Journal of Immune Based Therapies, Vaccines and Antimicrobials
Vol. 1 No. 2 (2012) , Article ID: 21263 , 7 pages DOI:10.4236/jibtva.2012.12002
Immune Responses to HSP65/60 in Periodontal Disease
1Unit of Periodontology, King’s College London Dental Institute, London, UK
2Queen Mary, University of London, London, UK
Email: adam.hasan@kcl.ac.uk
Received May 2, 2012; revised June 10, 2012; accepted July 5, 2012
Keywords: Heat Shock Proteins; Periodontitis; IgA; IgG
ABSTRACT
Chronic periodontitis (CP) is a chronic inflammatory condition which destroys the supporting tissues of teeth and increases in prevalence with age. Immune responses against heat shock proteins (HSP) can be cross-reactive among bacterial and human antigens. There is evidence that microbial HSP65 and human HSP60 are involved in periodontal disease. The aim of this study is to investigate immune responses to the human HSP60 and microbial HSP65 in patients with CP and relate these to the level of inflammation and smoking status. We collected serum samples from 30 patients with chronic gingivitis (CG) and 30 patients with CP. In each group, eight subjects were current smokers. ELISA was used to determine the levels of serum anti-HSP and C-reactive protein (CRP) in each group. Peripheral blood mono- nuclear cells were also isolated and stimulated with HSPs. Significant lymphoproliferation was seen in CP when stimulated with human HSP60. CRP and serum anti-human HSP60 IgG were elevated in CP compared to the CG, but not serum anti-microbial HSP 65 IgG. In view of the potential confounding effects of smoking in CP, a group of current smokers (n = 16) was also recruited to investigate whether smoking affects HSP immune responses. There was no significant difference in HSP-induced lymphoproliferation between smokers and non-smokers in either the CG or CP. There was a significant correlation between CRP and lymphoproliferative responses to Human HSP60 irrespective of smoking status. This study shows that serum anti-human HSP60 IgG and serum CRP are raised in untreated CP. In CP, serum CRP levels correlated significantly with human HSP60-induced lymphoprolife ration, but not with anti-HSP antibody levels.
1. Introduction
Chronic periodontitis (CP) is an inflammatory disease characterised by connective tissue destruction and bone resorption, affecting 10% - 15% of the developed world population and is the major cause of tooth loss in adults [1]. The aetiology of CP is associated with a number of bacteria, autoimmunity or microbial cross-reactivity [2]. There is evidence from cross-sectional studies implicating Porphyromonas gingivalis, Tannerella forsythia as well as other microbial species including Gram-negative and Gram-positive bacteria, some of which are unculturable [3]. These findings support the hypothesis that the aetiology of CP is polymicrobial. Serum concentration of CRP, a marker of systemic inflammation, is raised in patients with CP [4,5]. It has proved difficult to identify important antigens amongst the multitude of organisms implicated in periodontitis. HSPs are the most highly conserved group of proteins known in phylogeny with respect to biochemical function, mode of regulation and structure [6,7]. They are expressed in all eukaryotic and prokaryotic cells including Gram-positive and Gram-negative bacteria. The high degree of homology between microbial and human HSPs has led to the hypothesis that tissue damage can occur as a result of cross-reactivity between bacterial and human HSPs. Humoral and T cell responses to HSPs have been demonstrated in CP [5,8- 11], however smoking, a major risk factor and confounder in periodontitis, was not controlled for. The objectives of this investigation are to determine whether serum anti HSP60/65 IgG and IgA are associated with smokers or non-smokers with CG or CP and to determine if these responses are related to inflammation, as reflected by CRP levels, or to smoking status.
