Open Journal of Apoptosis
Vol.1 No.1(2012), Article ID:18750,8 pages DOI:10.4236/ojapo.2012.11001
Parkinson’s Disease—Apoptosis and Dopamine Oxidation
School of Pharmacy, University of Southern California, Los Angeles, USA
Email: jadams@usc.edu
Received March 2, 2012; revised April 3, 2012; accepted April 13, 2012
Keywords: Tyrosine Hydroxylase; Monoamine Oxidase; Aldehyde Dehydrogenase
ABSTRACT
Tyrosine hydroxylase, monoamine oxidase and aldehyde dehydrogenase all form oxygen radicals as part of their mechanisms of action. These oxygen radicals damage dopaminergic neurons in the substantianigra of the midbrain and cause them to die by a process of necrosis or apoptosis. Oxygen radicals quickly abstract hydrogen from DNA forming DNA radicals and causing DNA fragmentation, activation of DNA protective mechanisms, NAD depletion and cell death. Tyrosine hydroxylase is present in all dopaminergic neurons, is involved in the synthesis of dopamine and forms oxygen radicals in a redox mechanism involving its cofactor, tetrahydrobiopterin. Levodopa is used therapeutically in Parkinson’s disease patients since it is a precursor for dopamine, an inhibitor of tyrosine hydroxylase, and prolongs patient’s lives. Monoamine oxidase converts dopamine into 3,4-dihydroxyphenylacetaldehyde and forms oxygen radicals.Aldehyde dehydrogenase oxidizes the aldehyde and forms oxygen radicals and 3,4-dihydroxyphenylacetic acid. The treatment of Parkinson’s disease should involveinhibitors of oxygen radical formation in dopaminergic neurons and neuroprotective agents that stimulate DNA repair and prevent cell death.
1. Introduction
Even though drug therapy in Parkinson’s diseaseis effective treatment for the symptoms of patients in the early stages of the disease [1,2], the disease progresses. It has been known since the 1950’s that levodopa (Figure 1)
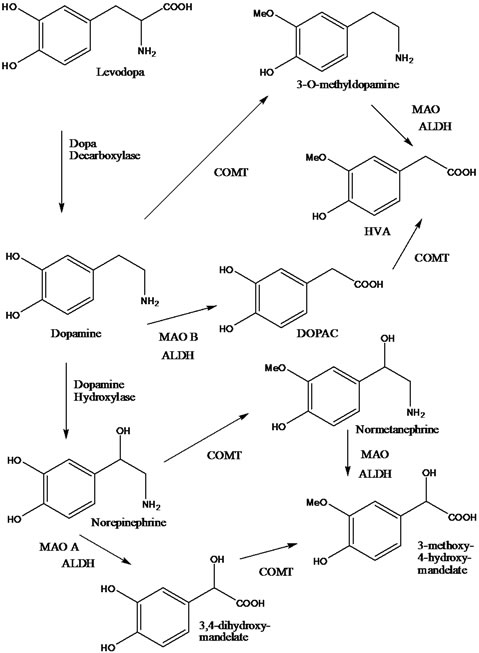
Figure 1. The metabolic oxidation of levodopa in the body. COMT is catechol-O-methyltransferase. MAO is monoamine oxidase. ALDH is aldehyde dehydrogenase. DOPAC is dihydroxyphenylacetic acid. HVA is homovanillic acid.
prolongs the lives of patients with Parkinson’s disease [1]. Many drugs are currently used in Parkinson’s disease including rotigotine and other dopamine agonists, entacapone which inhibits catechol-O-methyltransferase (COMT) and monoamine oxidase (MAO) inhibitors.
Newdrug therapy in Parkinson’s disease should involveneuroprotective agents that protect the brain from the damaging effects of oxygen radicals and slow down the progression of the disease [1]. It has been suggested that Parkinson’s disease, like Alzheimer’s disease, will affect most people who live long enough. It is known that, many people develop both Alzheimer’s and Parkinson’s diseases [3].
Parkinson’s disease is caused by the destruction of dopaminergic neurons, especially in the midbrain. Animal models of Parkinson’s disease have shown that dopaminergic neurons undergo apoptosis or necrosis [1]. Dopaminergic neurons die by a multifactorial process of oxidative stress involving oxygen radical generation by several mechanisms. The major source of oxygen radicals in dopaminergic neurons is the enzymatic oxidation of dopamine, not the nonenzymatic oxidation of dopamine with the formation of 6-hydroxydopamine [4], since this is a minor product of dopamine oxidation. Spontaneous oxidation of inherently unstable 6-hydroxydopamine produces superoxide radical anion, hydrogen peroxide and perhaps hydroxyl radical [5,6]. Iron or neuromelanin may be involved in the oxidation of 6-hydroxydopamine [7,8].
The major oxidation of dopamine occurs by MAO which produces oxygen radicals as part of its mechanism. These radicals attack DNA very rapidly [1]. Aging increases DNA fragmentation induced by oxygen radicals [9]. Neuroprotective agents that enhance DNA protective mechanisms may be able to slow down the progression of Parkinson’s disease.
