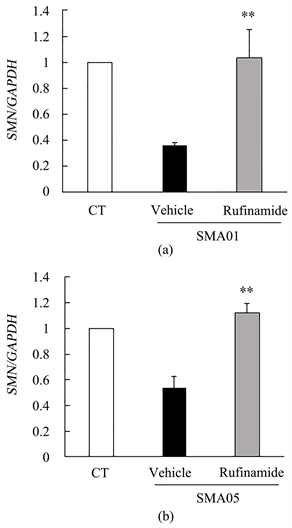
Figure 1. SMN mRNA was increased by rufinamide addition in type III SMA patient-derived fibroblasts. (a) Quantitative measurements of SMN mRNA in SMA 01. TIG-119 was used as the control human fibroblasts. Data are mean ± S.E.M. (n = 3). **p < 0.01 versus vehicle group (Student’s t-test). (b) Quantitative measurements of SMN mRNA in SMA 05. TIG-119 was used as the control human fibroblasts. Data are mean ± S.E.M. (n = 3). **p < 0.01 versus vehicle group (Student’s t-test). CT; control.

Figure 2. The expression level of SMN mRNA was not changed after rufinamide treatment in type I SMA patient-derived fibroblasts. (a) Quantitative measurements of SMN mRNA in SMA06. TIG-119 was used as the control human fibroblasts. Data are mean ± S.E.M. (n = 3, Student’s t-test). (b) Quantitative measurements of SMN mRNA in SMA08. TIG-119 was used as the control human fibroblasts. Data are mean ± S.E.M. (n = 3, Student’s t-test). (c) Quantitative measurements of SMN mRNA in SMA12. TIG-119 was used as the control human fibroblasts. Data are mean ± S.E.M. (n = 3, Student’s t-test). CT; control.
3.3. The Effect of Rufinamide on SMA-iPSCs Derived Spinal Motor Neuron
Degeneration of spinal motor neuron is the most distinctive feature of SMA. To evaluate the potential efficacy of rufinamide on spinal motor neuron, we used spinal motor neurons differentiated from SMA patients-derived iPSCs. SMA-iPSCs derived-spinal motor neuron (SMA-iPSCs-MNs) was treated by rufinamide for 2 weeks, then neurite outgrowth was evaluated to determine the effect of rufinamide. To evaluate neurite length, we performed immunostaining using anti-TUJ1 antibody. In type III SMA patient derived-iPSCs-MNs (SMA 01), rufinamide stimulated neurite elongation (Figure 3(a) and Figure 3(b)). On the other hand, rufinamide did not affect neurite outgrowth in type I SMA patients derived-iPSCs-MNs (SMA 06) (Figure 3(c)).
4. Discussion
In the present study, we first showed that rufinamide increased SMN mRNA in type III SMA patient-derived fibroblasts. Increasing SMN mRNA is effective strategy for SMA treatment. Nusinersen, the first approved drug for SMA, increases SMN mRNA by modulating the splicing of the SMN pre-mRNA transcript [14] [15] . Another candidate drug valproic acid also increases SMN mRNA in SMA patients derived fibroblasts [16] . Therefore, rufinamide may be effective for type III SMA patient.
