Journal of Immune Based Therapies, Vaccines and Antimicrobials
Vol.3 No.1(2014), Article ID:41055,9 pages DOI:10.4236/jibtva.2014.31001
Cytokines Elicited by HSP60 in Periodontitis with and without Coronary Heart Disease
1Unit of Periodontology, King’s College London Dental Institute, London, UK 2Queen Mary University of London, London, UK
Email: adam.hasan@kcl.ac.uk
Copyright © 2014 Adam Hasan et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Adam Hasan et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received September 29, 2013; revised October 29, 2013; accepted November 5, 2013
Heat Shock Proteins; Periodontitis; Cardiovascular Disease; Cytokines; IL-17ABSTRACT
The human 60 kDa and microbial 65 kDa heat shock proteins (HSP) have been implicated in the pathogenesis of chronic periodontitis (CP) and coronary heart disease (CHD). We have studied 100 subjects: Group (a) consisted of patients with gingivitis (n = 25), group (b) were patients with CP (n = 25), group (c) patients with CHD and gingivitis (n = 25) and group (d) patients with CHD and CP (n = 25). PBMCs separated from peripheral blood were stimulated with medium, PMA/ionomycin, human HSP60, microbial HSP65, or no stimulus for 18 hours before intracellular IL-2, IFN-γ, TNF-α, IL-4, IL-5, or IL-17 were detected by flow cytometry. The mean fluorescence intensity (MFLI) for intracellular TNF-α was significantly increased when PBMC were stimulated with human HSP60 amongst the four groups (p = 0.001, ANOVA); pairwise comparisons revealed significant differences in MFLI between the gingivitis group and the CP (p = 0.017); between gingivitis and ging/CHD (p = 0.001) as well; but no significant difference between the CP and CP/CHD (p = 0.442). There was no significant difference in intracellular expression of IL-17, or any of the other cytokines tested; and the MFLI for HSP-stimulated were comparable to unstimulated cultures. When heat-labile human HSP60 was heated, intracellular cellular TNF-α expression was abrogated. In contrast, heat-stable LPS elicited TNF-α expression from monocytes in bulk cultures in all groups. These results suggest that the cytokine expression was dependent on human HSP60 and not LPS. Serum CRP was significantly associated with MFLI of intracellular TNF-α in CP patients (rs = 0.665, p = 0.026) and CP/CHD (rs = 0.699, p = 0.011). We conclude that human HSP60 elicits increased monocytic expression of TNF-α in patients with CP, CP/CHD or ging/CHD compared to patients with gingivitis. Since the marker of inflammation, namely CRP correlates with CP with or without CHD and not with mild chronic gingivitis or ging/CHD, this suggests that human HSP60-induced production of TNF-α is associated with CP and not CHD. There was no significant difference in intracellular expression of IL-17.
1. Introduction
Chronic periodontitis (CP) is an inflammatory disease characterised by connective tissue destruction and bone resorption, affecting 10% - 15% of the developed world population and is the major cause of tooth loss in adults [1]. There is evidence from cross-sectional studies implicating a multitude of organisms [2,3]. HSPs are highly conserved proteins [4,5], expressed in all eukaryotic and prokaryotic cells and involved in both innate and adaptive immune responses, with innate responses helping to focus the cellular immune responses [6]. The high degree of homology between microbial and human HSPs has led to the hypothesis that tissue damage can occur as a result of cross-reactivity between bacterial and human HSPs. Humoral and T cell responses to HSP60/65 have been demonstrated in CP [4,5,7-9].
In CP, there is controversy as to whether cells with a TH1 or TH2 cytokine profile are associated with disease progression, as both TH1 and TH2 cytokines are produced [10]. TH1 cells produce IL-2 and IFN-γ and facilitate cell mediated immune responses and are pro-inflammatory. In contrast, TH2 cells produce IL-4, IL-5 and IL-10 [11] simultaneously inhibiting TH1 responses and promoting humoral immunity. The histological picture of stable periodontitis lesions resembles a type IV delayed hypersensitivity response and in progressive lesions changes to lesions dominated by B cells and plasma cells [12]. These findings have been previously interpreted as meaning the stable lesion is mediated by TH1 cells and the progressive lesion by TH2 cells [12]. Animal studies suggest that Th1 are involved in the progression of chronic periodontitis [13-16]. However, several studies using human cells have reported a predominance of TH2 responses in patients with CP compared to control patients [17-19]. These conflicting results may be due in part to differences in materials examined and methodology used.
There is an association between CP and atherosclerosis (cardiovascular disease) and an increased risk of developing cardiovascular disease in patients who have CP [20]. TH1 cytokines are found to be predominantly involved in the pathogenesis of atherosclerosis [21,22]. The TH1 subpopulation of CD4 + T cells predominates among atheroma plaque infiltrating lymphocytes [21,22] and becomes activated to secrete IFN-γ, IL-2, TNF-α, TNF-β [21-23], and IL-18 [23,24]. Upregulation of IFN- γ, IL-2, TNF-α, and TNF-β results in the activation of macrophages and vascular cells, promoting inflammation and cellular immunity [25]. IL-18 and its receptor act in concert to induce the production of IFN-γ, by smooth muscle cells [23]. Most human atherosclerotic lesions produce little TH2 type cytokines such as IL-4, IL-5 and IL-10 [21,22], except for IL-6 [26]. Furthermore, IL-6 correlates with CRP and plaque size in patients with acute myocardial infarction [27].
Various studies have demonstrated that HSP60/65 can activate monocytes/macrophages to produce pro-inflammatory cytokines such as TNF-α [28-31], IL-6 [32,33] and IL-8 [32]. In addition, human HSP60 has been shown to induce PBMC to produce IL-4, IL-10 [34] and IFN-γ [19,34] in patients with rheumatoid arthritis and chronic periodontitis [35].
Cytokines produced by T-cells cannot always be easily categorised as either TH1 or TH2. TH17 cells, which produce IL-17 and have been implicated in autoimmune and chronic inflammatory conditions such as rheumatoid arthritis, may offer an alternative to the TH1/TH2 paradigm, however, their role in atherosclerosis is similarly unclear [36]. We have previously found autoimmune or cross-reactive CD4+ class II-restricted T cell responses to the human HSP60 in CP and CHD [8]. Given the potential for innate immunity to focus adaptive immune responses, the objective of this study was to investigate the role of HSP-mediated induction of intracellular TNF- α, IFN-γ, and IL-2, IL-4 and IL-5, and IL-17 from PBMC of patients with chronic periodontitis and coronary heart disease, and to determine if these responses are related to inflammation, as reflected by CRP levels.
2. Material and Methods
2.1. Subjects
Patients with CG and CP were recruited from the Unit of Periodontology at Guy’s and St Thomas’ Foundation Trust and the Coronary Care Unit at St Thomas’ Hospital (Table 1). All subjects were aged between 38 and 58 years of age and had at least 15 standing teeth. Subjects were excluded from the study if they had systemic disease including diabetes mellitus, recurrent aphthous stomatitis, autoimmune disease, a history of malignancies, previous treatment for periodontitis, or a history of antibiotic therapy within the past six months prior to recruitment. Subjects were allocated into 4 groups: gingivitis, CP, Ging/CHD or CP/CHD. In each subject, probing depths and recession of all teeth were determined using a William’s probe. Recession was measured as the distance from the cemento-enamel junction to the gingival margin. Measurements were taken from 6 sites; probing attachment loss was then calculated as the sum of probing pocket depth and recession. Periodontitis was defined as probing attachment loss ³4 mm in at least 4 teeth and CG group as probing attachment loss <2 mm in all teeth. Ethical committee approval was obtained (code no 98/12/04) and subject consent obtained. 40 ml of venous blood were withdrawn from each subject.
The gingivitis subjects had minimal gingival inflammation and bleeding on probing (percentage of bleeding sites, mean ± s.d., 1.8 ± 0.8), compared with the diseased groups, with bleeding sites in CP (48.9 ± 11.7), coronary heart disease with gingivitis (Ging/CHD; 1.9 ± 0.7) and CP/CHD (45.9 ± 8.7), All Ging/CHD patients had CHD as determined by greater than 60% diameter stenosis in at least two major epicardial coronary arteries, and conversely only eight of the healthy control subjects were confirmed to be free of CHD based on angiography, the remaining members of the group denying any chest pain symptoms.
2.2. HSP
Recombinant HSP65 derived from Mycobacterium bovis was prepared at the National Institute of Public Health and Environmental Protection, Bilthoven, the Netherlands and used at a predetermined optimal concentration of 10 µg/ml. Human HSP60 was purchased from Stressgen (Victoria, Canada). The two HSPs were detoxified using Detoxi-gel columns (Pierce, Oxford, UK) and the endotoxin level was determined by Limulus Amoebocyte Lysate assay (Sigma-Aldrich, Poole, Dorset, UK). The concentration of endotoxin was < 0.007 U/µg or 7 pg endotoxin/µg for both HSPs.
