Neuroscience & Medicine
Vol.1 No.1(2010), Article ID:2675,13 pages DOI:10.4236/nm.2010.11001
Nanog Overexpression Allows Human Mesenchymal Stem Cells to Differentiate into Neural Cells*
——Nanog Transdifferentiates Mesenchymal Stem Cells
![]()
Biomolecular Science Center, Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, Orlando, USA.
Email: ksugaya@mail.ucf.edu
Received June 7th, 2010; revised July 15th, 2010; accepted July 20th, 2010.
Keywords: stem cells, dedifferentiation, transplantation, Nanog
ABSTRACT
Although stem cell therapies have been proposed as a candidate for treating neurological diseases, the best stem cell source and their therapeutic efficacy remain uncertain. Embryonic stem cells (ESCs) can efficiently generate multiple cell types, but pose ethical and clinical challenges, while the more accessible adult stem cells have a limited developmental potential. Following included-expression of Nanog, an ESC gene, adult human mesenchymal stem cells (HMSCs) are able to develop into cells exhibiting neural cell-like characteristics based on morphology, cell markers, and gene expressions. After expansion, Nanog overexpressed HMSCs differentiated into cells immunopositive for bIII-tubulin and glial fibrillary acidic protein, lineage markers for neurons and astrocytes, respectively, under the influence of conditional media from differentiated human neural stem cells. This result indicates that the Nanog expression increased the ability of HMSCs to become a neural cell lineage. We further demonstrated that Nanog-overexpressed HMSCs were able to survive, migrate, and undergo neural cell-like differentiation after transplantation in vivo. This data offers an exciting prospect that peripheral adult stem cells can be modified and used to treat neurological diseases.
1. Introduction
The central nervous system is one of the most limited systems in the human body in terms of regeneration and recovery after cell loss [1]. While many pharmacological treatments mediate symptoms, they fail to cure neurological diseases. Today, many researchers are investigating stem cells as potential therapeutics to overcome this issue.
The use of embryonic stem cells (ESCs) has been proposed as the most promising strategy for treatment of neurological diseases because of their pluripotency to become a variety of tissues, but concerns over ethics [2-4], immune response [5,6] and tumor formation [7-9] have been major barriers for their clinical use. Adult stem cells are known to exist throughout the body, and they can be harvested from a patient and autologously transplanted back to the patient. The autologous approach will eliminate the issues associated with the use of ESCs. However, the ability of adult stem cells to develop along multiple lineages is limited by their tissue origin. Thus to regenerate neural tissue, neural stem cells (NSCs) isolated from brain tissue are needed. However, finding the tissue within a patient from which to isolate them may be difficult. Utilization of other easily accessible adult stem cells, such as mesenchymal stem cells (MSCs) found in the bone marrow, could eliminate the difficulty of acquiring transplantable material. Although studies have claimed that human MSCs (HMSCs) transdifferentiate into cells outside their restricted germ layer, the transdifferentiation could have been from a very limited population of HMSCs [10,11] or due to the low frequencies of cell fusion, which allow HMSCs to acquire characteristics of multiple cell types by fusing to somatic cells [12,13]. Therefore, a strategy to increase the transdifferentiation abilities of adult stem cells is requisite for their use in neuroreplacement therapies.
In a previous study, we demonstrated that HMSCs treated with bromodeoxyuridine (BrdU), which is incorporated into the DNA as a thymidine analog, undergo neural differentiation following transplantation in the brains of rats and improve cognitive function [14]. This result indicates that epigenetic modification of the adult stem cells may increase their potency. However, efficiency of transdifferentiation and concerns with the nonspecific epigenetic modification, led us to explore other strategies.
One possible strategy is cell fusion that would alter the characteristics of adult stem cells based on the exogenous cell used for merging. This method could change the transdifferentiation ability of cells allowing them to develop into cells beyond their respective lineage [15-17]. The fusion of somatic cells to ESCs prompts expression of the ESC gene Oct-4 [17,18]. Thus, the expression of stem cell genes that regulate self-renewal and pluripotency may play an integral role in reprogramming the cell lineage.