Several mechanisms of oxygen radical formation in dopaminergic neurons are known. A minor mechanism is that dopamine may oxidize, nonenzymatically, forming oxygen radicals, dopaminequinones, dopamine semiquinones and neuromelanin [10]. Another minor pathway involves MAO formation of dopaminesemiquinone radicals and similar metabolites of dopamine [11-13].
Dopamine oxidation by MAO is a major factor in the progression of Parksinons’s disease. Dopamine autoxidation is clearly not the major mechanism involved the progression of Parkinson’s disease. Levodopa therapy increases brain dopamine levels, increases dopamine turnover and prolongs the lives of patients [14-16]. Levodopa greatly improves the quality of life and length of
life in Parkinson’s disease patients [1]. Dopamine autoxidation is not the major mechanism critical to disease progression, since levodopa treated patients do not die faster than untreated patients. Of course, levodopa may be able to induce toxicity in the midbrain after prolonged use, which may limit the long term use of levodopa.
Several antioxidants, that protect lipids from oxygen radical damage, have been examined with no success. Vitamin E is a very potent inhibitor of lipid peroxidation and is not effective at slowing the progression of the disease [1]. Clearly, protecting lipids in dopaminergic neurons is not the critical mechanism in Parkinson’s disease.
Current therapy involves dopaminergic agonists, pramipexole andropinirole, as the first therapy in Parkinson’s disease or as adjuncts to levodopa [17-19]. There is preliminary evidence that these agents may be able to slow down disease progression [20-22]. However, studies must be done to see if dopamine agonists really extend the lives of patients. Despite putative neuroprotection in a five year study with ropinirole and pramipexole, the motor scores of patients were worse than levodopa treated patients [23]. This result may indicate that levodopa slows disease progression whereas dopamine agonists do not. However, patients treated with pramipexole or ropinirole have a delayed requirement for levodopa therapy. This may imply that pramipexole and ropinirole slow down disease progression somewhat. Longevity studies are required to see if dopamine agonists actually do slow down disease progression.
Both pramipexole and ropinirole have toxicity problems in patients. They induce orthostatic hypotension and dizziness [24], which may lead to falling, hip fracture and potential death of patients. Apomorphine, a dopamine agonist, induces cardiac toxicity, including sudden death, myocardial infarction and angina, in 4% of patients [24]. All dopaminergic agonists induce hallucinations in a large portion of patients [25], which can make caring for patients a problem. As the disease progresses, the effects of dopamine agonists wane [26].Post marketing studies of pramipexole and ropinirole have reportedcardiac toxicity includingheart valve fibrosis [27], although at a lower incidence than for the dopaminergic agonists, cabergoline and pergolide [28]. Cardiac toxicity from dopamine agonists may limit their use in Parkinson’s disease.
2. Apoptosis or Necrosis
Dopaminergic neurons die through both necrotic and apoptotic mechanisms. Necrosis involves swelling and rupture of the nucleus, swelling and rupture of the cytoplasm, intranuclear vacuoles, loss of cytoplasmic organization, and occasionally mitochondrial swelling [29,30]. Apoptosis involves condensation of the nucleus, condensation of the cytoplasm, large cytoplasmic vacuoles, and mitochondrial shrinkage, leading to disintegration of the cell with the formation of apoptotic bodies [29-31].
Work with t-butylhydroperoxide, an oxidative stress inducing agent, has shown that the dose of oxidative stress determines whether the cells die predominantly from necrosis or apoptosis [29,30,32]. The presence of large amounts of reactive oxygen species causes predominantly necrosis. DNA is a primary target of oxygen radicals and fragments within minutes [30-32]. This activates poly(ADP-ribose) polymerase and other protective enzymes [30-32]. Normal cellular defense mechanisms, involving glutathione and other mechanisms, are overwhelmed [33-35]. The normal energy supply, involving ATP, NADH and NADPH, is exhausted [31]. This may allow cytoplasmic membrane channels to open with the influx of ions and water. The cell cannot survive and dies by necrosis.
Apoptosis involves a smaller dose of reactive oxygen species [29,30,32]. A small amount of DNA fragmentation occurs. Protective enzymes are activated, without the depletion of ATP, NADH and NADPH. DNA fragmentation may activate apoptotic programs that lead to delayed cell death. The apoptotic programs activated in Parkinson’s disease have been described [36].
2.1. Dopamine Oxidation by MAO
Free dopamine, in nerve terminals, can oxidize and producequinones, semiquinones and neuromelanin [10]. However, there is very little free dopamine in nerve terminals since it is captured and stored in vesicles to be reutilized. Some dopamine encounters mitochondria where it may be metabolized by MAO B and MAO A to produce hydrogen peroxide, superoxide and hydroxyl radical [37]. This is probably the major source of oxygen radicals from dopamine oxidation. Hydrogen peroxide is detoxified by glutathione peroxidase with glutathione oxidation, a sign of oxidative stress induction in neurons [38]. Dopamine oxidation by MAO produces 3,4-dihydroxyphenylacetaldehyde. MAO is a flavin protein found in all mitochondria and derives reducing equivalents from its substrate amines. One electron is donated from the amine to make the anionic semiquinoneflavin [39]. Dopamine is converted into an aminium radical cation that abstracts hydrogen from an adjacent carbon forming an imminium radical after loss of a proton. The imminium radical hydrolyzes to form the product aldehyde, and ammonia. However, the anionic semiquinoneflavin (Fl–) can interact with oxygen to make hydrogen peroxide or oxygen radicals (Figure 2)
.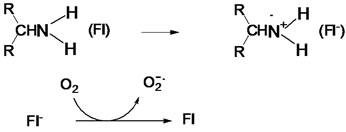
Figure 2. Oxygen radical formation by MAO. Fl is flavin.