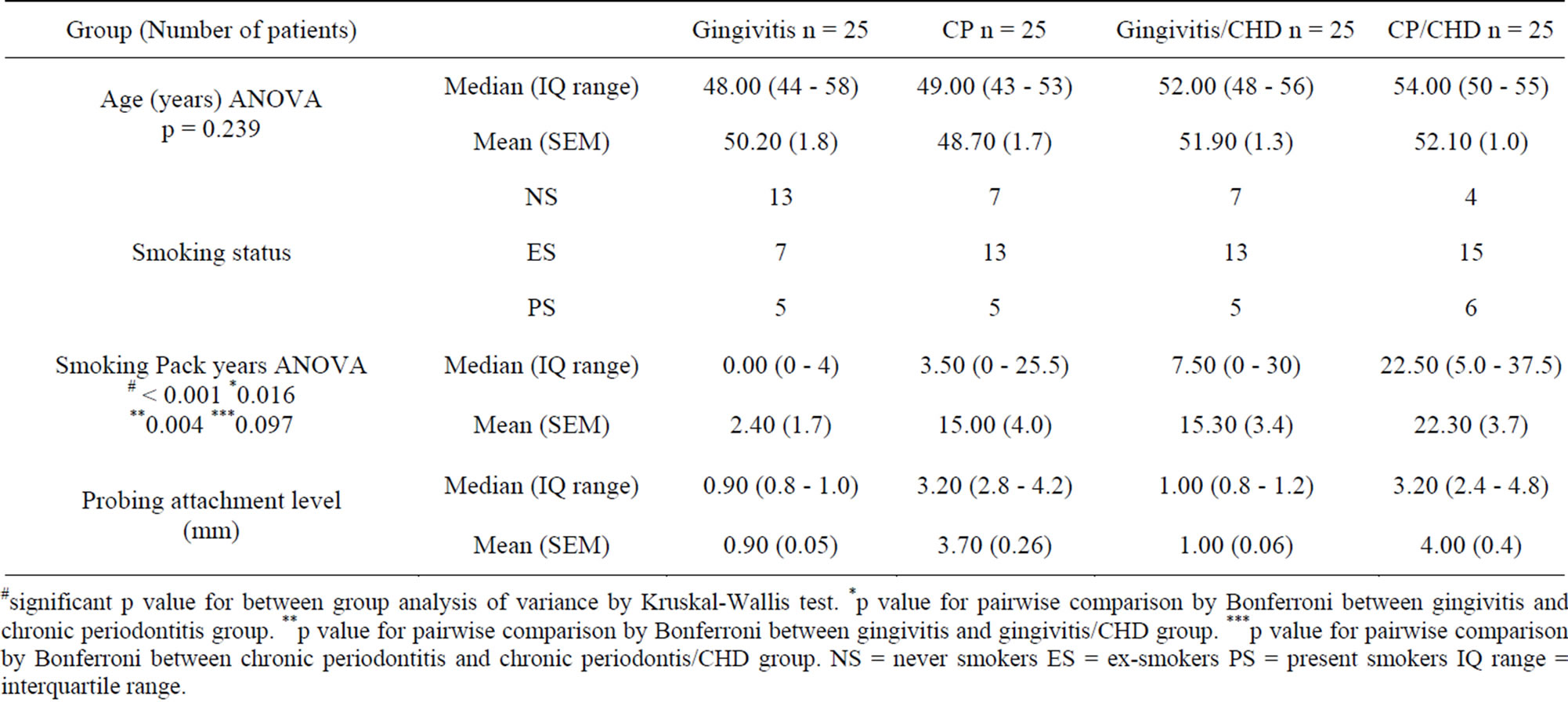
Table 1. Demographic data of patients showing median, interquartile range, mean and standard error of mean (SEM) of age, smoking status, smoking pack years and probing attachment level.
#significant p value for between group analysis of variance by Kruskal-Wallis test. *p value for pairwise comparison by Bonferroni between gingivitis and chronic periodontitis group. **p value for pairwise comparison by Bonferroni between gingivitis and gingivitis/CHD group. ***p value for pairwise comparison by Bonferroni between chronic periodontitis and chronic periodontis/CHD group. NS = never smokers ES = ex-smokers PS = present smokers IQ range = interquartile range.
2.3. Separation of PBMC
Peripheral blood mononuclear cells (PBMC) were seperated from defibrinated blood samples by density gradient centrifugation (Nycomed Pharma As, Oslo, Norway). Blood was diluted 1:1 with tissue culture medium (RPMI 1640, Sigma-aldrich, UK) containing penicillin 100 mg/ml, streptomycin 100 U/ml (Gibco, Paisley, Scotland, UK), 2 mM L-glutamine (Sigma-aldrich, Irvine UK) at room temperature. This mixture was then layered onto an equal volume of lymphoprep (Ficoll-Paque TM PLUS, Amersham Biosciences, Uppsala Sweden) in 50 ml plastic tubes (Bibby Sterilin Staffordshire UK) and centrifuged at 600 g for 30 minutes at 20˚C. Mononuclear cells were removed from the interface, and washed twice by spinning at 200 g for 10 minutes. The serum was retained and diluted with medium containing penicillin 100 mg/ml, streptomycin 100 U/ml (Gibco, Paisley, Scotland, UK), 2 mM L-glutamine (Sigma-aldrich, UK) to 10% and used in cell cultures.
2.4. Detection of Intracellular Cytokines by Flow Cytometry