Earlier studies have indicated that the expression of critical stem cell genes is capable of maintaining ESCs in a pluripotent state. The over-expression of ESC genes, including Nanog [19,20], Pem [21] and Rex1 [22], suppressed differentiation of ESCs, while the presence of elevated levels of Oct-4 was insufficient to guard against ESCs differentiation [23]. In this study, we tested our hypothesis that developmental ability of HMSCs can be increased by changing the gene expression profile through the overexpression of Nanog, and the resulting cells can be transdifferentiated into neural cells. This technology may allow us to perform autologous therapies to treat neurological diseases using patients’ own HMSCs.
2. Materials and Methods
Cell culture: Adult human bone marrow-derived HMSCs (Cambrex) were cultured in DMEM (Invitrogen) supplemented with 1% antibiotics (Invitrogen) and 10% FBS for improved HMSC growth (StemCell Technologies). Per Cambrex product information, mesenchymal stem cells are harvested and cultured from normal human bone marrow. Cell purity is far higher than cells from traditional Dexter cultures. Cells are tested for purity by flow cytometry and for their ability to differentiate into osteogenic, chondrogenic and adipogenic lineages. Cells are positive for CD105, CD166, CD29, and CD44. Cells test negative for CD14, CD34 and CD45. Media systems are available to support growth of HMSCs, and their differentiation into adipogenic, chondrogenic, and osteogenic lineages. Cells were cultured in T75 tissue culture treated flasks (BD Biosciences) and incubated in a CO2 chamber at 37℃ with 5% CO2 (NuAire). Co-culture experiments were carried out using differentiated NSCs in Falcon tissue culture treated 6-well plates (BD Biosciences). Prior to co-culture, fetal-derived human NSCs (Cambrex) were expanded in serum-free NSC medium of DMEM/F12 (Invitrogen) supplemented with B27 (1:50, Invitrogen), basic Fibroblast Growth Factor (bFGF, 20 ng/ml, R&D Systems), Epidermal Growth Factor (EGF, 20 ng/ml, R&D Systems), heparin (0.18 U/ml, Sigma), and 1% antibiotics (Invitrogen). Cells were allowed to spontaneously differentiate for one week in tissue culture treated 6-well plates containing serum-free neural basal medium.
For co-culture, cell culture inserts with a semi-permeable membrane with 0.4 µm pores (BD Biosciences) were used to separate the Nanog-transfected HMSCs from the differentiated HNSCs. This allowed for the dynamic exchange of secreted factors and eliminated direct cell contact to avoid possible cell fusion. Nanogor mock-transfected HMSCs were then transferred to co-culture to promote neural differentiation. To eliminate Nanog expression in viral-loxP-Nanog-transduced HMSCs prior to the co-culture, plasmids containing the Crerecombinase gene regulated by an EF1a promoter (Addgene, plasmid 11918) were transfected into the cells using the FuGene 6 reagent. Cells were allowed to differentiate for 10 days and then stained for early (bIIItubulin) and mature (MAP2) neuronal markers and astrocytic markers, GFAP and S100.
Cloning of Nanog gene: Nanog was originally cloned from male genomic DNA that was pre-digested with the restriction enzymes NotI, XbaI, and SpeI, then amplified by PCR using Nanog-specific primers, (CGTTCTGCTGGACTGAGCTGGTT, CGGGCGGATCACAAGGTCAG). PCR conditions consisted of pre incubation at 94℃ for three minutes, 30 cycles consisting of 94℃ for one minute, 52℃ for 30 seconds, and 72℃ for three minutes, and post dwells at 72℃ for 10 minutes. The PCR product was then placed into a mammalian expression vector (TopoHisMax 4.1, Invitrogen) according to manufacturer’s protocol. The cloned sequence was confirmed by DNA sequencing.
Production of lentivirus containing Nanog: The gene encoding for Nanog (gift from Austin Smith, MD University of Cambridge) was amplified using the Herculase II fusion DNA polymerase (Promega) and gene-specific primers containing a BamHI enzyme-cutting site in the forward primer and a SalI-cutting site in the reverse primer (ATAGGGATCCACATGAGTGTTGACCCAGCTT, ATAGGTCGACTCACACGTCTTCAGGTTGCA). The PCR amplified Nanog was sub-cloned into the pLox lentiviral vector (gift from Didier Trono, MD and Patrick Salmon, MD, LVPU, Centre MédicalUniversitaire, Genève, Switzerland).