A hallmark of Parkinson’s disease is low levels of dopamine in the striatum [1]. As dopamine levels decrease, the oxidation of dopamine may decrease such that the formation of oxygen radicals may decrease. However, the turnover of dopamine may increase during the disease process [40] such that oxygen radical formation may be high in some neurons.
Inhibition of MAO B is a therapeutic mechanism used in Parkinson’s disease with selegiline. The inhibition of MAO B could decrease oxygen radical formation in dopaminergic neurons and could be neuroprotective. The DATATOP study and studies by the Norwegian-Danish Study Group and the Swedish Parkinson Study Group have shown that selegilinedelays the need for levodopa therapy and slows down disease progression [41-43]. However, selegiline can also causepostural hypotension, arrhythmias, hypertension and the serotonin syndrome [44]. It interacts with meperidine and serotonin reuptake inhibitors such as fluoxetine to cause death or serious
<complications due to the serotonin syndrome [24]. Rasagiline is an irreversible MAO inhibitor, used in Parkinson’s disease, which appears to be selective for MAO B. Selegiline isan irreversible inhibitor of MAO B. When MAO B is irreversibly inhibited, it must be replaced by newly synthesized MAO B. This process may take 40 days [45]. It may make sense to use selegiline once every month. More frequent use of selegiline could result in overdosing and potential toxicity.
2.2. Aldehyde Dehydrogenase
Aldehyde dehydrogenase is a family of related enzymes that oxidize exogenous and endogenous aldehydes. The brain contains fairly abundant amounts of aldehyde dehydrogenase [46]. In some brain areas such as the striatum, the majority of the enzyme is mitochondrial. Aldehyde dehydrogenase is not aflavoprotein. The enzyme oxidizes aldehydes such as 3,4-dihydroxyphenylacetaldhyde (DOPAL) to produce 3,4-dihydroxyphenylacetic acid (DOPAC, Figure 1). Mitochondrial (ALDH2) and cytosolic (ALDH1) human aldehyde dehydrogenases have been characterized. They have 70% identity in their primary sequences and are expressed in the brain at the same levels [46].
A sulfhydryl and NAD+ are present inthe catalytic center of aldehyde dehydrogenase. The sulfhydryl is probably active as a thiolate anion (Figure 3).

Figure 3. Oxygen radical formation by ALDH.
The sulfhydryl binds aldehydes, which allows NAD+ to abstract hydrogen from the aldehyde forming a pyridinyl radical and a substrate radical [47]. The sulfhydryl group stabilizes the substrate radical through double bond formation. An electron is then transferred to the pyridinyl radical, which allows the formation of NADH. In general, sulfur radicals are fairly stable which may allow oxygen to interact forming superoxide. The sulfur radical may also transfer an electron to the pyridinyl moiety, forming the pyridinylradical, which is more stable than the sulfur radical. The pyridinyl radical can then interact with another molecule of oxygen forming more superoxide. It should be mentioned that the majority of the catalytic mechanism occurs through a two electron (H−) hydride transfer [48] from the substrate to NAD+. The sulfhydryl
bound ketone is formed, that hydrolyzes making the product acid.
Aldehyde dehydrogenase inhibitors could be considered for use in Parkinson’s disease. Aldehyde dehydrogenase inhibitors could slow down the progression of the disease, since they might inhibit oxygen radical formation. There is a major interaction of alcohol and aldehyde dehydrogenase inhibitors. Severe nausea and vomiting can result from this interaction and may seriously limit the use of aldehyde dehydrogenase inhibitors in Parkinson’s disease.
2.3. Tyrosine Hydroxylase
Tyrosine hydroxylase makes dopamine from tyrosine and is the rate limiting enzyme in the synthesis of dopamine [1]. The enzyme is found only in dopaminergic and catecholaminergic neurons and adrenal cells. The enzyme contains iron and biopterin as (6R,6S)-5,6,7,8-tetrahydrobiopterin, and forms oxygen radicals [49]. Tyrosine hydroxylase activity decreases with age due to enhanced deactivation [50]. The enzyme exists in four forms and is most abundant in human brain as TH1 and TH2 [51]. Dopamine binds to a specific site in the N-terminus and exerts feedback inhibition on the enzyme [49,52]. As dopamine levels decrease due to Parkinson’s disease, this feedback inhibition is released. This may result in increased dopamine synthesis in some neurons with increased oxygen radical formation by tyrosine hydroxylase. Levodopa therapy increases dopamine levels in neurons reestablishing the feedback inhibition and decreasing oxygen radical formation [46]. Therefore, it appears that dopamine inhibition of tyrosine hydroxylase is a critical mechanism in decreasing oxygen radical formation in Parkinson’s disease therapy.
Tyrosine hydroxylase performs both one and two electron reductions [49]. The major catalytic mechanism involves one electron processes with 4a-carbinolamine formation from tetrahydrobiopterin (Figure 4).