2. Material and Methods
2.1. Subjects
Patients with CG and CP were recruited from the Unit of Periodontology at Guy’s and St Thomas’ Foundation Trust (Table 1). All subjects were aged between 38 and 58 years of age and had at least 15 standing teeth. Eight smokers were also recruited and matched in age and condition to the 2 groups (Table 2). Subjects were excluded from the study if they had systemic disease including
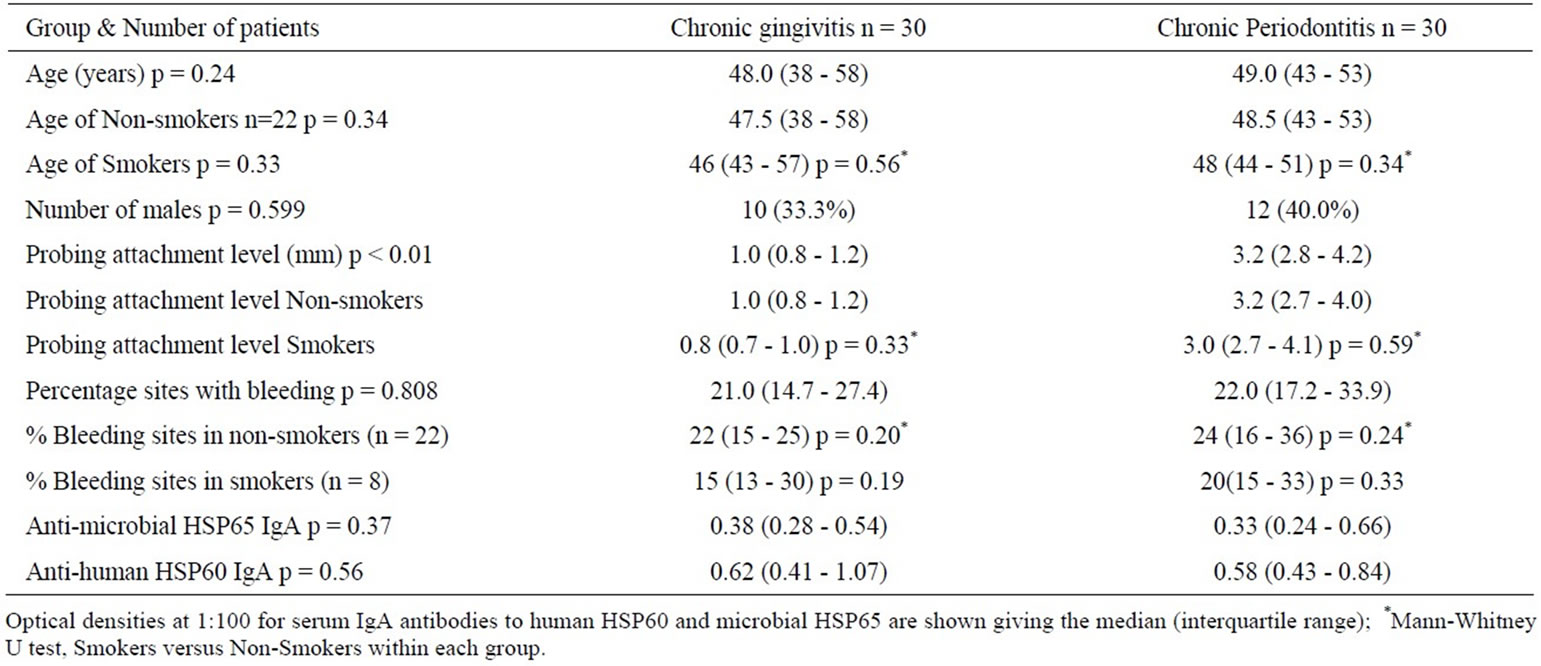
Table 1. Clinical data for the CG and CP, including smokers and non-smokers.

Table 2. Correlation between serum anti-human HSP60 IgG and serum CRP levels to serum anti-microbial HSP65 IgG.
diabetes mellitus, recurrent aphthous stomatitis and autoimmune disease, a history of malignancies, previous treatment for periodontitis, or a history of antibiotic therapy within the past six months prior to recruitment. Subjects were allocated into CG group or CP group. In each subject, probing depths and recession of all teeth were determined using a William’s probe. Recession was measured as the distance from the cemento-enamel junction to the gingival margin. Measurements were taken from 6 sites; probing attachment loss was then calculated as the sum of probing pocket depth and recession. Periodontitis was defined as probing attachment loss ³ 4mm in at least 4 teeth and CG group as probing attachment loss <2 mm in all teeth. Ethical committee approval was obtained (code no 98/12/04) and subject consent obtained. 50ml of venous blood were withdrawn from each subject.
2.2. HSP
Recombinant HSP65 derived from Mycobacterium bovis was prepared at the National Institute of Public Health and Environmental Protection, Bilthoven, the Netherlands and used at a predetermined optimal concentration of 10 µg/ml. Human HSP60 was purchased from Stressgen (Victoria, Canada). The two HSPs were detoxified using Detoxi-gel columns (Pierce, Oxford, UK) and the endotoxin level was determined by Limulus Amoebocyte Lysate assay (Sigma-Aldrich, Poole, Dorset, UK). The concentration of endotoxin was <0.007 U/µg or 7 pg endotoxin/µg for both HSPs.