Production of a lentiviral vector containing the Nanog sequence was carried out using a vector containing a LoxP site. The pLoxNanog vector, the packaging vector pCMVDR8.91 (AddGene) encoding for regulatory proteins Tat and Rev as well as the Gag and Pol precursors, and a vector for the envelope protein VSV-G (Clontech) were used for viral production. The aforementioned vectors and lentiviral vectors pLoxNanog and pLoxGFP, combined with the packaging and envelope plasmids at a ratio of 2:1:1 (pLox:pCMV∆R8.91:pVSV-G) [24,25], were transiently transfected into the HEK293T/17 cell line (ATCC) using Lipofectamine (Invitrogen) at a DNA (20mg) to Lipofectamine ratio of 1:2.5. The cell culture media was removed at 24 hours and collected every 12 hours thereafter for the next two days to harvest the viral supernatant.
Non-viral and viral gene delivery: For non-viral gene transfection, 75% confluent HMSCs were transfected with 3 µg of Nanog vector using two different reagents, Neuroporter (Gene therapy systems) or FuGene 6 (Roche), at DNA to reagent ratios of 1:15 and 1:3, respectively. Proliferative clusters began to emerge after one week and grew large enough for expansion typically by three weeks. Clustered cells that resembled Nanog-transfected HMSCs were passed by mechanical dissociation from the feeder layer and subsequently plated with a feeder cell layer of HMSCs.
For lentivirus-mediated transfection, viral supernatant was transferred to HMSC cultures for viral transduction. Delivery of Nanog was analyzed through fluorescent microscopy for positive green fluorescent protein (GFP) expression. Differentiation was induced through the deletion of the Nanog-containing proviral sequence with a vector encoding for Crerecombinase (Addgene pBS513) [26]. The Cre vector, which contains an EF1a promoter [26,27], was delivered to the cells through chemical transfection. Following Cre-transfection, cells were used for neural differentiation or gene expression analysis at 72 hours post-transfection. All recombinant DNA research was performed in accordance with NIH guidelines.
Gene expression analysis: RNA extraction was performed using TRIzol (Invitrogen) according to manufacturer’s instructions. Media was removed from cultured cells and incubated with 1ml of TRIzol for five minutes at room temperature. Reverse transcription was performed using an iScriptcDNA synthesis kit (Bio-Rad).
Primers used for RT-PCR were GAPDH (AGCCACATCGCTCAGACACC, GTACTCAGCGGCCAGCATCG), b-actin (TCCTGAGCGCAAGTACTCC, AAGCATTTGCGGTGGACGA), Nanog (ACAACTGGCCGAAGAATAGC, AGTGTTCCAGGAGTGGTTGC), Oct-4 (CTTGCTGCAGAAGTGGGTGGAGGAA, CTGCAGTGTGGGTTTCGGGCA), TERF1 (GCAACAGCGCAGAGGCTATTATT, AGGGCTGATTCCAAGGGTGTAA), Sox-2 (ATGCACCGCTACGACGTGA, CTTTTGCACCCCTCCCATTT), ZFP342 (GAAGGCATCACcCAAAAAGA, GCGGTTGAGCTTACTGCTCT), TERT (CGGAAGAGTGTCTGGAGCAA, GGATGAAGCGGAGTCTGGA), and eGFP (CCTGAAGTTCATCTGCACCA, GGTCTTGTAGTTGCCGTCGT).
Real-time two-step RT-PCR was performed using a SYBR green PCR mix (Bio Rad), carried out in a MyIQiCycler (Bio Rad) and then analyzed by the DCt method as previously described [28,29].
Stem cell transplantation: Two different transplantation studies were performed with C57/Black mice at four months of age in accordance with approved protocols from the University of Central Florida’s Institutional Animal Care and Use Committee.