PBMC from 100 patients were cultured (5 × 105 cells/ well) in 24 well flat bottomed plates (Corning 25,820). Cytokine secretion by PBMC was blocked by adding monensin 4 mg/ml (Sigma-aldrich, Irvine UK) and brefeldin A 10 mg/ml (Sigma-aldrich, Irvine UK). PBMC were then activated with phorbol myristate acetate (PMA) 10 ng/ml plus ionomycin 1 µg/ml (Sigma-aldrich, Irvine UK), human HSP60 10 µg/ml (NSP-540 Stressgen UK), microbial HSP 65 10 µg/ml (NSP581 stressgen UK) or medium alone and incubated at previously predetermined optimal time of 18 hours (Figure 1) in a humidified atmosphere (5% CO2 at 37˚C). To investigate the role of LPS contamination some of the patients PBMC were further incubated with heat treated LPS or HSP by boiling 100˚C for 30 minutes. After 18 hours of incubation, cells were transferred into 5 ml round-bottom tubes (Falcon 352,058). Cells were stained using CD4 fluorescein isothiocyanate (FITC) 5 µl (BD Pharmingen, San Diego), CD14 and CD8 peridinin chlorophyll-protein (PerCP) 5 µl (BD Pharmingen, San Diego), then incubated in the dark at room temperature for 30 minutes. Cells were then centrifuged at 500 g for 5 minutes and washed once in 2 mls of PBS/azide (×1 PBS, 0.5% BSA, 0.1% sodium azide). Cells were fixed with 1 ml FACS lysing solution containing <50% Diethylene glycol and < 15% formaldehyde (BD Pharmingen 349,202) diluted 1:10 with PBS/azide and incubated at room temperature for 10 minutes. The cells were washed in PBS/azide cells and then permeabilised with 500 µl of FACS permeabilising solution (BD pharmingen 340,973) containing 0.02% saponin, diluted 1:10 with deionised water. Cells were incubated for 10 minutes at room temperature and then centrifuged at 500 g for 5 minutes, washed once and resuspended in PBS/azide.