2.3. Serum ELISA Measurements
Antibodies to the HSP (human HSP60 and microbial HSP65) were detected by enzyme-linked immunosorbent assay. 96-well flat-bottomed polystyrene microtitre plates were coated (Immulon 4 HBX USA) with HSP60 (Stressgen, Victoria, Canada) or microbial HSP65 diluted (1 mg/ml) with phosphate buffered saline (PBS, pH 7.4) and left overnight at room temperature. HSPs were detoxified using Detoxigel columns (Pierce, Oxford, UK) and the endotoxin level was determined by Limulus Amoebocyte Lysate assay (Sigma-Aldrich, Poole, Dorset, UK). Uncoated sites were then blocked with 0.5% wt/vol BSA (Sigma-Aldrich Irvine UK) in PBS (200 ml/well) for 60 minutes at room temperature. In addition, human sero-negative and sero-positive samples were identified by running the whole series of patient serum samples and obtaining the most positive and negative samples as positive and negative controls respectively. Serum from rabbits previously immunised with microbial HSP65 and human HSP60 (KCL Dental Institute, London) was also used as positive control. Serum samples obtained from patients including positive and negative controls were then diluted as follows in PBS containing 0.5% BSA and 0.05% Tween 20; 2 fold dilution of serum at 1:100 for anti-human HSP60 IgG, anti-microbial HSP65 IgG and anti-HSP60 IgA, 2 fold dilution of serum at 1:50 for anti-microbial HSP65 IgA. 100 ml of each diluted sample or positive/negative control were then added in duplicates to each well and serially diluted to 1:6400, except for anti-microbial HSP65 IgA which had a final dilution of 1:3200.
Plates were then incubated at room temperature for 2 hours and washed 4 times with PBS containing 0.05% Tween 20. Secondary antibody goat anti-human IgG, or IgA Fc specific, alkaline phosphatase conjugates (Sigmaaldrich, UK) were diluted in diluent buffer to an optmal concentration. 100 ml were then added to each well and incubated at room temperature for 2 hours. Anti-human IgG alkaline phosphatase conjugate was used at 1:2000. Anti-human IgA alkaline phosphatase conjugate was used at 1:1000 for detection of serum anti-human HSP60 IgA and at 1:500 for the detection of serum antimicrobial HSP65 IgA. Plates were then washed 4 times with wash buffer and were developed with para-nitrophenylphosphate in diethanolamine buffer (pH 9.8) at room temperature. The absorbance was detemined using a microplate reader (Anthos 2001, Anthos labtec instruments UK) for IgG, and (Opsys MR DYNEX technologies) for IgA at 405 nm with wavelength correction at 620 nm. Inter-assay variation was monitored using a standard positive serum in each assay [12]. The variation of the titre of this positive serum was within one dilution step.
2.4. Separation of Cells
Peripheral blood mononuclear cells (PBMC) were separated from blood by density gradient centrifugation and cultured as described previously (Pervin et al. 1993). The results were assessed by calculating the stimulation index (SI), which is the ratio of antigen-stimulated to antigenunstimulated cultures [13].
2.5. Detection of C-Reactive Protein by ELISA
50 mL of pre-diluted standard and blank were added to a 96 well plate precoated with anti-serum CRP IgG (Kalon Biological Ltd). Serum samples were diluted 1:1000 with assay diluent (Kalon Biological Ltd.) and dispensed in duplicates to designated wells in CRP precoated plates. The Plate was then incubated at room temperature for 60 minutes. Plates were washed 4 times with wash buffer (Kalon Biological Ltd). 100 mL of CRP tracer (affinity purified sheep anti-CRP labelled with alkaline phosphatase, Kalon Biological Ltd UK) were then dispensed to each well and incubated uncovered for 30 minutes at room temperature. Plates were washed again 4 times with washing buffer (Kalon Biological Ltd, UK).
100 ml of substrate solution (4-nitrophenylphosphate in substrate buffer Kalon Biological Ltd.) were then dispensed to each well and incubated at room temperature for 30 minutes. The reaction was stopped with 100 ml of (120 g/L) sodium hydroxide. Optical densities were read at 405 nm with microplate reader (Anthos 2001, Anthos labtec instruments UK). A standard curve was constructed with standard points and curve fitted with four parameter logistic curve fitting software. Test serum values were then read off the standard curve.