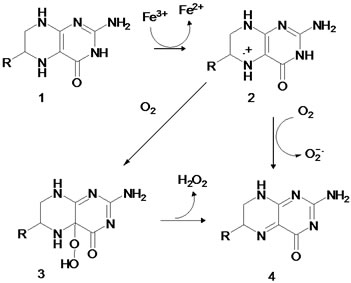
Figure 4. Oxygen radical formation by tyrosine hydroxylase. Tetrahydrobiopterin (1), radicalcation (2), hydroperoxide (3), quinonoiddihydrobiopterin (4).
This involves electron donation from tetrahydrobiopterin 1 to ferric iron making 2, the radical cation [49,53]. This is the major mechanism of the enzyme.Oxygen interacts with the radical cationforming an unstable intermediate such as a peroxyl radical or radical hydroperoxide 3. In addition, oxygen can interact with the radical cation to produce superoxide. Otherresearch has also found oxygen radical generation from tyrosine hydroxylase [54, 55].
Levodopa should remain the mainstay of Parkinson’s disease therapy [24]. Levodopa has two main mechanisms of action: as a precursor for dopamine; and as a feedback inhibitor of tyrosine hydroxylase, which decreases oxygen radical formation [49]. Therapeutic levels of levodopa, through dopamine, are adequate to fully
inhibit tyrosine hydroxylase anddecrease oxygen radical formation by the enzyme [49]. This is a neuroprotective effect of levodopa. Levodopa is the only agent shown to increase the life span of patients [14-16]. Prior to levodopa, patients only lived 10 - 15 years after diagnosis. Since the introduction of levodopa, patients live longer. Levodopa may also be more effective against symptoms than other agents [18,26].
Levodopa is clearly toxic to neurons in culture [56]. It has been suggested that levodopa cannonenzymaticallyoxidize leading to oxygen radical formation [56]. This could lead to dyskinesias and on-off phenomena noted after 5 years or more of levodopa treatment. However, studies have demonstrated that disease duration (progression), not therapy duration, correlates with dyskinesias and motor fluctuations [23]. Nonetheless, decreasing the dose of levodopa is a goal in Parkinson’s disease. This can be done with concomitant use of other agents such asdopamine agonists or selegiline.
Inhibitors of tyrosine hydroxylase, other than levodopa, should be examined in Parkinson’s disease. Patients on levodopa appear to exist for years with inhibited tyrosine hydroxylase. Other inhibitors might be useful in Parkinson’s disease. Tyrosine hydroxylase inhibitors could provide a new approach to the treatment of Parkinson’s disease.
3. Nicotinamide
Neuroprotection through protection of DNA in dopaminergic neurons is an approach to the treatment of Parkinson’s disease. Nicotinamide, a vitamin B3, has been shown to protect DNA in the midbrain and decrease cell death in a model of Parkinson’s disease [30]. Nicotinamide is a precursor for brain NAD, is taken up rapidly into the brain and results in 50% increases in brain NAD levels within a few hours [32]. NAD is a substrate for poly (ADP-ribose) polymerase (PARP), a critical enzyme in DNA protection, that is activated by DNA nicks and double strand breaks. PARP uses NAD to alter the activities of histones and other nuclear enzymes through poly (ADP-ribosylation). This ribosylation process activates several repair enzymes that rapidly restore DNA. However, the consequence of PARP activity is decreased levels of NAD, [32] an adenine containing compound. Depletion of NAD also causes ATP depletion [57] and the loss of cellular energy that leads to cell death. Nicotinamide is a precursor for NAD and protects cellular NAD and ATP levels during DNA damage and repair [57].
Nicotinamide has been shown to decrease cell death that occurs through apoptotic or necrotic mechanisms [9, 30,57-60]. In animals treated with t-butylhydroperoxide, apoptosis decreased in the brains of animals treated with nicotinamide [58]. In a stroke model, the brain had a smaller infarct volume, less necrosis and less apoptosis in animals treated with nicotinamide [59,60]. Nicotinamide could be easily tested in clinical trials since it is a well known vitamin.
Up to 30% of elderly people are deficient in nicotinamide [61]. Severe nicotinamide deficiency causes fatal neurodegeneration, known as Pellagra. Clinical trials have found good results with NAD therapy in Alzheimer’s disease and Parkinson’s disease [62,63]. NAD is degraded to nicotinamide in the gut such that NAD is a delivery form for nicotinamide. Nicotinamide itself may be useful therapy in Parkinson’s disease.
The toxicity of nicotinamide is mild. Nicotinamideis associated with none of thetoxicity of niacin. Nicotinamidehas been shown to produce vasodilation, induce several enzymes and inhibit the synthesis of other enzymes [31]. The major metabolic product of nicotinamide is NAD, in the brain and other organs [31]. A minor metabolite of nicotinamide is N-methylnicotinamide [31], which interacts with complex 1 in mitochondria to produce oxygen radicals and destroy complex 1 [64]. Injection of N-methylnicotinamide into the rodent midbrain decreases striatal dopamine [64]. The formation of Nmethylnicotinamide from nicotinamide in the brain should be examined. Nicotinamide in excess can shorten the life span of some cells in culture possibly by inhibiting Sir2 enzymes [65]. The possible importance of this mechanism should be examined in the brain.