Cells were then stained for intracellular cytokines, using 1 µg/ml of the following monoclonal antibodies conjugated to phycoerythrin (PE): anti-human IL-2 phycoerythrin (PE) conjugate (554,566 BD Pharmingen, San Diego), anti-human IL-4PE conjugate (554,516 BD Pharmingen, San Diego), anti-human IL-6PE conjugate (554,545 BD Pharmingen, San Diego), anti-human IFN-γ PE conjugate (554,701 BD Pharmingen, San Diego), anti-humanTNF-α PE conjugate (554,513 BD Pharmin-
 (a)
(a)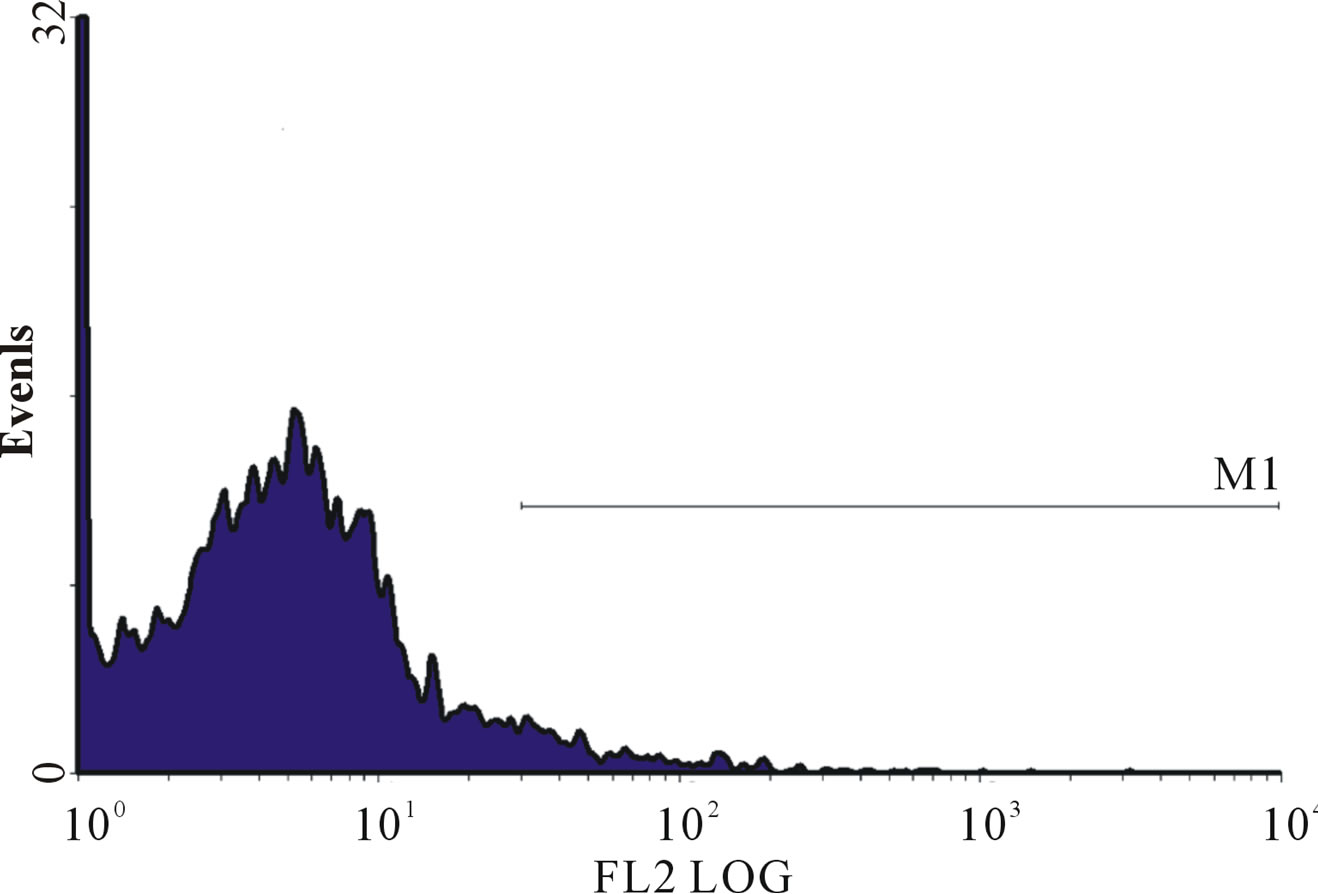 (b)
(b)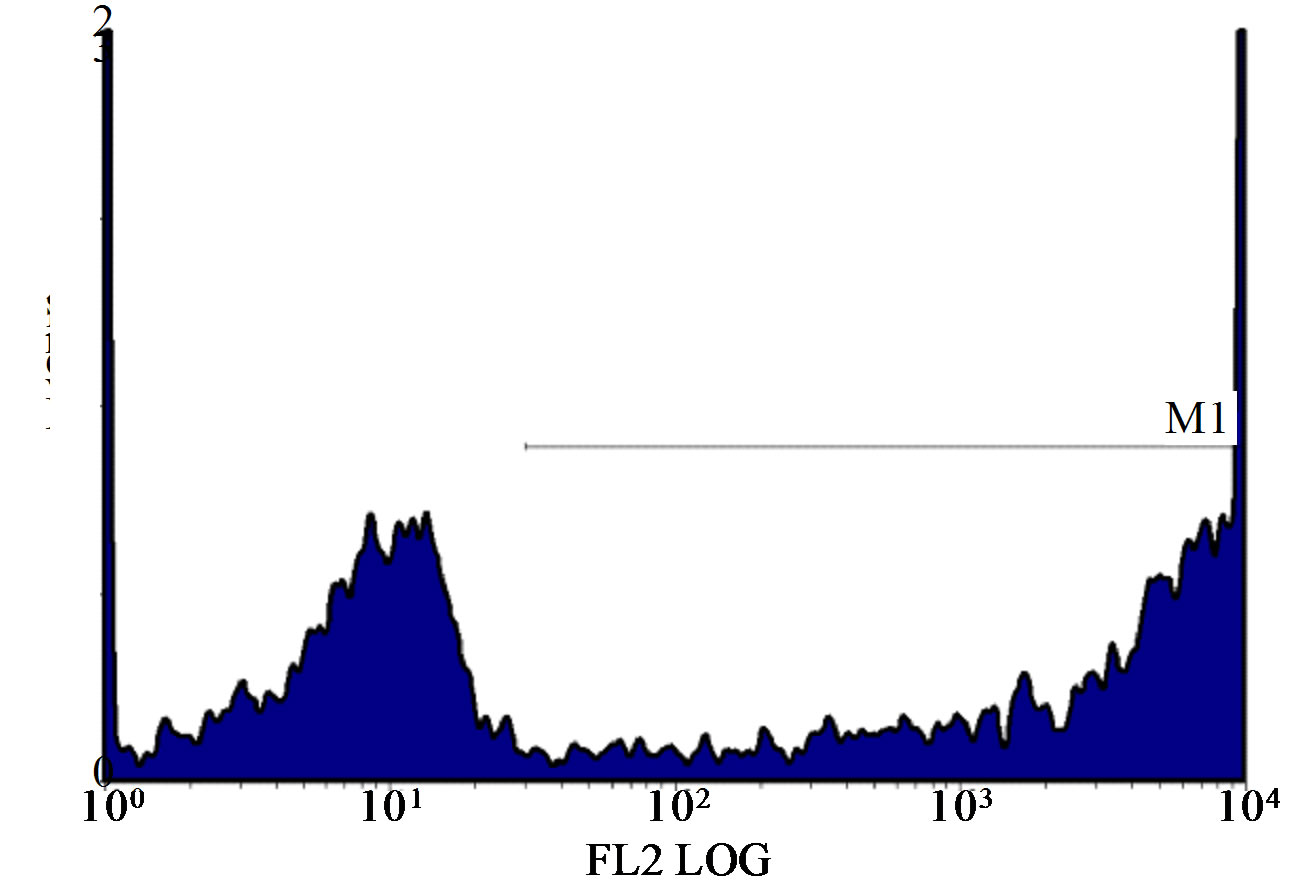 (c)
(c)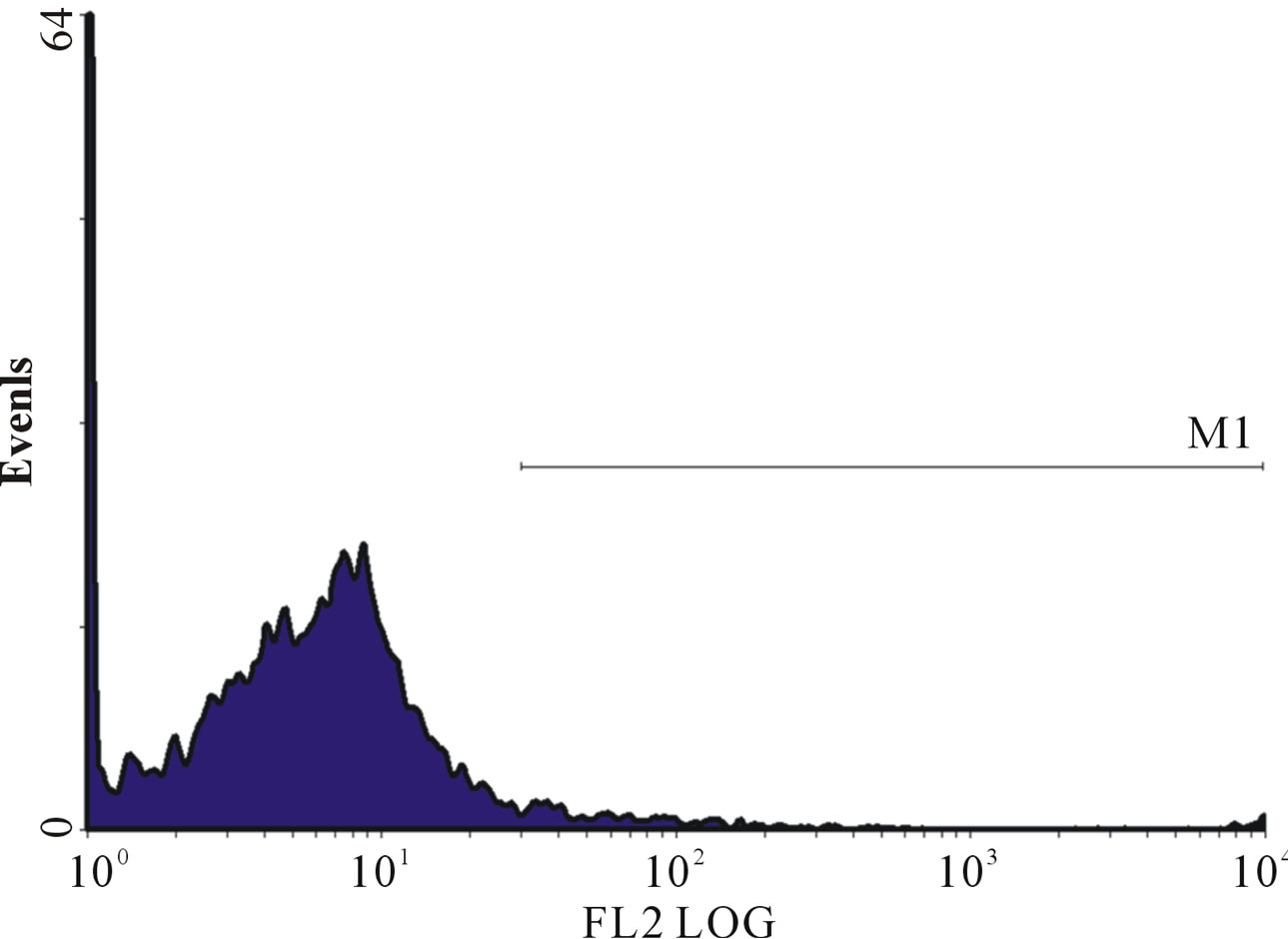 (d)
(d)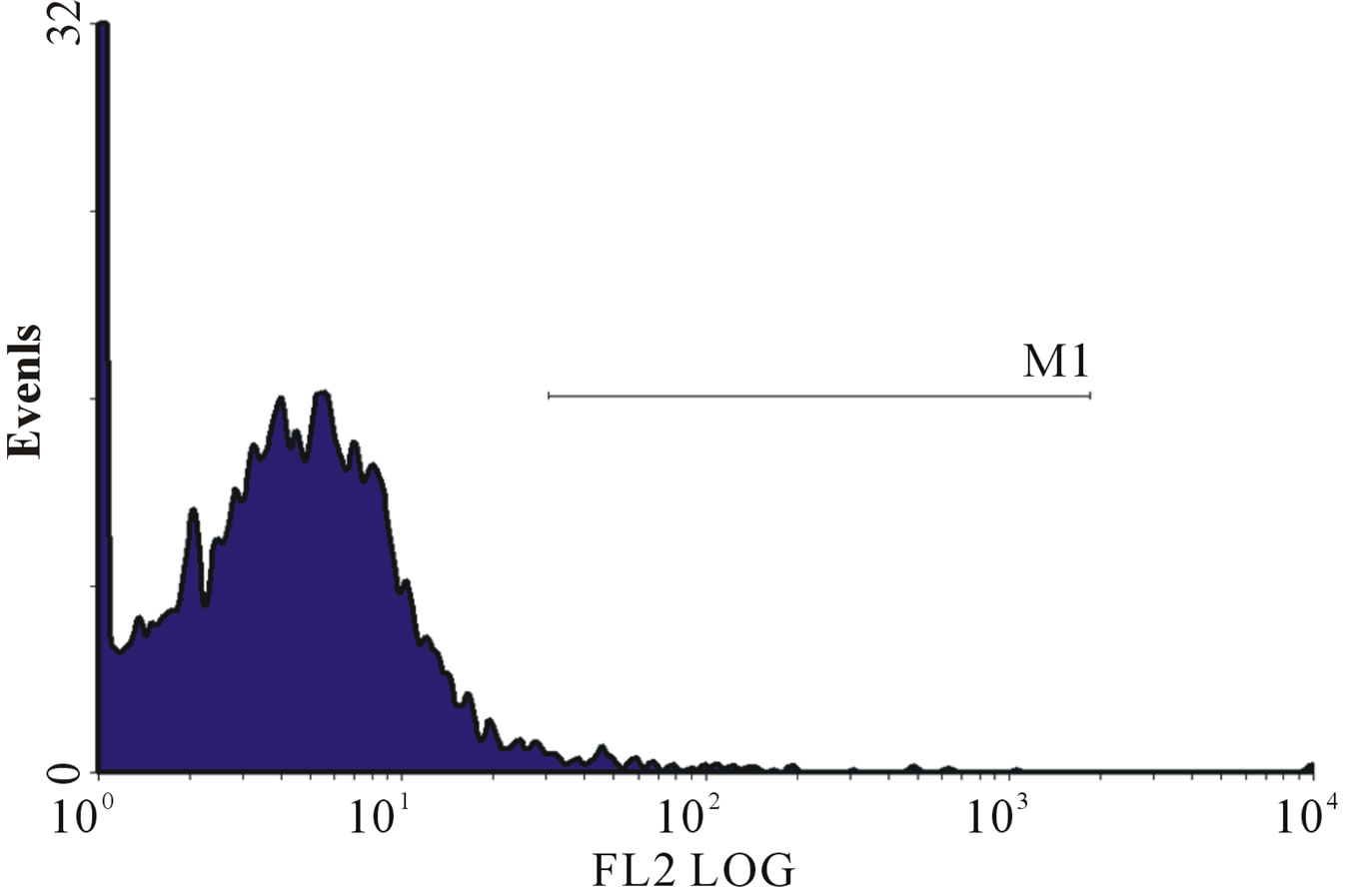 (e)
(e)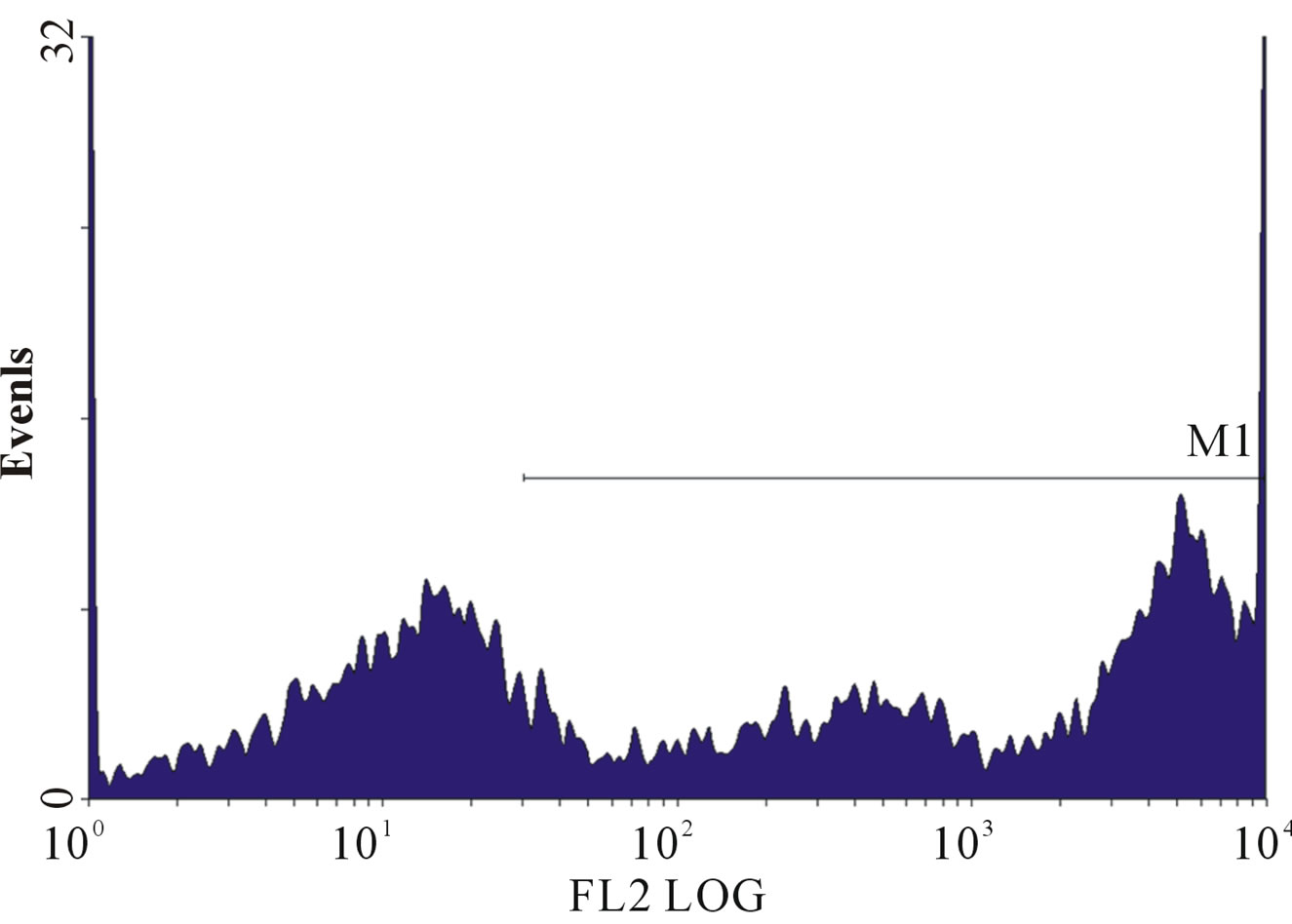 (f)
(f)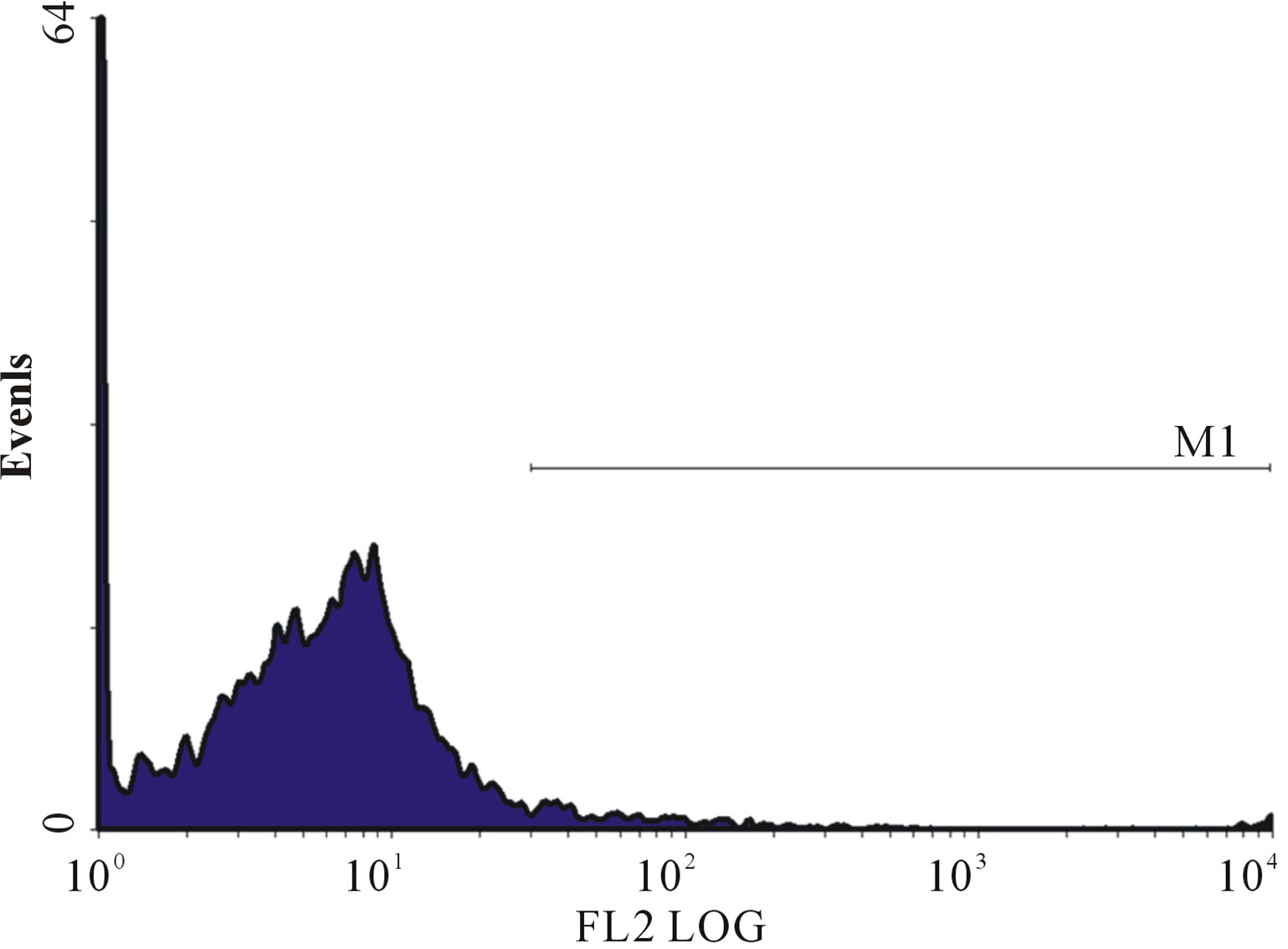 (g)
(g)
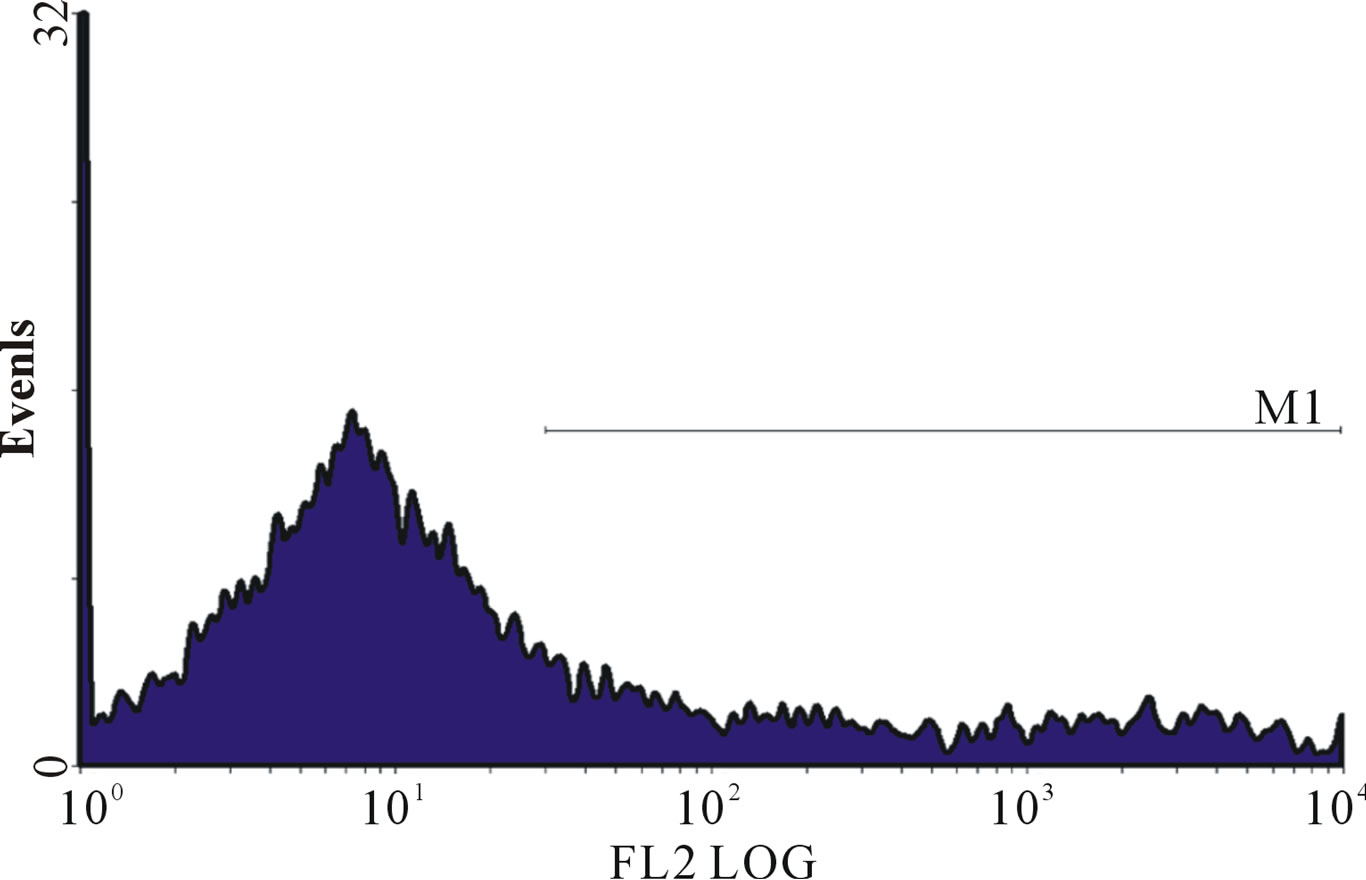
Figure 1. (a)-(g): Histogram of flow cytometric intracellular cytokine expression in monocytes stained with PE-labelled anticytokine monoclonal in a patient with chronic periodontitis antibody showing events versus mean fluorescence intensity. (a) TNF-µ expression following stimulation with LPS; (b) TNF-µ expression following stimulation with microbial HSP65; (c) TNF-µ expression following stimulation with Human HSP60; (d) TNF-µ expression following heat treatment with LPS; (e) TNF-µ expression following stimulation with heat treated microbial HSP65; (f) TNF-µ expression following stimulation with heat treated human HSP60; (g) TNF-µ expression following incubation with medium only. (a) LPS TNF-µ; (b) HSP65 TNF-µ; (c) HSP60 TNF-µ; (d) LPS TNF-a following heat treatment; (e) Heat denatured microbial HSP65 TNF-µ; (f) Heat denatured human HSP60 TNF-µ; (g) Medium TNF-µ.