The animals were fixed in a stereotaxic apparatus and approximately 1x105 cells in 10 µl of phosphate-buffered saline (PBS) were slowly injected into the right lateral ventricle (coordinates: AP -1.4, ML 1.8, DV 3.8). Experiments were carried out independently using HMSCs dedifferentiated through non-viral transfection or lentiviral transduction.
Brain sample preparation: Animals were deeply anesthetized and perfused using a 10% sucrose solution followed by fixation with a 4% paraformaldehyde PBS (pH 7.2). Following fixation, brains were removed and placed inside a 20% sucrose/4% paraformaldehyde solution and left overnight at 4℃. When the brain settled to the bottom of the container, it was froze in isopentane pre-cooled by submerging the beaker into liquid nitrogen. The brains were mounted using a cryomedium, sliced into 20 mm sections using a cryostat at –20℃ and collected in PBS and stored at 4℃ until antibody staining.
Immunocytochemistry and immunohistochemistry: Cultured cells were washed with PBS then fixed with a 4% buffered paraformaldehyde (Sigma) solution overnight at 4℃. Following fixation, cells were washed with PBS (Sigma) then permealized with PBS-Tween (Sigma) containing 0.1% Triton-X (Fisher Scientific) for one hour at room temperature. Brain sections were washed with PBS then permealized by incubation in PBS-Tween with 0.1% Triton-X at room temperature for one hour. The samples were then incubated for one hour at room temperature in a blocking solution of PBS-Tween with 3% donkey serum (Jackson ImmunoResearch). Primary antibodies TRA-1-60 (MAB4360), SSEA-3 (mab4303), Sox-2 (AB5603), MAP2 (AB5622), and Oct-4 (mab4305) all from Chemicon, Nanog (AF1997, R&D Systems), bIII-tubulin (T8660, Sigma), S100 (S2644, Sigma), and GFAP (G9269, Sigma) were added to blocking solution and incubated overnight at 4℃. The next day, samples were washed with PBS and incubated in the dark with FITCor TRITC-conjugated secondary antibodies at room temperature. Samples were washed with PBS, cover-sliped with water-based mounting solution containing DAPI (Vector Laboratories), and sealed using clear nail polish.
3. Results
Cloning of Nanog: Sequence analysis of the clone established from genomic DNA showed over 99% sequence identity with Nanog but did not contain introns, suggesting that it may be Nanogpseudogene 8 (NANOGP8) [30], one of twelve Nanog variants [30-32]. The high homology and intact coding region suggests that the cloned sequence should be indistinguishable from Nanog and the translated product virtually identical to the actual Nanog protein, with the exception of substitutions occuring in residues 16 and 253, changing alanine and glutamine for glutamate and histidine, respectively. The cloned gene sequence can be segmented into seven distinct regions: the 5’ untranslated region (UTR), N-terminal domain, homeodomain, C1 domain, Cw domain, C2 domain, and the 3’ UTR. The 5’ region contains binding sites for ESC genes Oct-4 and Sox-2, which are part of a transcriptional regulatory loop [33-35]. The 5’ region also contains a p53-binding site within the Nanog promoter region that facilitates ESC differentiation [36] and is possibly responsible for the shift in replication timing observed with neural differentiation [37]. The N-terminal region of Nanog has transcriptional activity [38] and encodes for a sequence containing a SMAD-binding domain [31,39]. The homeodomain portion is similar to the NK-2 and ANTP family of homeodomain transcription factors, but comparing 120 different homeodomain proteins using BLOcksSUbstitution Matrix (BLOSUM) and Point Accepted Mutation (PAM) matrices suggests that Nanog represents a distinct protein family divergent from both the NK-2 and distal-less gene family (data not shown). The C-terminal domain contains no apparent transactivation motifs, but has greater transactivation activity compared to the N-terminal and homeodomain [38,40].
The C-terminal domain can be subdivided into three regions: the portion immediately following the homeodomain region (C1), the subregion containing a unique repeated motif of tryptophan flanked with four polar-uncharged amino acids (Cw), and a more distal sequence (C2). Cloning of Nanog inside the pLoxlentiviral vector was successful and DNA sequencing confirmed a match for the actual Nanog gene. The Nanog sequence was properly inserted into the vector containing a LoxP sequence within the long tandem repeat, allowing for efficient proviral deletion following delivery of Crerecombinase [41,42].