4. Conclusion
Many important mechanisms of oxygen radical formation exist in dopaminergic neurons. MAO makes oxygen radicalsandis important in Parkinson’s disease. MAO B inhibition is a widely used therapy in Parkinson’s disease, although the benefits may be mild. The formation of oxygen radicals by aldehyde dehydrogenase is not a current therapeutic target in Parkinson’s disease. Inhibition of aldehyde dehydrogenase can produce very unpleasant interactions with alcohol. Tyrosine hydroxylase induced oxygen radical formation is already a mainstay in Parkinson’s disease therapy with levodopa. Levodopa causes dopamine levels to increase in dopaminergic neurons, which can inhibit tyrosine hydroxylase through feed back inhibition. Nicotinamide protects neuronal DNA, is neuroprotective and should be explored as a means of decreasing the progression of Parkinson’s disease.
REFERENCES
- J. D. Adams, “Agents Used in Neurodegenerative Disorders,” In: M. E. Wolff, Ed., Burger’s Medicinal Chemistry and Drug Discovery, John Wiley and Sons, New York, 1996, pp. 261-319.
- J. D. Adams, “Antiparkinsonian Drug Therapy,” American Journal of Pharmacy Education, Vol. 55, 1991, pp. 173-176.
- J. D. Adams, L. K. Klaidman, I. N. Odunze, H. C. Shen and C. A. Miller, “Alzheimer’s and Parkinson’s Disease: Brain Levels of Glutathione, Glutathione Disulfide and Vitamin E,” Molecular and Chemical Neuropathology, Vol. 14, No. 3, 1991, pp. 213-226. doi:10.1007/BF03159937
- R. Heikkila and G. Cohen, “Inhibition of Biogenic Amine Uptake by Hydrogen Peroxide: A Mechanism for Toxic Effects of 6-Hydroxydopamine,” Science, Vol. 172, No. 3989, 1971, pp. 1257-1258. doi:10.1126/science.172.3989.1257
- R. Heikkila and G. Cohen, “In Vivo Generation of Hydrogen Peroxide from 6-Hydroxyl-dopamine,” Experientia, Vol. 28, No. 10, 1972, pp. 1197-1198. doi:10.1007/BF01946168
- R. Heikkila and G. Cohen, “6-Hydroxydopamine: Evidence for Superoxide Radical as an Oxidative Intermediate,” Science, Vol. 81, No. 4098, 1973, pp. 456-457. doi:10.1126/science.181.4098.456
- A. Slivka and G. Cohen, “Hydroxyl Radical Attack on Dopamine,” Journal of Biological Chemistry, Vol. 260, No. 29, 1985, pp. 15466-15472.
- M. Elstner S. Muller, L. Leidolt, C. Laub, L. Krieg, F. Schlaudraff, B. Liss, C. Morris, D. Turnbull, E. Masliah, H. Prokisch, T. Klopstock and A. Bender, “Neuromelanin, Neurotransmitter Status and Brainstem Location Determine the Differential Vulnerability of Catecholaminergic Neurons to Mitochondrial DNA Deletions,” Molecular Brain, Vol. 4, No. 1, 2011, p. 43. doi:10.1186/1756-6606-4-43
- J. D. Adams, S. K. Mukherjee, L. K. Klaidman, M. L. Chang and R. Yasharel, “Apoptosis and Oxidative Stress in the Aging Brain,” Annals of the New York Academy of Science, Vol. 786, 1996, pp. 135-151. doi:10.1111/j.1749-6632.1996.tb39058.x
- D. G. Graham, “On the Origin and Significance of Neuromelanin,” Archives of Pathology and Laboratory Medicine, Vol. 103, No. 7, 1979, pp. 359-362.
- D. G. Anderson, S. V. Santhana Mariappan, G. R. Buettner and J. A. Doorn, “Oxidation of 3,4-Dihydroxyphenylacetaldehyde, a Toxic Dopaminergic Metabolite, to a Semiquinone Radical and an Ortho-Quinone,” Journal of Biological Chemistry, Vol. 286, No. 30, 2011, pp. 26978-26986. doi:10.1074/jbc.M111.249532
- D. G. Graham, S. M. Tiffany, W. R. Bell and W. F. Gutknecht, “Autoxidation versus Covalent Binding of Quinones as the Mechanism of Toxicity of Dopamine, 6-Hydroxydopamine, and Related Compounds toward C1300 Neuroblastoma Cells in Vitro,” Molecular Pharmacology, Vol. 14, No. 4, 1978, pp. 644-653.
- D. M. A. Mann and P. O. Yates, “Pathogenesis of Parkinson’s Disease,” Archives of Neurology, Vol. 39, No. 9, 1982, pp. 545-549. doi:10.1001/archneur.1982.00510210015004
- M. M. Hoehn and M. D. Yahr, “Parkinsonism: Onset, Progression and Mortality,” Neurology, Vol. 17, No. 5, 1967, pp. 427-442.