gen, San Diego), anti-human IL-17PE conjugate (560,438 BD Pharmingen) or CD64PE conjugate (Serotec Oxford). After 30 minutes cells were washed twice and resuspended in PBS/Azide. Samples were analysed in a Coulter Epics FACS machine, at least 10,000 events were analysed. Results were expressed as the mean fluorescence intensity (MFLI).
2.5. Statistical Method
Data recorded on dental charts and data obtained from the different experiments were transcribed onto computer records and analysed using SPSS-11.0 for windows (2001). All continuous variables were examined to establish whether the data conformed to a normal distribution with the Lilliefors (Kolmogorov-Smirnov) test for normality. Skewness and kurtosis were also examined. Analysis revealed non-normal distribution for all variables tested which were not amenable to log or square root transformation. Therefore the non-parametric KruskalWallis ANOVA test was used to assess differences between the groups established. The significance level was set at p < 0.05. Post ANOVA pairwise comparisons between the gingivitis group, CP group, ging/CHD group and CP with CP/CHD group were carried out with MannWhitney U test, using the Bonferroni correction. Interrelations between two variables were tested using the Spearman ranked correlation coefficients (rs); when the p value was <0.05, correction for smoking pack years was carried out. Non-parametric data are displayed in tables as median (1st and 3rd quartile). Data were also displayed as mean ± standard error of mean (SEM) to facilitate comparison with previously reported findings in the literature.
2.6. Detection of C-Reactive Protein by ELISA
Pre-diluted standard (50 mL) and blank were added to a 96-well plate pre-coated with anti-serum CRP IgG (Kalon Biological Ltd). Serum samples were diluted 1:1000 with assay diluent (Kalon Biological Ltd.) and dispensed in duplicates to designated wells in CRP precoated plates. The plate was then incubated at room temperature for 60 minutes. Plates were washed 4 times with wash buffer (Kalon Biological Ltd); 100 mL of CRP tracer (affinity purified sheep anti-CRP labelled with alkaline phosphatase, Kalon Biological Ltd UK) were then dispensed to each well and incubated uncovered for 30 minutes at room temperature. Plates were washed again 4 times with washing buffer (Kalon Biological Ltd, UK).
Substrate solution (100 ml of 4-nitrophenylphosphate in substrate buffer Kalon Biological Ltd.) was then dispensed to each well and incubated at room temperature for 30 minutes. The reaction was stopped with 100 ml of (120 g/L) sodium hydroxide. Optical densities were read at 405 nm with microplate reader (Anthos 2001, Anthos labtec instruments UK). A standard curve was constructed with standard points and curve fitted with four parameter logistic curve fitting software. Test serum values were then read off the standard curve.
3. Results
3.1. Detection of Intracellular Cytokines
Unstimulated cells incubated in medium showed relatively low mean fluorescence intensity (MFLI) (Figure 2(g)) in all intracellular cytokines studied; no statistically significant difference was found amongst the 4 groups (ANOVA, p = 0.803, Table 2).
There was a statistically significant difference amongst the 4 groups in MFLI for intracellular TNF-α by ANOVA (p = 0.001) (Table 2, Figures 2 and 3); also between the gingivitis group compared to the CP group (p = 0.017) and between the gingivitis group and the ging/CHD
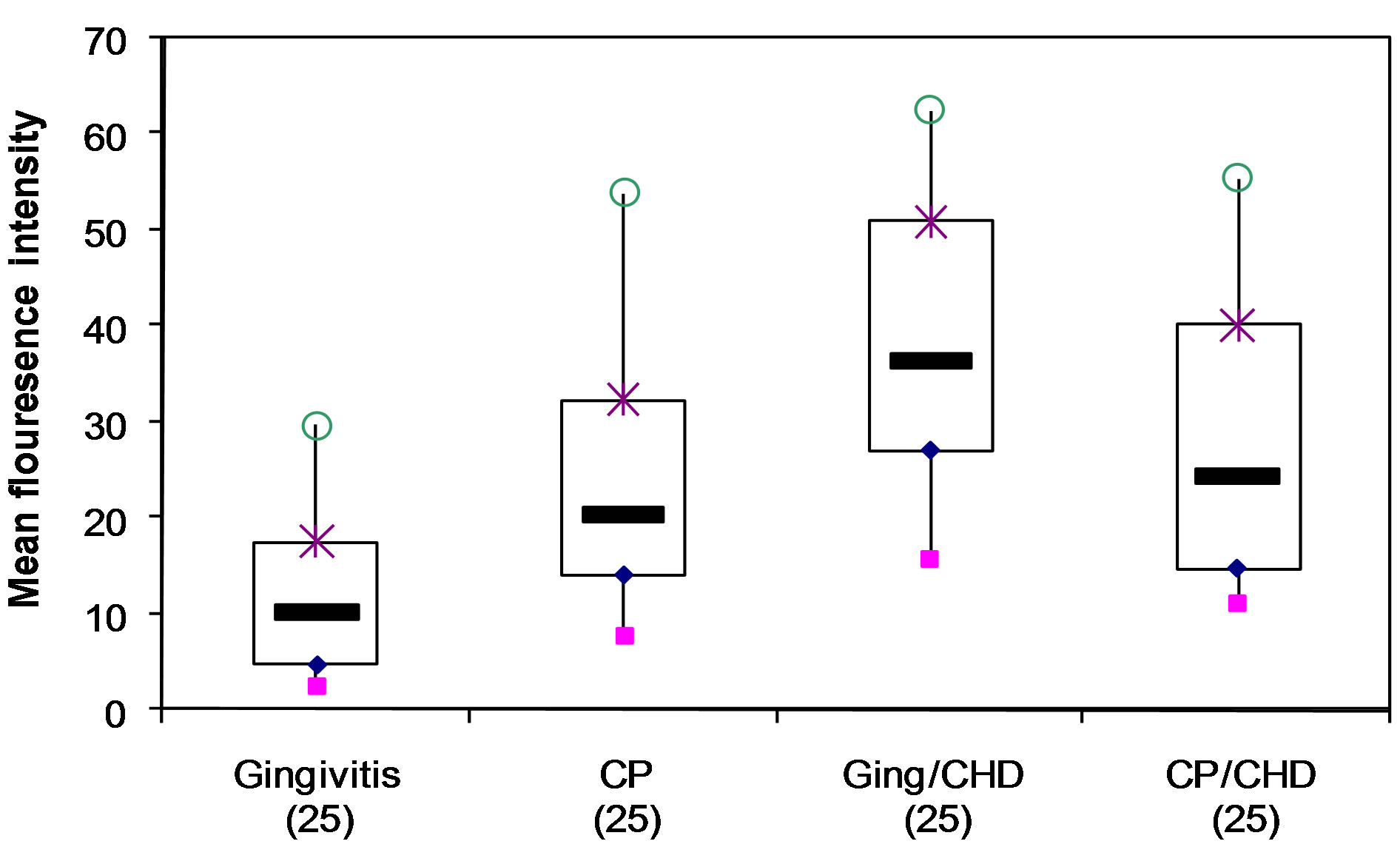
Figure 2. Detection of intracellular TNF-a in monocytes stimulated with human HSP60 showing median, interquartile range, mean and standard error of mean (SEM) of MFLI for each of 4 groups of patients (ANOVA p = 0.001) o maximum; × 75th percentile; minimum; 25th percentile;median.
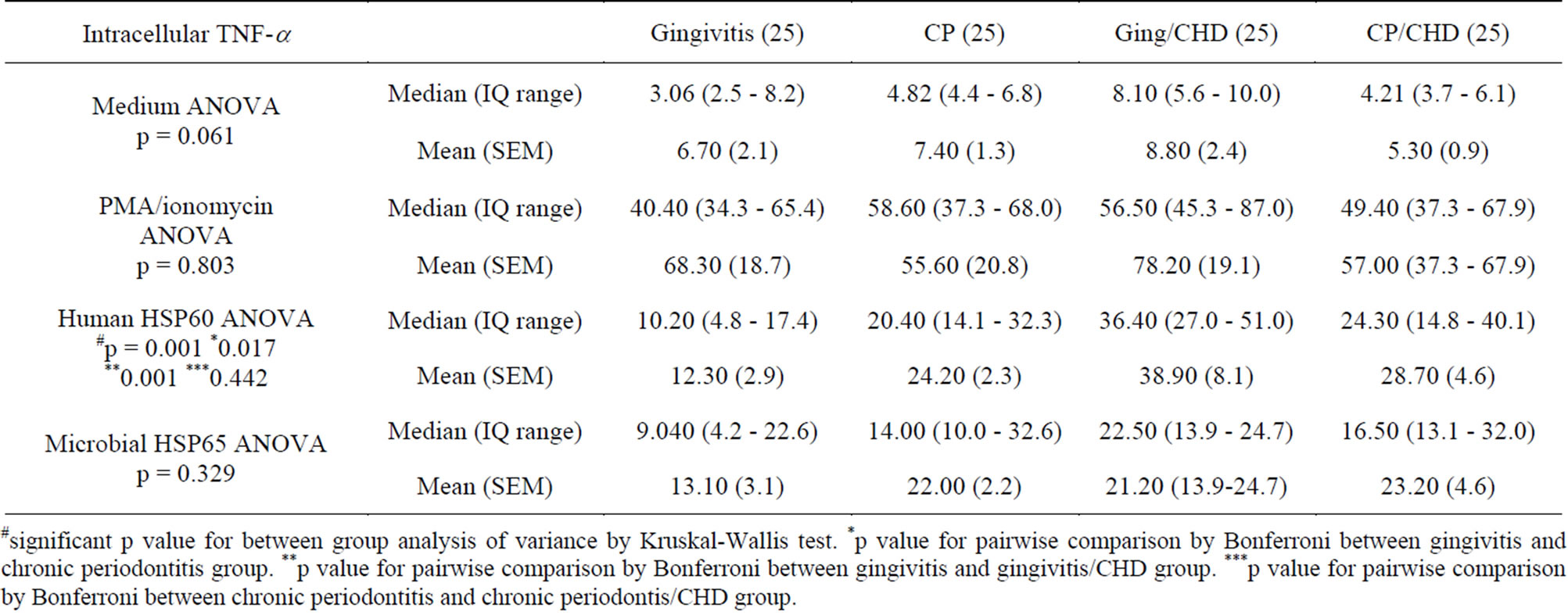
Table 2. Detection of intracellular TNF-a in monocytes stimulated with phorbol myristate acetate(PMA)/ionomycin, human HSP60, microbial HSP65 or no stimulus showing median, interquartile range, mean and standard error of mean (SEM) of MFLI.