On the other hand, the expression level of SMN mRNA was not changed after rufinamide treatment in type I SMA patient-derived fibroblasts. We consider that the reason of responsive difference between type III and type I patient-derived fibroblasts is the number of SMN2 gene copy. Each type III SMA patient in this study has 3 copies of SMN2 gene, and type I SMA patients have 2 copies. These findings indicate that rufinamide activated transcription of SMN2
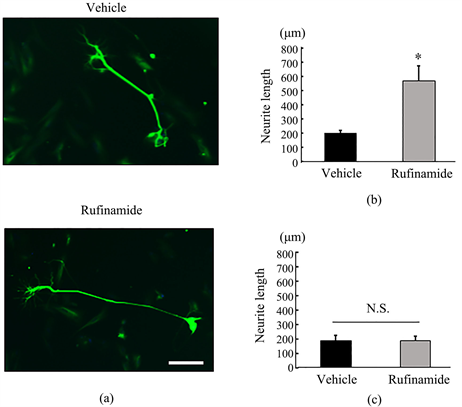
Figure 3. Rufinamide stimulated neurite elongation in SMA-iPSCs-MNs. (a) Representative images of SMA-iPSCs-MNs neurite (SMA 01) after 14 days vehicle or rufinamide addition. A scale bar is 200 µm. (b) Quantitative measurements of neurite length of SMA-iPSCs-MNs (SMA 01). Data are mean ± S.E.M. (n = 4 or 5). *p < 0.05 versus vehicle group (Student’s t-test). (c) Quantitative measurements of neurite length of SMA-iPSCs-MNs after 14 days vehicle or 5 µM rufinamide addition (SMA 06). Data are means ± S.E.M. (n = 4 or 5).
gene, therefore cells that have high copy number of SMN2 gene is more sensitive for rufinamide. Other factors which may affect the sensitivity for rufinamide are splicing factors which are related to SMN2 exon 7 splicing. The previous report showed that that high expression level of Tra2-β1 is responsible for increased SMN2 exon 7 inclusion [17] . Therefore, individual difference of expression level of splicing factors including Tra2-β1 may affect the sensitivity for rufinamide. On the other hand, the relationship between Tra-b1 and rufinamide is unknown. Therefore, further investigations are needed for the well understanding the sensitivity for rufinamide.
Next, we examined the effect of rufinamide on SMA-iPSCs-MNs. In this study, rufinamide at 5 µM was used. The mean plasma concentration of rufinamide between 1 and 9 within 12 h after administration is about 70 µM, and rufinamide shows high intracerebral transferability [18] [19] . Therefore, we thought that rufinamide would be suitable for SMA treatment. We determined 5 µM as the valid and effective concentration. In this study, the effect of rufinamide on the length of neurite was determined, because the neurite length reflects neural survival and function. In accord with the result in fibroblasts, rufinamide stimulated neurite elongation in type III SMA patient derived-iPSCs-MNs. On the other hand, rufinamide did not affect the neurite outgrowth in type I SMA patients derived-iPSCs-MNs. These results suggest that rufinamide has a protective effect on type III SMA patient derived-iPSCs-MNs, but did not affect in type I SMA patients derived-iPSCs-MNs. Considering the copy number of SMN2 gene, high copy number of SMN2 gene is more sensitive factor for rufinamide. SMN mRNA plays an important role in neural elongation in motor neurons [20] . Taken together, these findings suggest that the effect of rufinamide on neural elongation is related to SMN mRNA.
Rufinamide shows an antiepileptic effect by modulating sodium-dependent neuronal refractory period. Liu et al. reported that SMA patient-derived motor neurons exhibit enhanced sodium-channel activities which lead to hyperexcitability [21] . Therefore, the neuroprotective effect of rufinamide on SMA-iPSCs may be partially due to modulating sodium dependent neuronal activity.
5. Conclusion
In conclusion, rufinamide may be one of the potential candidate drugs for mild type of SMA.
Acknowledgements
We greatly appreciate the patients and their family who cooperated with this study. We thank for Dr. Toshio Saito (Toneyama National Hospital) and Dr. Hisahide Nishio (Kobe Gakuin University) for providing the SMA patients’ fibroblasts. We received generous technical support from Junko Seki and Chizuru Kawase (Nagara Medical Center).
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this paper.