- H. Zumstein and J. Siegfried, “Mortality among Parkinson Patients Treated with L-Dopa Combined with a Decarboxylase Inhibitor,” European Neurology, Vol. 14, No. 5, 1976, pp. 321-328. doi:10.1159/000114756
- R. J. Uitti, J. E. Ahlskog, D. M.Maraganore, M. D. Muenter, E. J. Atkinson, R. H. Cha and P. C. O’Brien, “Levodopa Therapy and Survival in Idiopathic Parkinson’s Disease: Olmstead County Project,” Neurology, Vol. 43, No. 10, 1993, pp. 1918-1926.
- Parkinson Study Group, “Safety and Efficacy of Pramipexole in Early Parkinson’s Disease. A Randomized Dose Ranging Study,” Journal of the American Medical Association, Vol. 278, No. 2, 1997, pp. 125-130. doi:10.1001/jama.1997.03550020057038
- C. E. Clarke and M. Guttman, “Dopamine agonist monotherapy in Parkinson’s Disease,” Lancet, Vol. 360, No. 9347, 2002, pp. 1767-1769. doi:10.1016/S0140-6736(02)11668-0
- M. Gerlach, K. Double, H. Reichmann and P. Riederer, “Arguments for the Use of Dopamine Receptor Agonists in Clinical and Preclinical Parkinson’s Disease,” Journal of Neural Transmission, Vol. 65, 2003, pp. 167-183.
- Parkinson Study Group, (2002) “Dopamine Transporter Brain Imaging to Assess the Effects of Pramipexolevs Levodopa on Parkinson Disease Progression,” Journal of the American Medical Association, Vol. 287, No. 13, 1999, pp. 1653-1661. doi:10.1001/jama.287.13.1653
- J. S. Rakshi, N. Pavese, T. Uema, K. Ito, P. K. Morrish, D. L. Bailey and D. J. Brooks, “A Comparison of the Progression Of Early Parkinson’s Disease in Patients Started on Ropinirole or L-Dopa: An 18F-Dopa PET Study,” Journal of Neural Transmission, Vol. 109, No. 12, 2002, pp. 1433-1443. doi:10.1007/s00702-002-0753-0
- A. L. Whone, R. L. Watts, A. J. Stoessl, M. Davis, S. Reske, C. Nahmias, A. E. Lang, O. Rascol, M. J. Ribeiro, P. Remy, W. H. Poewe, R. A. Hauser and D. J. Brooks, “Slower Progression of Parkinson’s Disease with Ropinirole versus Levodopa: The REAL-PET Study,” Annals of Neurology, Vol. 54, No. 1, 2003, pp. 93-101. doi:10.1002/ana.10609
- J. E. Ahlskog, “Slowing Parkinson’s Disease Progression,” Neurology, Vol. 60, No. 3, 2003, pp. 381-389.
- Drug Facts and Comparisons, “Facts and Comparisons,” Drug Facts and Comparisons, St. Louis, 2012.
- J. E. Ahlskog, “Parkinson’s Disease: Is the Initial Treatment Established?” Current Neurology and Neuroscience Report, Vol. 3, No. 4, 2003, pp. 289-295.
- K. W. Lange, “Clinical Pharmacology of Dopamine Agonists in Parkinson’s Disease,” Drugs of Aging, Vol. 13, No. 5, 1998, pp. 381-389. doi:10.2165/00002512-199813050-00004
- S. Perez-Lloret and O. Rascol, “Dopamine Receptor Agonists for the Treatment of Early or Advanced Parkinson’s Disease,” CNS Drugs, Vol. 24, No. 11, 2010, pp. 941-968. doi:10.2165/11537810-000000000-00000
- A. Antonini and W. Poewe, “Fibrotic Heart Valve Reactions to Dopamine Agonist Treatment in Parkinson’s Disease,” Lancet Neurology, Vol. 6, No. 9, 2007, pp. 826-829. doi:10.1016/S1474-4422(07)70218-1
- J. D. Adams P. Kalivas and C. Miller, “The Acute Histopathology of MPTP in the Mouse CNS,” Brain Research Bulletin, Vol. 23, No. 1-2, 1989, pp. 1-17. doi:10.1016/0361-9230(89)90157-3
- S. K. Mukherjee, L. K. Klaidman, R. Yasharel and J. D. Adams, “Increased Brain NAD Prevents Apoptosis in Vivo,” European Journal of Pharmacology, Vol. 330, No. 1, 1997, pp. 27-34. doi:10.1016/S0014-2999(97)00171-4
- J. D. Adams, L. K. Klaidman, M. Morales, K. Moran, B. Schiavoni, J. R. Hsu and S. K. Mukherjee, “Nicotinamide and Neuroprotection,” In: S. Bondy, Ed., Chemicals and Neurodegenerative Diseases, Prominent Press, Scottsdale, 1999, pp. 231-262.