#significant p value for between group analysis of variance by Kruskal-Wallis test. *p value for pairwise comparison by Bonferroni between gingivitis and chronic periodontitis group. **p value for pairwise comparison by Bonferroni between gingivitis and gingivitis/CHD group. ***p value for pairwise comparison by Bonferroni between chronic periodontitis and chronic periodontis/CHD group.
group (p = 0.001) (Table 2). However, there was no statistical difference between the MFLI for the CP group and the CP/CHD group (p = 0.442) (Table 2, Figures 2 and 3).
There were no statistically significant differences amongst the groups in the MFLI for any of the remaining intracellular cytokines (IFN-γ, IL-2, IL-4, IL-5, IL-6, IL-17) following stimulation with human HSP60 or microbial HSP65.
There were no significant differences in intracellular IFN-γ, IL-2, IL-4, or IL-6 amongst the 4 groups when monocytes were stimulated with either human HSP60 or microbial HSP65.
3.2. CRP
Serum CRP was significantly associated with MFLI of intracellular TNF-α in CP patients (rs = 0.665, p = 0.026) and CP/CHD (rs = 0.699, p = 0.011) even after correction for smoking pack years in chronic periodontitis (rs = 0.649, p = 0.002) and chronic periodontitis/CHD (rs = 0.603, p = 0.011) (Table 3). However, serum CRP levels were not significantly associated with MFLI of intracellular TNF-α production in the gingivitis group (rs = 0.596, p = 0.053) and the gingivitis/CHD group (rs = 0.515, p = 0.128). There was no direct association be-
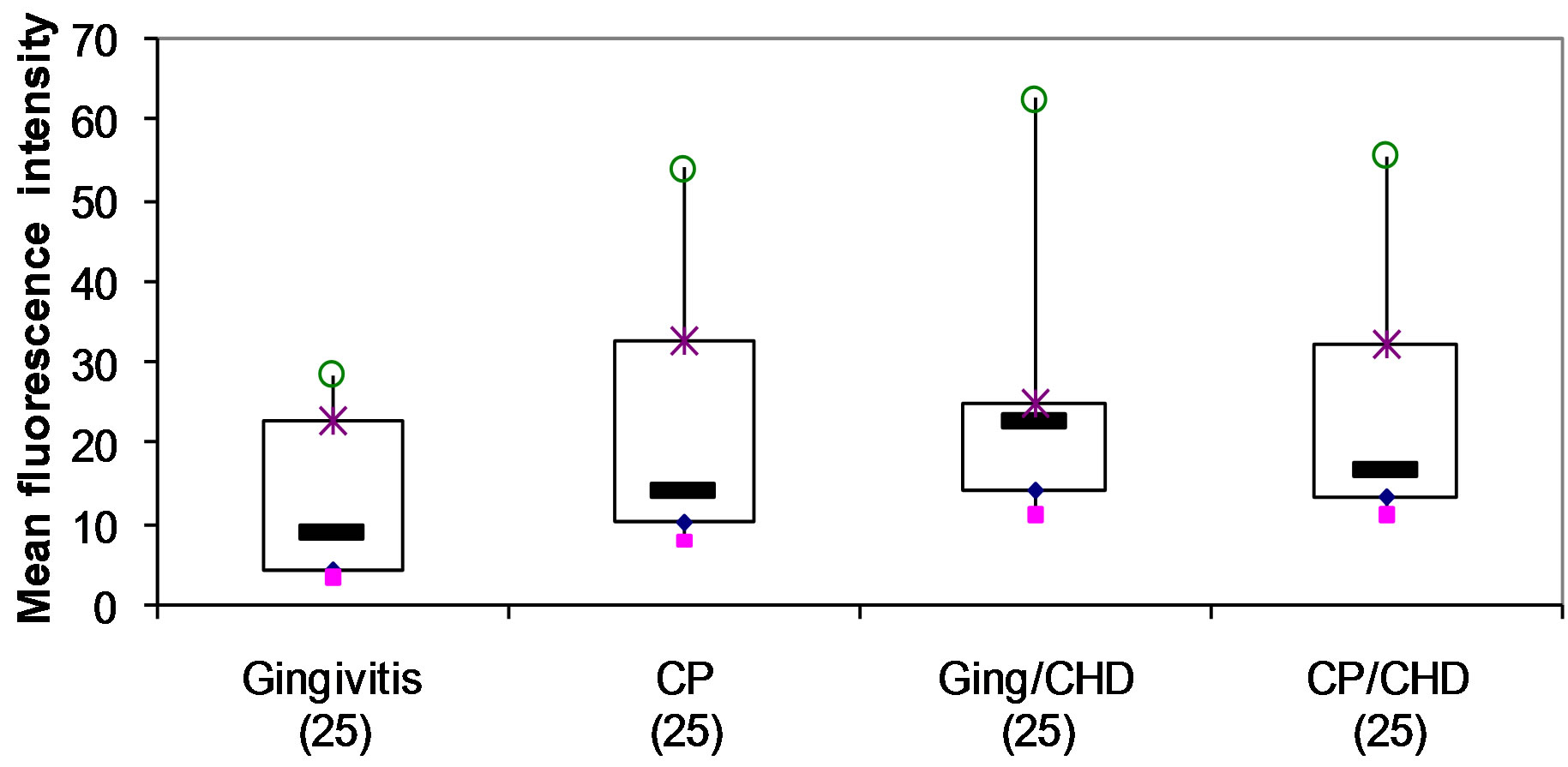
Figure 3. Detection of intracellular TNF-α in monocytes following stimulation with microbial HSP65 in 4 groups of patients (ANOVA p = 0.329).
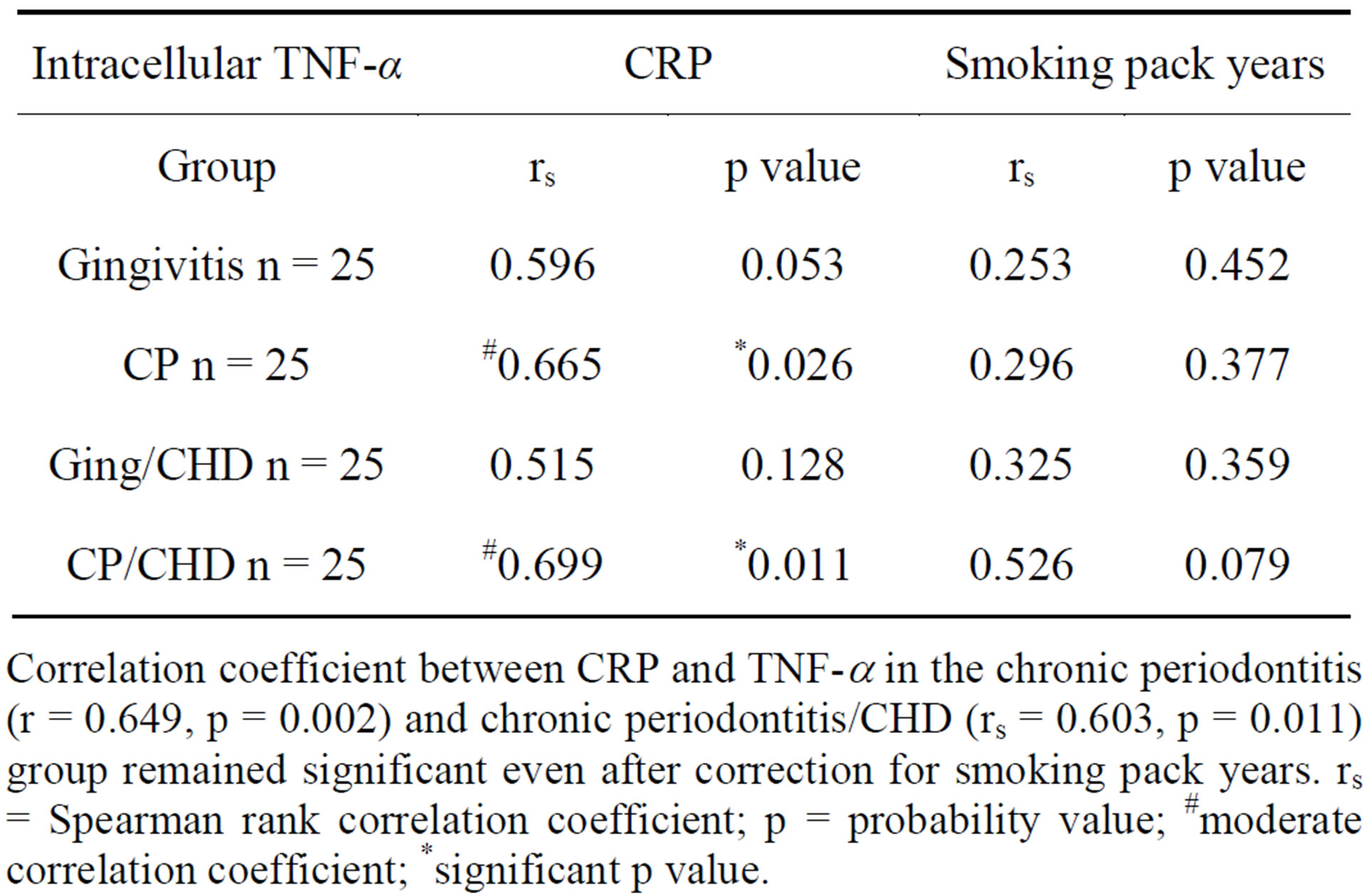
Table 3. Association of serum CRP and smoking pack years to human HSP60-induced monocytic production of intracellular TNF-α in 4 groups of patients.