Transfection of Nanog: In the current study, human bone marrow HMSCs were cultured and grown to 75% confluency and then treated with either a plasmid containing Nanog or a mock-transfected control. Following optimization, we achieved transfection rates of less than 5% using non-viral transfection. Nanog transfection altered the morphology of cells, producing smaller, proliferative cells that themselves formed clusters (Figure 1 (a)).
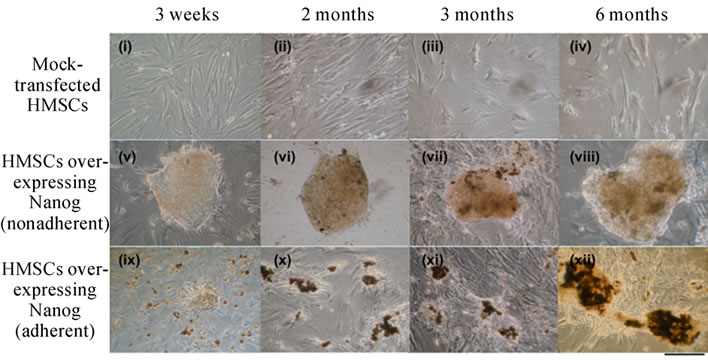
Two basic cell types were observed; namely, the proliferative clusters tended to form either an adherent mass of cells, resembling the morphology of an ES cell cluster (Figure 1(a), v-viii); or, more spherical, non-adherent/loosely adherent clumps, somewhat resembling embryoid bodies morphology (Figure 1(a), ix-xii). The former cells extensively proliferated for a long period. The latter type cells originated as small, scattered clumps, but formed larger aggregates within weeks. These larger clusters seemed to be mainly the result of clump aggregation rather than cell proliferation. Normal MSCs have a doubling time of approximately 70 hours in our conditions. After Nanog transfection of HMSCs, their doubling time changed to approximately 16 hours.
Transfection with the neuroporter reagent appeared to have more toxicity and was more likely to produce more HMSCs; therefore FuGene 6 was the preferred transfec-
 (a)
(a)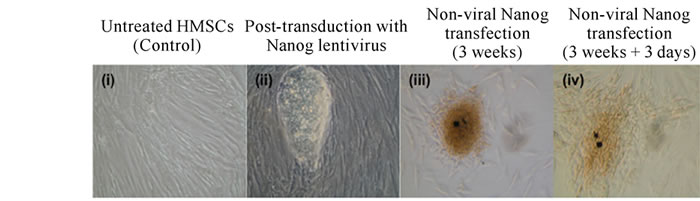 (b)
(b)
Figure 1. (a) HMSCs over-expressing Nanog displayed ESC-like (v-viii) or EB-like morphology (ix-xii). Morphological changes seen at three weeks (i,v,ix), two (ii,vi,x), three (iii,vii,xi), and six months (iv,viii,xii) post-transfection. (b) HMSCs nine days post-transduction with Nanog lentivirus (ii). Three weeks following non-viral Nanog transfection (iii) and three days later (iv). Untreated HMSCs showed as a control (i).
tion reagent. Cells that displayed the flattened, ES celllike morphology were detected as early as one week, but were usually distinct at two to three weeks. The number of colonies produced did not appear to directly correspond to transfection rates. Following one week, one or two colonies could be observed in the wells. No colonies were able to expand without a feeder layer, and only a few colonies were able to expand into larger colonies of thousands of cells for subsequent passaging.
Moreover, the colonies resembling morphology of an ES cell cluster were only found within the Nanog-transfected HMSC cultures. Both of the previously defined cell types either adhered and differentiated or underwent cell death when transferred to separate culturing flasks with no feeder layer (data not shown). The inability of isolated colonies to continually proliferate on their own indicates that the majority of non-transfected HMSCs served as a feeder layer, helping provide growth factors and aid in cell survival. Nanog-transfected HMSC colonies were less homologous and displayed greater propensity for differentiation than has been reported with ESCs (Figure 1(a)).