- L. K. Klaidman, S. K. Mukherjee and J. D. Adams, “Oxidative Changes in Brain Pyridine Nucleotides and Neuroprotection Using Nicotinamide,” Biochimica et Biophysica Acta, Vol. 1525, No. 1-2, 2001, pp. 136-148. doi:10.1016/S0304-4165(00)00181-1
- J. D. Adams, B. Wang, L. K. Klaidman, C. P. LeBel, I. N. Odunze and D. Shah, “New Aspects of Brain Oxidative Stress Induced by Tert-Butylhydroperoxide,” Free Radicals in Biology and Medicine, Vol. 15, No. 2, 1993, pp. 195-202. doi:10.1016/0891-5849(93)90059-4
- M. L. Chang, L. Klaidman and J. D. Adams Jr., “Age Dependent Effects of t-BuOOH on Glutathione Disulfide Reductase Glutathione Peroxidase and Malondialdehyde in the Brain,” Molecular and Chemical Neuropathology, Vol. 26, No. 2, 1995, pp. 95-106. doi:10.1007/BF02815008
- M. L. Chang and J. D. Adams, “The Effects of Oxidative Stress on in Vivo Brain GSH Turnover in Young and Mature Mice,” Molecular and Chemical Neuropathology, Vol. 30, No. 3, 1997, pp. 187-198. doi:10.1007/BF02815097
- M. Sanchez and F. Cardozo-Pelaez, “Intracellular Signaling Pathways in Parkinson’s Disease,” In: J. D. Adams and K. Parker, Eds., Extracellular and Intracellular Signaling, Royal Society of Chemistry, Cambridge, 2011.
- J. D. Adams, L. K. Klaidman and A. C. Leung, “MPP+ and MPDP+ Induced Oxygen Radical Formation with Mitochondrial Enzymes,” Free Radicals in Biology and Medicine, Vol. 15, No. 2, 1993, pp. 181-186. doi:10.1016/0891-5849(93)90057-2
- G. Cohen, R. Farooqui and N. Kesler, “Parkinson’s Disease: A New Link between Monoamine Oxidase and Mitochondrial Electron Flow,” Proceedings of the National Academy of Science of USA, Vol. 94, No. 10, 1997, pp. 4890-4894. doi:10.1073/pnas.94.10.4890
- C. Kay, H. El Mkami, G.Molla, L. Pollegioni and R. Ramsay, “Characterization of the Covalently Bound Anionic Flavin Radical in Monoamine Oxidase A by Electron Paramagnetic Resonance,” Journal of the American Chemical Society, Vol. 129, No. 51, 2007, pp. 16091-16097. doi:10.1021/ja076090q
- J. D. Adams, “Parkinson’s Disease and Oxygen Free Radicals,” Neurology Forum, Vol. 4, 1993, pp. 2-14.
- J. P. Larsen, J. Boas and J. E. Erdal, “Doesselegiline Modify the Progression of Early Parkinson’s Disease? Results from a Five Year Study,” European Journal of Neurology, Vol. 6, No. 5, 1999, pp. 539-547. doi:10.1046/j.1468-1331.1999.650539.x
- Parkinson Study Group, “Effects of Tocopherol and Deprenyl on the Progression of Disability in Early Parkinson’s Disease,” New England Journal of Medicine, Vol. 328, 1993, pp. 176-183. doi:10.1056/NEJM199301213280305
- S. Palhagen, E. H. Heinonen, J. Hagglund, T. Kaugesaar, H. Kontants, O. Maki-Ikola, R. Palm and J. Turunen, “Selegiline Delays the Onset of Disability in De Novo Parkinsonian Patients,” Neurology, Vol. 51, No. 2, 1998, pp. 520-525.
- Anonymous, “Selegiline: A Second Look. Six Years Later: Too Risky in Parkinson’s Disease,” Prescrire International, Vol. 11, No. 60, 2002, pp. 108-111.
- J. S. Fowler, N. D.Volkow, J. Logan, G. J.Wang, R. R. MacGregor, D. Schyler, A. P. Wolf, N. Pappas, D. Alexoff and C. Shea, “Slow Recovery of Human Brain MAO B after L-Deprenyl (Selegiline) Withdrawal,” Synapse, Vol. 28, No. 2, 1994, pp. 86-93. doi:10.1002/syn.890180203
- J. D. Adams, J. Yang and L. Klaidman, “Parkinson’s Disease, Using Drug Therapy to Slow Down Disease Progression,” In: M. J. Willow, Ed., Focus on Parkinson’s Disease Research, Nova Science Publishers Inc., New York, 2006, pp. 79-96.
- J. D. Adams and L. K. Klaidman, “Acrolein Induced Oxygen Radical Formation,” Free Radicals in Biology and Medicine, Vol. 15, No. 2, 1993, pp. 187-193. doi:10.1016/0891-5849(93)90058-3
- C. J. Mann and H. Weiner, “Differences in the Roles of Conserved Glutamic Acid Residues in the Active Site of Human Class 3 and Class 2 Aldehyde Dehydrogenase,” Protein Science, Vol. 8, No. 10, 1999, pp. 1922-1929. doi:10.1110/ps.8.10.1922
- J. D. Adams, L. K. Klaidman and P. Ribeiro, “Tyrosine Hydroxylase: Mechanisms of Oxygen Radical Formation,” Redox Report, Vol. 3, No. 5-6, 1997, pp. 273-279.