Correlation coefficient between CRP and TNF-a in the chronic periodontitis (r = 0.649, p = 0.002) and chronic periodontitis/CHD (rs = 0.603, p = 0.011) group remained significant even after correction for smoking pack years. rs = Spearman rank correlation coefficient; p = probability value; #moderate correlation coefficient; *significant p value.
tween smoking pack years to human HSP60 induced intracellular TNF-α production in the control (rs = 0.253, p = 0.452), CP (rs = 0.296, p = 0.377), ging/CHD (rs = 0.325, p = 0.359) and CP/CHD (rs = 0.526, p = 0.079) (Table 3).
3.3. Excluding Any Activity Due to Contamination of HSP with LPS
To exclude the possibility of LPS contamination affecting the production of intracellular cytokine, the microbial HSP65 and human HSP60 was heat-treated as previously described [8]. HSP is heat labile whereas, LPS is heat stable [8]. Stimulation of cultures with heat-treated microbial HSP65 (Figure 1(e)) and human HSP60 (Figure 1(f)) abolished the production of intracellular TNF-α. However, LPS stimulation of PBMC producing TNF-α (Figure 1(a)) was not affected by heat treatment (Figure 1(d)). The abrogation of TNF-α production makes it unlikely that LPS contamination was responsible for the intracellular cytokine expression.
4. Discussion
Unstimulated cells incubated in medium showed relatively low mean fluorescence intensity (Table 2). This is not surprising as unstimulated cells usually produce little or no measurable cytokines [37] except in patients with progressive malignancies [38]. PMA/ionomycin stimulation of cells resulted in an increase in the mean fluorescence intensity of TH1 cytokines in both controls and patients with CP and CHD (Table 2). Although this increased mean fluorescence intensity was not statistically significant, there was a trend towards higher mean fluorescence intensities for TNF-α and IFN-γ in the patient groups compared to the control group (Table 2). There was little or no increase in the mean fluorescence intensity for IL-4 and IL-5 following stimulation with PMA/ionomycin in the patient groups. Perhaps the stimulation protocol employed in this study was not sensitive enough to detect IL-4 and IL-5 [39]; as PMA/ionomycin stimulation of IL-4 and IL-10 was insufficient for their intracellular detection, often requiring secondary culture with cytokines followed by re-stimulation with mitogen [40]. However, some studies have shown an induction of intracellular IL-4 with only primary stimulation with PMA/ionomycin [38].
Stimulation of cell cultures with human HSP60 [38,41] resulted in a statistically significant increase in mean fluorescence intensity for intracellular TNF-a in the CP and CHD patients compared with gingivitis group (Table 2, Figure 2), but no significant differences were seen amongst the 4 groups for intracellular IFN-γ, IL-2, IL-4 IL-5 and IL-17. In order to ensure HSP60 and not LPS was eliciting the cytokine response, we denatured HSP60 before addition to the culture. Denaturing HSP completely abolished the cytokine response (Figure 1(f)). Since LPS is heat stable, this suggests that HSP is eliciting the cytokine production, and not LPS. Either CD14 or TLR4 or both receptors have been implicated in HSP recognition and are thought to mediate activation of monocytes/macrophages in response to human HSP60-stimulation [6,42]. As HSP60 has been implicated in the pathogenesis of chronic inflammatory diseases such as rheumatoid arthritis [43] and atherosclerosis [44], the abundant expression of HSP and TLR-4 within periodontitis lesions [42,45] together with HSP’s role in eliciting TNF-α production , suggests that HSP60 is also likely to be playing a role in chronic inflammatory periodontal disease. The highest intracellular TNF-a levels were in patients with CHD with or without periodontal disease and CP, suggesting that the cytokine levels are related to disease severity, however, the smoking status was not the same across the groups potentially confounding the results.
Although microbial HSP65 cultures showed an increase in mean fluorescence intensity for intracellular TNF-a compared to unstimulated samples, there was no statistically significant difference amongst the 4 groups (Table 2, Figure 2). Additionally, no significant differences were seen amongst the groups in the mean fluorescence intensity of intracellular IFN-a, IL-2, IL-4 and IL-5 (data not shown).
These results are consistent with previous studies showing that TNF-a can be induced by HSP60 in monocyte/macrophages [28-30]. The lack of any significant difference in the mean fluorescence intensity of intracellular TNF-a amongst groups with periodontal disease with/without coronary artery disease following stimulation with microbial HSP65 is consistent with the lymphoproliferative responses which showed no difference between the 4 groups. Most subjects respond to microbial HSP65 and this may be a result of the ability of microbial HSP65 to stimulate both naïve and primed cells [8,31, 46].
Smoking has been a confounding variable in studies of the association of CP and CHD [47]. Exposure to smoking has been associated with reduced release of TNF-a in human alveolar macrophages [48,49]. In this study there was significant difference in the smoking pack years amongst the patient group compared to the gingivitis group (Table 2). There was no correlation of smoking pack years to human HSP60-induced intracellular TNF-a production in this study (Table 2).
CRP a marker of inflammation was significantly associated with human HSP 60 induced intracellular TNF-a production in the CP group with or without CHD, but not in the gingivitis group or the ging/CHD (Table 2). The lack of significant association between serum CRP and TNF-a in the gingivitis and ging/CHD groups may simply reflect the complexity of inflammation and its many pathways and HSP60-induced TNF-a may be an indirect association with CRP and that CP induced inflammation drives TNF-a production. There is evidence for this from previous studies showing that CRP and TNF-a are both increased in patients with CP [48-50] and reduced following anti-microbial and non-surgical treatment of chronic periodontitis [50].
5. Conclusion
This study suggests that human HSP60 but not microbial HSP65 induces significant production of intracellular TNF-a in patients with CP and CHD. Since the marker of inflammation, namely CRP correlates with CP with or without CHD and not with mild chronic gingivitis or ging/CHD, this suggests that human HSP60-induced production of TNF-a is associated with CP and not CHD. There was no direct link between smoking pack years and the production of human HSP60 induction of TNF-a, however this merits further investigation.
REFERENCES
- D. Locker, G. D. Slade and H. Murray, “Epidemiology of Periodontal Disease among Older Adults: A Review,” Periodontology 2000, Vol. 16, No. 1, 1998, pp. 16-33. http://dx.doi.org/10.1111/j.1600-0757.1998.tb00113.x
- P. S. Kumar, et al., “New Bacterial Species Associated with Chronic Periodontitis,” Journal of Dental Research, Vol. 82, No. 5, 2003, pp. 338-344. http://dx.doi.org/10.1177/154405910308200503
- M. A. Listgarten and P. M. Loomer, “Microbial Identification in the Management of Periodontal Diseases. A Systematic Review,” Annals of Periodontology, Vol. 8, No. 1, 2003, pp. 182-192. http://dx.doi.org/10.1902/annals.2003.8.1.182
- D. E. Lopatin, et al., “Humoral Immunity to Stress Proteins and Periodontal Disease,” Journal of Periodontology, Vol. 70, No. 10, 1999, pp. 1185-1193. http://dx.doi.org/10.1902/jop.1999.70.10.1185
- G. Schett, et al., “Salivary Anti-hsp65 Antibodies as a Diagnostic Marker for Gingivitis and a Possible Link to Atherosclerosis,” International Archives of Allergy and Immunology, Vol. 114, No. 3, 1997, pp. 246-250. http://dx.doi.org/10.1159/000237675
- C. A. Colaco, et al., “Heat Shock Proteins: Stimulators of Innate and Acquired Immunity,” BioMed Research International, Vol. 2013, 2013, p. 11.
- K. Buhlin, et al., “Risk Factors for Cardiovascular Disease in Patients with Periodontitis,” European Heart Journal, Vol. 24, No. 23, 2003, pp. 2099-2107. http://dx.doi.org/10.1016/j.ehj.2003.09.016
- A. Hasan, et al., “The Immune Responses to Human and Microbial Heat Shock Proteins in Periodontal Disease with and without Coronary Heart Disease,” Clinical & Experimental Immunology, Vol. 142, No. 3, 2005, pp. 585- 594.
- K. Tabeta, et al., “Elevated Humoral Immune Response to Heat Shock Protein 60 (hsp60) Family in Periodontitis Patients,” Clinical & Experimental Immunology, Vol. 120, No. 2, 2000, pp. 285-293. http://dx.doi.org/10.1046/j.1365-2249.2000.01216.x
- I. Ishikawa, et al., “Induction of the Immune Response to Periodontopathic Bacteria and Its Role in the Pathogenesis of Periodontitis,” Periodontology 2000, Vol. 14, 1997, pp. 79-111. http://dx.doi.org/10.1111/j.1600-0757.1997.tb00193.x
- T. R. Mosmann, et al., “Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins,” Journal of Immunology, Vol. 136, No. 7, 1986, pp. 2348-2357.
- E. Gemmell, K. Yamazaki, and G. J. Seymour, “Destructive Periodontitis Lesions Are Determined by the Nature of the Lymphocytic Response,” Critical Reviews in Oral Biology & Medicine, Vol. 13, No. 1, 2002, pp. 17-34. http://dx.doi.org/10.1177/154411130201300104
- J. W. Eastcott, et al., “Adoptive Transfer of Cloned T Helper Cells Ameliorates Periodontal Disease in Nude Rats,” Oral Microbiology and Immunology, Vol. 9, No. 5, 1994, pp. 284-289. http://dx.doi.org/10.1111/j.1399-302X.1994.tb00072.x
- M. A. Taubman and T. Kawai, “Involvement of T-Lymphocytes in Periodontal Disease and in Direct and Indirect Induction of Bone Resorption,” Critical Reviews in Oral Biology & Medicine, Vol. 12, No. 2, 2001, pp. 125-135. http://dx.doi.org/10.1177/10454411010120020301
- K. Yamashita, et al., “Effect of Adoptive Transfer of Cloned Actinobacillus Actinomycetemcomitans-Specific T Helper Cells on Periodontal Disease,” Infection and Immunity, Vol. 59, No. 4, 1991, pp. 1529-1534.