2.6. Statistical Analysis
Relationships between variables were analysed using Kruskal-Wallis ANOVA or Mann-Whitney U test. A power calculation was used to determined how many smokers needed to be recruited to show a difference in OD of 0.2 with a standard deviation of 0.17 (80% power; alpha = 0.05); this indicated 8 subjects were required. ELISA for serum anti-HSP IgG or IgA was analysed by using data consisting of optical density readings for the performed test. The mean optical density was then analysed at 1:100. Inter-relationships between serum antiHSP60 IgG and serum CRP levels were tested using Spearman ranked correlation coefficients (rs). Non-parametric data are displayed in graphs as median (1st and 3rd quartile, minimum, maximum).
3. Results
3.1. Clinical Data
We recruited 60 patients (44 non-smokers, 16 current smokers) divided into 2 groups. The median (interquartile range) age of the subjects in the gingivitis group was 48 (38 - 58) years, and 49 (43 - 53) years in the CP group and there was no statistically significant difference between the groups (p = 0.24, Table 1). The current smokers were matched for age, gender, and disease status with nonsmokers from the 2 groups. There were no significant differences in the age and periodontal status of nonsmokers and smokers in the 2 groups (p > 0.05). For the non-smokers, the median (interquartile range) probing attachment level in the CG group was 1.0 (0.8 - 1.2) mm, and in the CP group 3.2 (2.7 - 4.0) mm (Table 1). Serum cotinine levels were determined by ELISA to help ensure no current smokers were included in the non-smoking groups. Cotinine levels were significantly lower for nonsmokers (median [interquartile range], 0.1 [0.10 - 0.12] in CG and 0.1 [0.10 - 0.11 in CP] respectively) compared to current smokers (253 [177 - 351] in CG, p < 0.001; and 255 [176 - 351] in CP respectively; p < 0.001).
3.2. IgA Antibodies to Microbial HSP65 and Human HSP60
The OD at 1:100 dilution of serum for anti-microbial HSP65 IgA was 0.38 (0.28 - 0.54) in the CG group and 0.33 (0.24 - 0.66) in the CP group; this difference was not significant (p = 0.36, Table 1). Similarly, there was no difference in serum anti-human HSP60 IgA (Table 1). The median (interquartile range) OD at 1:100 dilution of serum was 0.62 (0.41 - 1.07) in the gingivitis group and 0.58 (0.43 - 0.84) in the CP group (p = 0.56, Table 1).
3.3. IgG Antibodies to Microbial HSP65 and Human HSP60
The median (interquartile range) optical density at 1:100 dilution of serum for anti-microbial HSP65 IgG was 0.34 (0.17 - 0.63) in the gingivitis group, and 0.46 (0.25 - 0.85) in the CP group (Figure 1(a)). There was no significant difference amongst the 2 groups in serum anti-HSP65 IgG titres (p = 0.661). The median (interquartile range) OD of 1:100 dilution of serum for anti-human HSP60 IgG was 0.26 (0.17 - 0.34) in the gingivitis group and 0.87 (0.42 - 1.27) in the CP group and this was statistically significant (p < 0.001) (Figure 1(b)).
Serum levels of anti-microbial HSP65 IgG were only significantly associated with serum levels of anti-human HSP60 IgG in the CP group (rs = 0.538, p = 0.005) (Table 2). No statistically significant relationship was revealed between serum anti-microbial HSP65 IgG and serum anti-human HSP60 IgG in the CG group (rs = 0.005, p = 0.983) (Table 2).
3.4. Lymphoproliferative Responses to Human HSP60 in Smokers and Non-Smokers
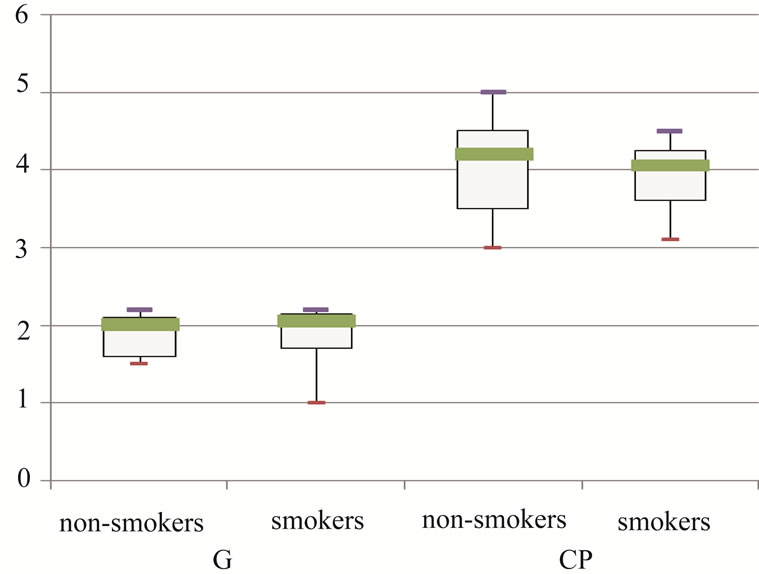
An analysis of lymphoproliferative responses of smokers and non-smokers in each group yielded no significant differences within either group (Figure 2). All CP groups responded significantly to human HSP60, in contrast to the CG which yielded a median (interquartile range) stimulation index of 2.0 (1.65 - 2.10) (p < 0.001). Smoking does not appear to influence HSP60-induced proliferation in CP.