There was little difference in the morphologies between Nanog-transfected HMSCs and ESCs for up to two months in culture, and while they were able to proliferate, they did not appear to grow past 1000 mm. However, by three months, gradual changes became evident as heterogeneity within the structures became more apparent. It is uncertain whether this phenomenon is the result of cells undergoing differentiation, reaching a proliferative limit, or the result of changes in the underlying feeder layer of un-transfected HMSCs. Control HMSCs showed changes in morphology and displayed little or no proliferation at three months in culture.
Beyond three months, the number of Nanog-transfected HMSCs diminished, and mock-transfected HMSCs showed age-related alterations. HMSCs could be cultured for longer periods of time through continuous passages, but late-passaged HMSCs displayed changes in morphology, including increased cell size, larger cytoplasm, and no detectable proliferation. Following one year of culturing and expansion, HMSCs failed to survive and few Nanog transfected-HMSC remained.
Cells co-transduced with Nanog lentiviruses showed prominent cluster formation (Figure 1(b), ii). Colonies formed by transduction with Nanog were easier to maintain and grew much larger than HMSCs chemically transfected with Nanog. Colonies produced through chemical transfection were difficult to maintain as the colonies tended to disperse (Figure 1(b), iii, iv).
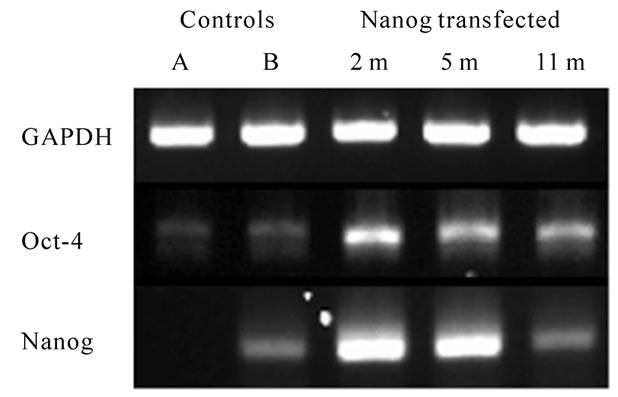
Gene expression and immnohistochemistry of Nanogtransfected HMSCs: Exploring biochemical changes following Nanog transfection, we performed RT-PCR for Nanog and Oct-4 to compare with mock-transfected HMSCs, as well as immunostaining for known ESC markers. Expression levels of Nanog and Oct-4 were absent or low in two different batches of mock-transfected HMSCs (Figure 2(a)).
This illustrates the heterogeneity of HMSCs in culture and is consistent with the data showing a subpopulation of pluripotent HMSCs [10,11,43,44]. Following Nanog transfection, expression of both Nanog and Oct-4 were highly elevated at two, five, and eleven months. It was unexpected that either Nanog or Oct-4 would be expressed following long-term expansion since the few remaining cell clusters did not appear to proliferate at one year. Interestingly, levels of Oct-4 did not directly correlate with expression of Nanog, which is consistent with findings that Oct-4 is not directly controlled by Nanog [33,45,46].
Quantitative gene expression analysis was difficult given the heterogeneous population and relatively low frequency of Nanog-transfected cells. We therefore at-
 (a)
(a)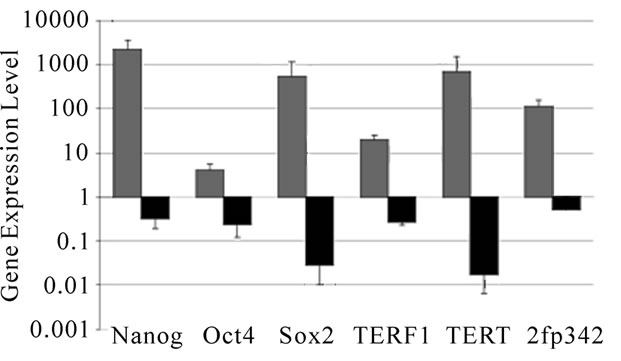 (b)
(b)
Figure 2. (a) RT-PCR shows little or no expression of Nanog and Oct-4 in mock-transfected HMSCs but up-regulation of both at two, five, and eleven months following Nanog transfection up to 11 month in a culture. GAPDH was a control. (b) qRT-PCR of Nanog lentiviral-transduced cells. Up-regulation of multiple ESC genes after Nanog transfection (grey) were observed and these genes were down-regulated after (black) delivery of Cre recombinase vectors to remove Nanog expression.