Cite this paper
Ando, S., Sato, A., Funato, M., Ohuchi, K., Inagaki, S., Nakamura, S., Shimazawa, M., Kaneko, H. and Hara, H. (2019) The Effects of Rufinamide on in Vitro Spinal Muscular Atrophy Model. Pharmacology & Pharmacy, 10, 159-168. https://doi.org/10.4236/pp.2019.104014
References
- 1. Crawford, T.O. and Pardo, C.A. (1996) The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiology of Disease, 3, 97-110. https://doi.org/10.1006/nbdi.1996.0010
- 2. Lefebvre, S., Bürglen, L., Reboullet, S., Clermont, O., Burlet, P., Viollet, L., Benichou, B., Cruaud, C., Millasseau, P., Zeviani, M., et al. (1995) Identification and Characterization of a Spinal Muscular Atrophy-Determining Gene. Cell, 80, 155-165. https://doi.org/10.1016/0092-8674(95)90460-3
- 3. Lorson, C.L., Hahnen, E., Androphy, E.J. and Wirth, B. (1999) A Single Nucleotide in the SMN Gene Regulates Splicing and is Responsible for Spinal Muscular Atrophy. Proceedings of the National Academy of Sciences of the United States of America, 96, 6307-6311.
- 4. Prior, T.W., Snyder, P.J., Rink, B.D., Pearl, D.K., Pyatt, R.E., Mihal, D.C., Conlan, T., Schmalz, B., Montgomery, L., Ziegler, K., Noonan, C., Hashimoto, S. and Garner, S. (2010) Newborn and Carrier Screening for Spinal Muscular Atrophy. American Journal of Medical Genetics Part A, 152A, 1608-1616. https://doi.org/10.1002/ajmg.a.33474
- 5. Zerres, K., Wirth, B. and Rudnik-Schoneborn, S. (1997) Spinal Muscular Atrophy—Clinical and Genetic Correlations. Neuromuscular Disorders, 7, 202-207.
- 6. Finkel, R.S., Chiriboga, C.A., Vajsar, J., Day, J.W., Montes, J., De Vivo, D.C., et al. (2016) Treatment of Infantile-Onset Spinal Muscular Atrophy with Nusinersen: A Phase 2, Open-Label, Dose-Escalation Study. The Lancet, 388, 3017-3026. https://doi.org/10.1016/S0140-6736(16)31408-8
- 7. Gresham, J., Eiland, L.S. and Chung, A.M. (2010) Treating Lennox-Gastaut Syndrome in Epileptic Pediatric Patients with Third Generation Rufinamide. Neuropsychiatric Disease and Treatment, 6, 639-645.
- 8. Park, J.A. and Lee, C.H. (2018) Effect of Rufinamide on the Kainic Acid-Induced Excitotoxic Neuronal Death in the Mouse Hippocampus. Archives of Pharmacal Research, 41, 776-783. https://doi.org/10.1007/s12272-018-1043-1
- 9. Swoboda, K.J., Scott, C.B., Reyna, S.P., Prior, T.W., LaSalle, B., Sorenson, S.L., et al. (2009) Phase II Open Label Study of Valproic Acid in Spinal Muscular Atrophy. PLoS ONE, 4, e5268. https://doi.org/10.1371/journal.pone.0005268
- 10. Darbar, I.A., Plaggert, P.G., Resende, M.B., Zanoteli, E. and Reed, U.C. (2011) Evaluation of Muscle Strength and Motor Abilities in Children with Type II and III Spinal Muscle Atrophy Treated with Valproic Acid. BMC Neurology, 21, 686.