3.5. CRP
The median (interquartile range) of serum CRP was 0.64
(0.18 - 1.86) mg/L in the CG group, and 1.27 (1.01 - 2.83) mg/L in the CP group (Figure 2, p = 0.007). Serum CRP levels were not significantly associated with serum anti-human HSP60 IgG levels in the CG group (rs = 0.115, p = 0.583), or the CP group (rs = –0.194, p = 0.352) (Table 2). In smokers and non-smokers with CP, HSP proliferative responses correlated moderately with CRP levels (rs = 0.550, p = 0.004 and rs = 0.553, p = 0.005).
4. Discussion
The results show that serum anti-human HSP60 IgG but not serum anti-microbial HSP65 IgG was elevated in patients with CP compared to CG irrespective of smoking status. No significant differences were noted in the level of serum anti-human HSP60 IgA and serum antimicrobial HSP65 IgA between CG group and patients with CP. These findings contrast with a study showing an elevation of serum anti-HSP65 IgG [11] and another study showing depression of serum anti-HSP65 IgA with no difference in serum anti-human HSP60 IgG and serum
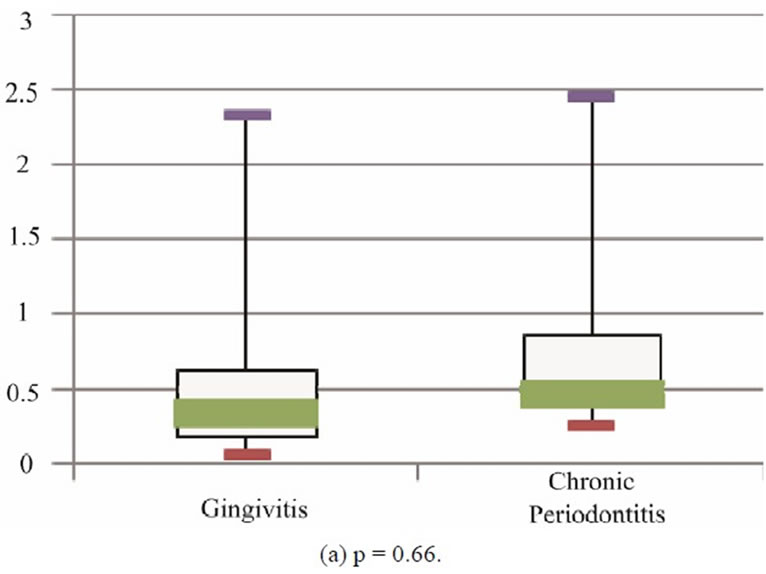
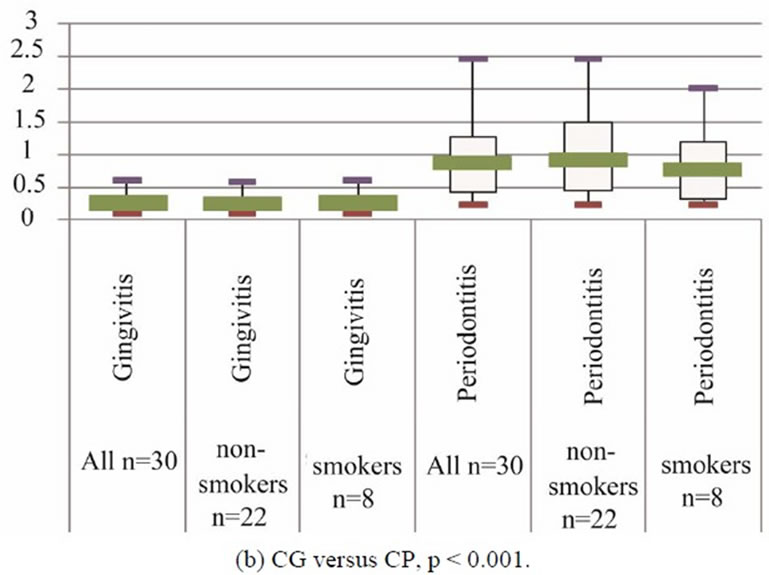
Figure 1. Boxplots showing median, interquartile range, minimum and maximum OD for serum anti-microbial HSP65 IgG.