tempted to select out Nanog-dedifferentiated cells using a lentiviral system co-expressing GFP. Using lentivirus to deliver Nanog and GFP, we created identifiable clusters of cells resembling the morphology of ES C clusters that were more homogenous, highly proliferative, and easily expandable. In fact, the cells were able to grow with or without feeder cells for over 40 passages.
Quantitative real-time PCR was performed on lentiviral-transduced cells and showed a dramatic increase in most of the ESC genes tested. We were able to detect low levels of both Nanog and Oct-4 in two of three HMSC batches tested, but telomerase expression was absent. Following forced expression of Nanog, we measured dramatic increases in Nanog, Sox-2, zinc-finger protein 342, TERF1 and telomerase. Given the earlier lack of telomerase expression, we assigned the lowest value for detection in order to perform an analysis that does not allow for “zero” expression. We observed only a modest, yet statistically significant, increase in levels of Oct-4 to four times the normal level (Figure 2 (b)). The measured changes, particularly the sudden expression of telomerase and increase in TERF1, demonstrate fundamental changes in the HMSCs following delivery of Nanog. Previous work has revealed that HMSCs fail to express telomerase and have a unique telomerase biology compared to other stem cells [47,48], so the link between Nanog and telomerase is an exciting area that warrants exploration.
Removing Nanog and GFP using a vector encoding for the Cre recombinase enzyme should reverse gene expression changes in the viral-transduced cells. Recombination and gene excision were successful, as most viral-transduced cells were negative for GFP post Cre transfection, allowing for neural differentiation. Additionally, real-time PCR reveals an 89% decrease in GFP expression in viral-transduced cells 72 hours following delivery of Cre (data not shown). We compared changes in gene expression in Cre-transfected, virally-de-differentiated cells and found reduction in most stem cells genes. Nanog, Oct4 and TERF1 showed a 70% to 80% decrease in expression, while Sox-2 and telomerase showed decreases of approximately 98% each (Figure 2(b)).
Immunohistochemical staining was performed using antibodies against the ESC markers Nanog, Oct-4, stagespecific embryonic antigen-3 (SSEA3), and keratan sulphate-associated antigen TRA1-60 (Figure 3). If Nanog increases the transdifferentiation ability of HMSCs, transfected cells should stain positive for these markers. The vast majority of untreated cells failed to stain for any of the markers, but a small population (approximately 1%) of cells, did show positive staining for transcription factors Oct-4 and Nanog, while faint staining for surface markers was seen in about 5% of cells (Figure 3). Alternatively, Nanog-transfected cells did display positive staining for the transcription factors Nanog and Oct-4 within the proliferative cell clusters, although not in the surrounding feeder layer of HMSCs. Nanog-transfected HMSCs clusters also showed positive expression of surface markers SSEA3 and TRA1-60 (Figure 3).
This staining pattern was more apparent with the use of the GFP and Nanoglentiviral-transduced cells. Following transduction with Nanog and GFP, large colonies morphologically similar to ES cell colonies began to form. GFP expression appeared localized to these colonies and showed positive staining for Nanog and Sox-2, unlike non-treated HMSCs (Figure 4). Taken together, it appears that forced expression of Nanog results in the increased transdifferentiation ability of HMSCs and induces the expression of genes related to pluripotency. Immunostaining against ESC markers in HMSCs has previously been done [49].