- C. P. De La Cruz, E. Revilla, J. L. Venero, A. Ayala, J. Cano and A. Machado, “Oxidative Inactivation of Tyrosine Hydroxylase in Substantianigra of Aged Rat,” Free Radicals in Biology and Medicine, Vol. 20, No. 1, 1996, pp. 53-61. doi:10.1016/0891-5849(95)02025-X
- T. Nagatsu, “Genes for Human Catecholamine Synthesizing Enzymes,” Neuroscience Research, Vol. 12, No. 2, 1991, pp. 315-345. doi:10.1016/0168-0102(91)90001-F
- A. Nakashima, K. Mori, T. Suzuki, H. Kurita, M. Otani, T. Nagatsu and A. Ota, “Dopamine Inhibition of Human Tyrosine Hydroxylase Type 1 Is Controlled by the Specific Portion in the N-Terminus of the Enzyme,” Journal of Neurochemistry, Vol. 72, No. 5, 1999, pp. 2145-2153. doi:10.1046/j.1471-4159.1999.0722145.x
- G. Eberlein, T. C. Bruice, R. A. Lazarus, R. Henrie and S. J. Benkovic, “The interconversion of the 5,6,7,8-Tetrahydro, 7,8-Dihydro and Radical Forms of 6,6,7,7-Tetramethyldihydropterin. A Model for the Biopterin Center of Aromatic Amino Acid Mixed Function Oxidases,” Journal of the American Chemical Society, Vol. 106, No. 25, 1984, pp. 7916-7924. doi:10.1021/ja00337a047
- J. Haavik, B. Almas and T. Flatmark, “Generation of Reactive Oxygen Species by Tyrosine Hydroxylase: Possible Contribution to the Degeneration of Dopaminergic Neurons,” Journal of Neurochemistry, Vol. 68, No. 1, 1997, pp. 328-332. doi:10.1046/j.1471-4159.1997.68010328.x
- D. M. Kuhn, R. E. Arthur, D. M. Thomas and L. A. Elferink, “Tyrosine Hydroxylase Is Inactivated by CatecholQuinones and Converted to a Redox-Cycling Quinoprotein: Possible Relevance to Parkinson’s Disease,” Journal of Neurochemistry, Vol. 73, No. 3, 1999, pp. 1309-1317. doi:10.1046/j.1471-4159.1999.0731309.x
- P. F. VonVoigtlander, G. J. Fici and J. S. Althaus, “Pharmacological Approaches to Counter the Toxicity of Dopa,” Amino Acids, Vol. 14, No. 1-3, 1998, pp. 189-196. doi:10.1007/BF01345261
- J. Yang, L. K. Klaidman, A. Nalbandian, J. Oliver, M. L. Chang, P. H. Chan and J. D. Adams, “The Effects of Nicotinamide on Energy Metabolism Following Transient Focal Cerebral Ischemia in Wistar Rats,” Neuroscience Letters, Vol. 333, No. 2, 2002, pp. 91-94. doi:10.1016/S0304-3940(02)01005-4
- L. K. Klaidman, S. Mukherjee, T. Hutchin and J. D. Adams, “Nicotinamide as a Precursor for NAD prevents Apoptosis in the Mouse Brain Induced by t-Butylhydroperoxide,” Neuroscience Letters, Vol. 206, No. 1, 1996, pp. 5-8. doi:10.1016/0304-3940(96)12446-0
- J. Yang, L. K. Klaidman, M. L. Chang, S. Kem, T. Sugawara, P. Chan and J. D. Adams, “Nicotinamide Therapy Protects against Both Necrosis and Apoptosis in a Stroke Model,” Pharmacology Biochemistry and Behavior, Vol. 73, No. 4, 2002, pp. 901-910. doi:10.1016/S0091-3057(02)00939-5
- [61] M. L. Chang, J. Yang, S. Kem, L. Klaidman, T. Sugawara, P. Chan and J. D. Adams, “Nicotinamide and Ketamine Reduce Infarct Volume and DNA Fragmentation in Rats after Brain Ischemia and Reperfusion,” Neuroscience Letters, Vol. 322, No. 3, 2002, pp. 137-140. doi:10.1016/S0304-3940(01)02520-4
- [62] A. Bianchetti, R. Rozzini, C. Carabellese, O. Zanetti and M. Trabucchi, “Nutritional Intake, Socioeconomic Conditions, and Health Status in a Large Elderly Population,” Journal of the American Geriatric Society, Vol. 38, No. 5, 1990, pp. 521-526.
- [63] J. G. D. Birkmayer, “Coenzyme Nicotinamide Adenine Dinucleotide New Therapeutic Approach for Improving Dementia of the Alzheimer Type,” Annals of Clinical and Laboratory Science, Vol. 26, No. 1, 1996, pp. 1-9.
- [64] J. G. D. Birkmayer, C. Vrecko, D. Volc and W. Birkmayer, “Nicotinamide Adenine Dinucleotide: A New Therapeutic Approach to Parkinson’s Disease,” Acta Neurologica Scandinavica, Vol. 87, No. 146, 1993, pp. 32-35.
- [65] T. Fukushima, A. Kaetsu, H. Lim and M. Moriyama, “Possible Role of 1-Methylnicotinamide in the Pathogenesis of Parkinson’s Disease,” Experimental Toxicology and Pathology, Vol. 53, No. 6, 2002, pp. 469-473. doi:10.1078/0940-2993-00214
- [66] J. Yang, L. K. Klaidman and J. D. Adams, “Update to Medicinal Chemistry of Nicotinamide in the Treatment of Ischemia and Reperfusion,” Medical and Chemical Reviews Online, Vol. 1, No. 1, 2004, pp. 1-5. doi:10.2174/1567203043480403