 (a)
(a)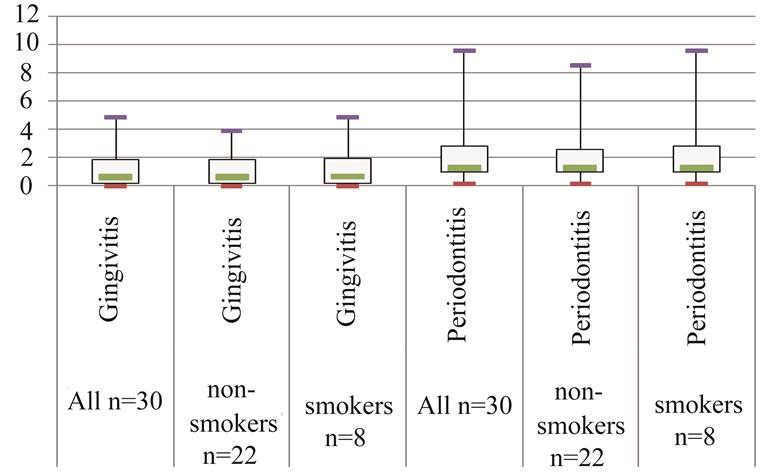 (b)
(b)
Figure 2. (a) Boxplots showing median, interquartile range, minimum and maximum stimulation index of Human HSP60-induced lymphoproliferative responses in non-smokers and smokers(n = 8 for each group). CP responded signifycantly to HSP60 (ANOVA, p < 0.001); (b) Boxplots showing median, interquartile range, minimum and maximum level of serum C-reactive protein (mg/L) amongst the 2 groups of patients (There is a significant difference between CG and CP by ANOVA, p = 0.002).
anti-microbial HSP65 IgG in CP patients compared to the CG [5]. However these studies are difficult to compare directly as there are significant differences in demographical and clinical parameters. In one of these studies the patient group had a mean age of 37.5 ± 12.6 years range 26 - 65 years [11]; with this age range it is likely that more than one form of periodontitis was includeeed. Our study included only patients with moderate-advanced chronic periodontitis or chronic gingivitis. The median age (interquartile range) of our patients with chronic periodontitis was 49 (43 - 53). Younger aged patients with advanced periodontitis may represent a different diagnostic category or a different spectrum of response from older patients suffering from chronic periodontitis [14], and may mount different immune responses [15].
The recruitment of patients who are undergoing treatment for chronic periodontitis [5] is problematic and may yield conflicting results as treatment of CP has a modulating effect on humoral immune responses to HSP [16] and instrumentation may immunise patients with plaque antigens. To minimise the confounding effect of treatment in our results we recruited untreated cases. Our findings are in agreement with studies where the mean ages of untreated chronic periodontitis were similar [9, 10]. These studies demonstrated a significant elevation in serum anti-human HSP60 IgG compared to a control group [10]. In our study there was a moderate correlation between serum anti-human HSP60 IgG and serum antimicrobial HSP65 IgG in patients with CP, and therefore the possibility of cross-reactivity exists. Once the epitopes recognised in CP have been mapped, the role of T and B cell epitopes in relation to inflammatory and antiinflammatory responses can be elucidated.
Smoking is a risk factor for chronic periodontitis [17] and has been suggested to be a confounding variable in CP [18]. Smoking is known to modulate immune responses and in chronic periodontitis there is a direct correlation between serum cotinine levels and serum levels of intracellular adhesion molecule [19]. The role of smoking on the cellular and humoral immune responses to HSP in patients with chronic periodontitis has not been fully elucidated although, exposure to smoking reduces the release of TNF-α in macrophages and PBMC [20,21]. We found no difference between smokers and nonsmokers in any of the subject groups suggesting that at least HSP-induced proliferation is not affected by smoking. It however remains to be determined whether smoking affects recognition of HSP-epitopes and alters HSPinduced cytokine production in CP.
Serum CRP levels in patients with CP when compared to CG group were significantly elevated. Previous studies have shown that serum CRP levels are elevated in CP [5,22,23]. No significant correlation was shown between CRP and serum anti-HSP60 IgG levels. This is in agreement with previous reports in the literature which do not demonstrate significant relationship between CRP and serum anti-human HSP60 levels [24,25]. The correlation of CRP and HSP-proliferative responses suggests that inflammation may play a role in human HSP 60-induced lymphoproliferation in CP. We have previously demonstrated that HSP60/65 lymphoproliferation in male patients with chronic periodontitis involves class II restricted CD4+ T cells. Our findings support a previous study demonstrating significant proliferative responses of PBMC to human HSP60 in CP [26].