- 11. Brichta, L., Hofmann, Y., Hahnen, E., Siebzehnrubl, F.A., Raschke, H., Blumcke, I., et al. (2003) Valproic Acid Increases the SMN2 Protein Level: A Well-Known Drug as a Potential Therapy for Spinal Muscular Atrophy. Human Molecular Genetics, 12, 2481-2489. https://doi.org/10.1093/hmg/ddg256
- 12. Merlini, L., Solari, A., Vita, G., Bertini, E., Minetti, C., Mongini, T., Mazzoni, E., Angelini, C. and Morandi, L. (2003) Role of Gabapentin in Spinal Muscular Atrophy: Results of a Multicenter, Randomized Italian Study. Journal of Child Neurology, 18, 537-541. https://doi.org/10.1177/08830738030180080501
- 13. Ohuchi, K., Funato, M., Kato, Z., Seki, J., Kawase, C., Tamai, Y., Ono, Y., Nagahara, Y., Noda, Y., Kameyama, T., Ando, S., Tsuruma, K., Shimazawa, M., Hara, H. and Kaneko H. (2016) Established Stem Cell Model of Spinal Muscular Atrophy Is Applicable in the Evaluation of the Efficacy of Thyrotropin-Releasing Hormone Analog. STEM CELLS Translational Medicine, 5, 152-163. https://doi.org/10.5966/sctm.2015-0059
- 14. Hua, Y., Vickers, T.A., Okunola, H.L., Bennett, C.F. and Krainer, A.R. (2008) Antisense Masking of an hnRNP A1/A2 Intronic Splicing Silencer Corrects SMN2 Splicing in Transgenic Mice. American Journal of Human Genetics, 82, 834-848. https://doi.org/10.1016/j.ajhg.2008.01.014
- 15. Hua, Y., Sahashi, K., Rigo, F., Hung, G., Horev, G., Bennett, C.F. and Krainer, A.R. (2011) Peripheral SMN Restoration is Essential for Long-Term Rescue of a Severe Spinal Muscular Atrophy Mouse Model. Nature, 478, 123-126. https://doi.org/10.1038/nature10485
- 16. Sumner, C.J., Huynh, T.N., Markowitz, J.A., Perhac, J.S., Hill, B., Coovert, D.D. Schussler, K., Chen, X., Jarecki, J., Burghes, A.H., Taylor, J.P. and Fischbeck, K.H. (2003) Valproic Acid Increases SMN Levels in Spinal Muscular Atrophy Patient Cells. Annals of Neurology, 54, 647-654. https://doi.org/10.1002/ana.10743
- 17. Chen, Y.C., Chang, J.G., Jong, Y.J., Liu, T.Y. and You, C.Y. (2015) High Expression Level of Tra2-β1 Is Responsible for Increased SMN2 Exon 7 Inclusion in the Testis of SMA Mice. PLoS ONE, 10, e0120721. https://doi.org/10.1371/journal.pone.0120721
- 18. Ohtsuka, Y., Yoshinaga, H., Shirasaka, Y., Takayama, R., Takano, H. and Iyoda, K. (2014) Rufinamide as an Adjunctive Therapy for Lennox-Gastaut Syndrome: A Randomized Double-Blind Placebo-Controlled Trial in Japan. Epilepsy research, 108, 1627-1636. https://doi.org/10.1016/j.eplepsyres.2014.08.019
- 19. Gáll, Z., Vancea, S., Szilágyi, T., Gáll, O. and Kolcsár, M. (2015) Dose-Dependent Pharmacokinetics and Brain Penetration of Rufinamide Following Intravenous and Oral Administration to Rats. European Journal of Pharmaceutical Sciences, 68, 106-113. https://doi.org/10.1016/j.ejps.2014.12.012
- 20. Rathod, R., Havlicek, S., Frank, N., Blum, R. and Sendtner, M. (2012) Laminin Induced Local Axonal Translation of β-Actin mRNA Is Impaired in SMN-Deficient Motoneurons. Histochemistry and Cell Biology, 138, 737-748. https://doi.org/10.1007/s00418-012-0989-1
- 21. Liu, H., Lu, J., Chen, H., Du, Z., Li, X.J. and Zhang, S.C. (2015) Spinal Muscular Atrophy Patient-Derived Motor Neurons Exhibit Hyperexcitability. Scientific Reports, 5, Article No. 12189. https://doi.org/10.1038/srep12189
NOTES
*Contributed equally.