- H. Yoshie, et al., “Periodontal Bone Loss and Immune Characteristics after Adoptive Transfer of ActinobacillusSensitized T Cells to Rats,” Journal of Periodontal Research, Vol. 22, No. 6, 1987, pp. 499-505. http://dx.doi.org/10.1111/j.1600-0765.1987.tb02061.x
- T. Aoyagi, et al., “Interleukin 4 (IL-4) and IL-6-Producing Memory T-Cells in Peripheral Blood and Gingival Tissue in Periodontitis Patients with High Serum Antibody Titers to Porphyromonas Gingivalis,” Oral Microbiology and Immunology, Vol. 10, No. 5, 1995, pp. 304- 310. http://dx.doi.org/10.1111/j.1399-302X.1995.tb00159.x
- Y. Tokoro, et al., “Relevance of Local Th2-Type Cytokine mRNA Expression in Immunocompetent Infiltrates in Inflamed Gingival Tissue to Periodontal Diseases,” Clinical & Experimental Immunology, Vol. 107, No. 1, 1997, pp. 166-174. http://dx.doi.org/10.1046/j.1365-2249.1997.d01-880.x
- K. Yamazaki, et al., “IL-4- and IL-6-Producing Cells in Human Periodontal Disease Tissue,” Journal of Oral Pathology & Medicine, Vol. 23, No. 8, 1994, pp. 347-353. http://dx.doi.org/10.1111/j.1600-0714.1994.tb00074.x
- A. Blaizot, et al., “Periodontal Diseases and Cardiovascular Events: Meta-Analysis of Observational Studies,” International Dental Journal, Vol. 59, No. 4, 2009, pp. 197-209.
- J. Frostegard, “Immunity, Atherosclerosis and Cardiovascular Disease,” BMC Medicine, Vol. 11, 2013, p. 117. http://dx.doi.org/10.1186/1741-7015-11-117
- J. Frostegard, et al., “Cytokine Expression in Advanced Human Atherosclerotic Plaques: Dominance of Pro-Inflammatory (Th1) and Macrophage-Stimulating Cytokines,” Atherosclerosis, Vol. 145, No. 1, 1999, pp. 33-43. http://dx.doi.org/10.1016/S0021-9150(99)00011-8
- N. Gerdes, et al., “Expression of Interleukin (IL)-18 and Functional IL-18 Receptor on Human Vascular Endothelial Cells, Smooth Muscle Cells, and Macrophages: Implications for Atherogenesis,” Journal of Experimental Medicine, Vol. 195, No. 2, 2002, pp. 245-257. http://dx.doi.org/10.1084/jem.20011022
- Z. Mallat, et al., “Expression of Interleukin-18 in Human Atherosclerotic Plaques and Relation to Plaque Instability,” Circulation, Vol. 104, No. 14, 2001, pp. 1598-1603. http://dx.doi.org/10.1161/hc3901.096721
- G. K. Hansson, et al., “Innate and Adaptive Immunity in the Pathogenesis of Atherosclerosis,” Circulation Research, Vol. 91, No. 4, 2002, pp. 281-291. http://dx.doi.org/10.1161/01.RES.0000029784.15893.10
- K. Kato, et al., “Elevated Levels of Pro-Inflammatory Cytokines in Coronary Artery Thrombi,” International Journal of Cardiology, Vol. 70, No. 3, 1999, pp. 267-273. http://dx.doi.org/10.1016/S0167-5273(99)00093-5
- M. Suzuki, et al., “Relation of C-Reactive Protein and Interleukin-6 to Culprit Coronary Artery Plaque Size in Patients with Acute Myocardial Infarction,” American Journal of Cardiology, Vol. 91, No. 3, 2003, pp. 331-333. http://dx.doi.org/10.1016/S0002-9149(02)03162-4
- C. Habich, et al., “Different Heat Shock Protein 60 Species Share Pro-Inflammatory Activity but Not Binding Sites on Macrophages,” FEBS Letters, Vol. 533, No. 1-3, 2003, pp. 105-109. http://dx.doi.org/10.1016/S0014-5793(02)03772-9
- A. Kol, et al., “Cutting Edge: Heat Shock Protein (HSP) 60 Activates the Innate Immune Response: CD14 Is an Essential Receptor for HSP60 Activation of Mononuclear Cells,” Journal of Immunology, Vol. 164, No. 1, 2000, pp. 13-17.
- K. Ohashi, et al., “Cutting Edge: Heat Shock Protein 60 Is a Putative Endogenous Ligand of the Toll-Like Receptor-4 Complex,” Journal of Immunology, Vol. 164, No. 2, 2000, pp. 558-561.
- W. E. Peetermans, et al., “Mycobacterial 65-Kilodalton Heat Shock Protein Induces Tumor Necrosis Factor Alpha and Interleukin 6, Reactive Nitrogen Intermediates, and Toxoplasmastatic Activity in Murine Peritoneal Macrophages,” Infection and Immunity, Vol. 63, No. 9, 1995, pp. 3454-3458.
- J. S. Friedland, R. J. Shattock and G. E. Griffin, “Phagocytosis of Mycobacterium tuberculosis or Particulate Stimuli by Human Monocytic Cells Induces Equivalent Monocyte Chemotactic Protein-1 Gene Expression,” Cytokine, Vol. 5, No. 2, 1993, pp. 150-156. http://dx.doi.org/10.1016/1043-4666(93)90054-9
- P. Launois, et al., “IL-6 Production in Response to Purified Mycobacterial Heat-Shock Proteins and to Antigen 85 in Leprosy,” Cell Immunology, Vol. 148, No. 2, 1993, pp. 283-290. http://dx.doi.org/10.1006/cimm.1993.1112
- L. M. Macht, et al., “Relationship between Disease Severity and Responses by Blood Mononuclear Cells from Patients with Rheumatoid Arthritis to Human Heat-Shock Protein 60,” Immunology, Vol. 99, No. 2, 2000, pp. 208- 214. http://dx.doi.org/10.1046/j.1365-2567.2000.00966.x
- K. Yamazaki, et al., “Accumulation of Human Heat Shock Protein 60-Reactive T Cells in the Gingival Tissues of Periodontitis Patients,” Infection and Immunity, Vol. 70, No. 5, 2002, pp. 2492-2501. http://dx.doi.org/10.1128/IAI.70.5.2492-2501.2002
- S. Taleb, A. Tedgui and Z. Mallat, “Interleukin-17: Friend or Foe in Atherosclerosis?” Current Opinion in Lipidology, Vol. 21, No. 5, 2010, pp. 404-408. http://dx.doi.org/10.1097/MOL.0b013e32833dc7f9
- C. Prussin and D. D. Metcalfe, “Detection of Intracytoplasmic Cytokine Using Flow Cytometry and Directly Conjugated Anti-Cytokine Antibodies,” Journal of Immunological Methods, Vol. 188, No. 1, 1995, pp. 117-128. http://dx.doi.org/10.1016/0022-1759(95)00209-X
- E. D. Rossmann, et al., “Intracellular T Cell Cytokines in Patients with B Cell Chronic Lymphocytic Leukaemia (B-CLL),” European Journal of Haematology, Vol. 68, No. 5, 2002, pp. 299-306. http://dx.doi.org/10.1034/j.1600-0609.2002.01612.x
- E. M. Caraher, et al., “Flow Cytometric Analysis of Intracellular IFN-Gamma, IL-4 and IL-10 in CD3(+)4(+) T-Cells from Rat Spleen,” Journal of Immunological Methods, Vol. 244, No. 1-2, 2000, pp. 29-40. http://dx.doi.org/10.1016/S0022-1759(00)00249-0
- B. Sander, et al., “Similar Frequencies and Kinetics of Cytokine Producing Cells in Murine Peripheral Blood and Spleen. Cytokine Detection by Immunoassay and Intracellular Immunostaining,” Journal of Immunological Methods, Vol. 166, No. 2, 1993, pp. 201-214. http://dx.doi.org/10.1016/0022-1759(93)90361-A
- I. M. Chalmers, et al., “Intracellular Cytokine Profile of Cord and Adult Blood Lymphocytes,” Blood, Vol. 92, No. 1, 1998, pp. 11-18.
- K. Ueki, et al., “Self-Heat Shock Protein 60 Induces Tumour Necrosis Factor-Alpha in Monocyte-Derived Macrophage: Possible Role in Chronic Inflammatory Periodontal Disease,” Clinical & Experimental Immunology, Vol. 127, No. 1, 2002, pp. 72-77. http://dx.doi.org/10.1046/j.1365-2249.2002.01723.x
- H. de Jong, et al., “Pan-DR-Binding Hsp60 Self Epitopes Induce an Interleukin-10-Mediated Immune Response in Rheumatoid Arthritis,” Arthritis & Rheumatism, Vol. 60, No. 7, 2009, pp. 1966-1976. http://dx.doi.org/10.1002/art.24656
- G. Almanzar, et al., “Autoreactive HSP60 Epitope-Specific T-Cells in Early Human Atherosclerotic Lesions,” Journal of Autoimmunity, Vol. 39, No. 4, 2012, pp. 441- 450. http://dx.doi.org/10.1016/j.jaut.2012.07.006
- K. Tabeta, et al., “Toll-Like Receptors Confer Responsiveness to Lipopolysaccharide from Porphyromonas Gingivalis in Human Gingival Fibroblasts,” Infection and Immunity, Vol. 68, No. 6, 2000, pp. 3731-3735. http://dx.doi.org/10.1128/IAI.68.6.3731-3735.2000
- J. M. Ramage, et al., “T Cell Responses to Heat-Shock Protein 60: Differential Responses by CD4+ T Cell Subsets According to Their Expression of CD45 Isotypes,” Journal of Immunology, Vol. 162, No. 2, 1999, pp. 704- 710.
- P. P. Hujoel, et al., “Periodontal Disease and Coronary Heart Disease Risk,” JAMA, Vol. 284, No. 11, 2000, pp. 1406-1410. http://dx.doi.org/10.1001/jama.284.11.1406
- M. I. Ryder, et al., “Effects of Tobacco Smoke on the Secretion of Interleukin-1beta, Tumor Necrosis FactorAlpha, and Transforming Growth Factor-Beta from Peripheral Blood Mononuclear Cells,” Oral Microbiology and Immunology, Vol. 17, No. 6, 2002, pp. 331-336. http://dx.doi.org/10.1034/j.1399-302X.2002.170601.x
- E. Yamaguchi, et al., “Release of Tumor Necrosis FactorAlpha from Human Alveolar Macrophages Is Decreased in Smokers,” Chest, Vol. 103, No. 2, 1993, pp. 479-483. http://dx.doi.org/10.1378/chest.103.2.479
- Y. Iwamoto, et al., “Antimicrobial Periodontal Treatment Decreases Serum C-Reactive Protein, Tumor Necrosis Factor-Alpha, but Not Adiponectin Levels in Patients with Chronic Periodontitis,” Journal of Periodontology, Vol. 74, No. 8, 2003, pp. 1231-1236. http://dx.doi.org/10.1902/jop.2003.74.8.1231