HSP60-reactive PBMC produced significant levels of gamma interferon (IFN-gamma) in patients with CP [26].
Analysis of the nucleotide sequences of the TCR by the authors demonstrated that human HSP60-reactive T-cell clones and periodontitis lesion-infiltrating T-cells have the same receptors, suggesting that HSP60-reactive T-cells accumulate in periodontitis lesions [26]. The T cells did not however respond to Porphyromonas gingivalis HSP65 [26]. In contrast, another study showed depressed responses of PBMC to human HSP60 and microbial HSP65 in patients with periodontitis with reduced production of interferon gamma in the patient group compared to the control group [27]. These conflicting results probably reflect differences in disease duration, severity, and patient age.
5. Conclusion
This study demonstrated that serum anti-human HSP60 IgG but not serum anti-microbial HSP65 IgG is elevated in patients with CP indicating there are autoantibodies to human HSP60 in CP and together with our previous findings of autoimmune T cell response to human HSP provides further support for cross-reactivity in the pathogenesis of CP. Serum CRP levels were also significantly raised in the CP, but there was no significant relationship between serum CRP levels with serum anti-human HSP 60 IgG levels. These HSP responses need to be further characterisied to identify immunodominant epitopes and their relationship to inflammatory cytokine production so that potential therapeutic interventions can be explored.
REFERENCES
- D. Locker, G. D. Slade and H. Murray, “Epidemiology of Periodontal Disease among Older Adults: A Review,” Periodontology 2000, Vol. 16, No. 1, 1998, pp. 16-33. doi:10.1111/j.1600-0757.1998.tb00113.x
- M. A. Listgarten and P. M. Loomer, “Microbial Identification in the Management of Periodontal Diseases. A Systematic Review,” Annals of Periodontology, Vol. 8, No. 1, 2003, pp. 182-192.
- P. S. Kumar, et al., “New Bacterial Species Associated with Chronic Periodontitis,” Journal of Dental Research, Vol. 82, No. 5, 2003, pp. 338-344. doi:10.1177/154405910308200503
- S. Amar, et al., “Periodontal Disease Is Associated With Brachial Artery Endothelial Dysfunction and Systemic Inflammation,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 23, No. 7, 2003, pp. 1245-1249. doi:10.1161/01.ATV.0000078603.90302.4A
- K. Buhlin, et al., “Risk Factors for Cardiovascular Disease in Patients with Periodontitis,” European Heart Journal, Vol. 24, No. 23, 2003, pp. 2099-2107. doi:10.1016/j.ehj.2003.09.016
- S. Jindal, et al., “Primary Structure of a Human Mitochondrial Protein Homologous to the Bacterial and Plant Chaperonins and to the 65-Kilodalton Mycobacterial Antigen,” Molecular and Cellular Biology, Vol. 9, No. 5, 1989, pp. 2279-2283.
- J. E. R. Thole, et al., “Antigenic Relatedness of a Strongly Immunogenic 65 kDA Mycobacterial Protein Antigen with a Similarly Sized Ubiquitous Bacterial Common Antigen,” Microbial Pathogenesis, Vol. 4, No. 1, 1988, pp. 71-83. doi:10.1016/0882-4010(88)90049-6
- A.Hasan, et al., “The immune responses to human and microbial heat shock proteins in periodontal disease with and without coronary heart disease,” Clinical & Experimental Immunology, Vol. 142, No. 3, 2005, pp. 585-594.
- D. E. Lopatin, et al. “Humoral Immunity to Stress Proteins and Periodontal Disease,” Journal of Periodontology, Vol. 70, No. 10, 1999, pp. 1185-1193. doi:10.1902/jop.1999.70.10.1185
- K. Tabeta, et al., “Elevated Humoral Immune Response to Heat Shock Protein 60 (hsp60) Family in Periodontitis Patients,” Clinical & Experimental Immunology, Vol. 120, No. 2, 2000, pp. 285-293. doi:10.1046/j.1365-2249.2000.01216.x
- G. Schett, et al., “Salivary Anti-hsp65 Antibodies as a Diagnostic Marker for Gingivitis and a Possible Link to Atherosclerosis,” International Archives of Allergy and Immunology, Vol. 114, No. 3, 1997, pp. 246-250.
- H. Direskeneli, et al., “Recognition of B-Cell Epitopes of the 65 kDa HSP in Behçet’s Disease,” Scandinavian Journal of Immunology, Vol. 43, No. 4, 1996, pp. 464- 471. doi:10.1046/j.1365-3083.1996.d01-53.x
- K. Pervin, et al., “T Cell Epitope Expression of Mycobacterial and Homologous Human 65-Kilodalton Heat Shock Protein Peptides in Short Term Cell Lines from Patients with Behcet’s Disease,” The Journal of Immunology, Vol. 151, No. 4, 1993, pp. 2273-2282.
- R. R. Ranney, “Classification of Periodontal Diseases,” Periodontology 2000,” Vol. 2, No. 1, 1993, pp. 13-25. doi:10.1111/j.1600-0757.1993.tb00216.x
- J. C. Gunsolley, et al., “Serum antibodies to periodontal bacteria,” Journal of Periodontology, Vol. 61, No. 7, 1990, pp. 412-419. doi:10.1902/jop.1990.61.7.412
- K. Yamazaki, et al., “Effect of Periodontal Treatment on the Serum Antibody Levels to Heat Shock Proteins,” Clinical & Experimental Immunology, Vol. 135, No. 3, 2004, pp. 478-482. doi:10.1111/j.1365-2249.2003.02375.x
- R. Genco, S. Offenbacher and J. Beck, “Periodontal Disease and Cardiovascular Disease,” The Journal of the American Dental Association, Vol. 133, 2002, pp. 14S- 22S.
- P. P. Hujoel, et al., “Periodontal Disease and Coronary Heart Disease Risk,” The Journal of the American Medical Association, Vol. 284, No. 11, 2000, pp. 1406-1410. doi:10.1001/jama.284.11.1406
- R. M. Palmer, et al., “Potential Mechanisms of Susceptibility to Periodontitis in Tobacco Smokers,” Journal of Periodontal Research, Vol. 34, No. 7, 1999, pp. 363-369. doi:10.1111/j.1600-0765.1999.tb02267.x
- M. I. Ryder, et al., “Effects of Tobacco Smoke on the Secretion of Interleukin-1β, Tumor Necrosis Factor-α, and Transforming Growth Factor-β from Peripheral Blood Mononuclear Cells,” Oral Microbiology and Immunology, Vol. 17, No. 6, 2002, pp. 331-336. doi:10.1034/j.1399-302X.2002.170601.x
- E. Yamaguchi, et al., “Release of Tumor Necrosis Factor-Alpha from Human Alveolar Macrophages is Decreased in Smokers,” Chest, Vol. 103, No. 2, 1993, pp. 479-483. doi:10.1378/chest.103.2.479
- M. Ide, et al., “Effect of Treatment of Chronic Periodontitis on Levels of Serum Markers of Acute-Phase Inflammatory and Vascular Responses,” Journal of Clinical Periodontology, Vol. 30, No. 4, 2003, pp. 334-340. doi:10.1034/j.1600-051X.2003.00282.x
- F. D’Aiuto, et al., “Periodontitis and Systemic Inflammation: Control of the Local Infection is Associated with a Reduction in Serum Inflammatory Markers,” Journal of Dental Research, Vol. 83, No. 2, 2004, pp. 156-160. doi:10.1177/154405910408300214
- A. Veres, et al., “Relationship of Anti-60 kDa Heat Shock Protein and Anti-Cholesterol Antibodies to Cardiovascular Events,” Circulation, Vol. 106, No. 22, 2002, pp. 2775-2780. doi:10.1161/01.CIR.0000038890.39298.8D
- J. Zhu, et al., “Antibodies to Human Heat-Shock Protein 60 Are Associated With the Presence and Severity of Coronary Artery Disease: Evidence for an Autoimmune Component of Atherogenesis,” Circulation, Vol. 103, No. 8, 2001, pp. 1071-1075. doi:10.1161/01.CIR.103.8.1071
- K. Yamazaki, et al., “Accumulation of Human Heat Shock Protein 60-Reactive T Cells in the Gingival Tissues of Periodontitis Patients,” Infection and Immunity, Vol. 70, No. 5, 2002, pp. 2492-2501. doi:10.1128/IAI.70.5.2492-2501.2002
- M. D. A. Petit, et al., “Depressed Responsiveness of Peripheral Blood Mononuclear Cells to Heat-shock Proteins in Periodontitis Patients,” Journal of Dental Research, Vol. 78, No. 8, 1999, pp. 1393-1400. doi:10.1177/00220345990780080401