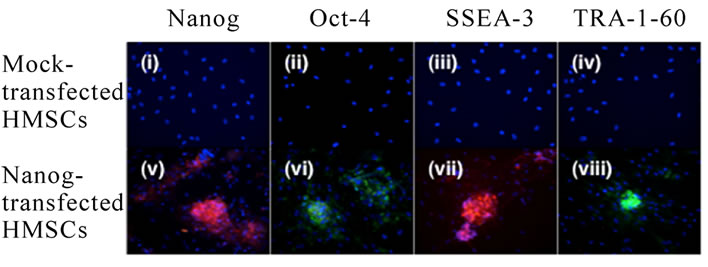
Figure 3. Immunocytochemistry of Nanog transfected HMSC colonies showed strong immunoreaction for ES cell markers after15 weeks in a culture (v-viii) while there is no expression of ES cell markers in mock-transfected HMSCs (i-iv). The cells were immunostained for Nanog (i, v), Oct-4 (ii, vi), SSEA-3 (iii, vii), and TRA-1-60 (iv, vii).
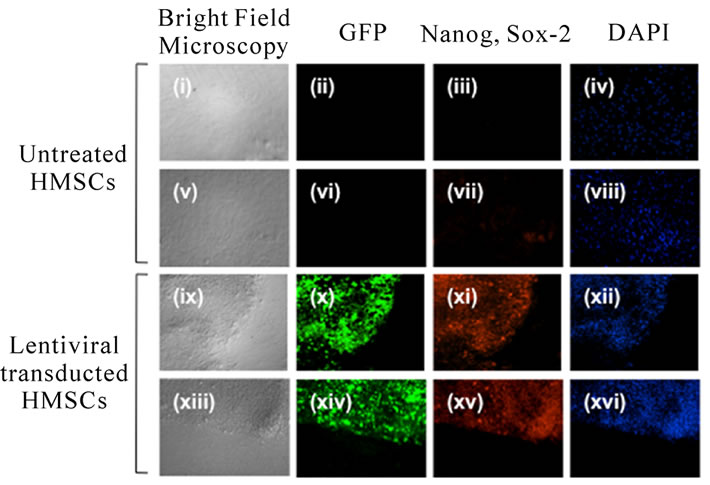
Figure 4. With mock-transfected HMSCs showed no ESClike colony formation (i,v) nor GFP expressions (ii,vi) and immunoreactivities for Nanog (iii) and Sox-2 (vii) are not detected. Lentiviral transduction with Nanog and GFP induced colony formation (ix, xiii) and GFP expression (x, xiv), and positive for Nanog (xi) and Sox-2 (xv) immunoreactivities. DAPI is used counter stain of nuclei (iv, viii, xii, xvi).
We next tested the ability of Nanog-transfected cells to undergo neural differentiation using a previously established co-culture system. Cells were placed in co-culture consisting of a feeder cell layer separated by a semi-permeable membrane to eliminate direct cell contact. The feeder cells used were neurons and glial cells derived from HNSCs, and were grown as neural spheres and cultured in serum-free basal media. This system allows for the exchange of growth factors and eliminates the concern over cell fusion since it prevented direct cell contact between the HMSCs and underlying feeder cells. Cell clusters adhered to the membrane surface and differentiation occurred as cells radiated outwards. Control HMSCs adhered to the membrane surface but failed to differentiate into neurons and astrocytes. The differentiation pattern was tested by immunostaining against bIII-tubulin and GFAP, early neuronal and astrocytic lineage markers, respectively (Figure 5, i-iii). LentiviralNanog-transfected HMSCs were able to undergo neural differentiation following transfection using a Crerecombinase vector. The un-transfected HMSCs did not show positive staining for the neuronal early lineage marker bIII-tubulin, but approximately two percent of the cells did show weak expression of GFAP (Figure 5, i). This may represent a subpopulation of HMSCs [10,43] that is capable of differentiating into astrocytes. Modified cells formed spherical clusters with a similar appearance to differentiated NSCs, forming a web-like network of neurons and astrocytes that stained positive for both bIII-tubulin and GFAP (Figure 5, ii). Since, bIII-tubulin and GFAP are early lineage commitment markers, so we examined the expression of MAP2 and S100 to determine if these mature neuronal and astrocytic markers would be expressed following neural differentiation to our modified cells. Cells were cultured for two weeks and stained for mature neural markers. Cells stained positive for MAP2 and S100, indicating the cells were able to express mature neuronal and astrocytic makers, respectively (Figure 5, iv). Induction of differentiation in the non-viral transduced cells was achieved by first transfecting the cells with 3mg of Nanog vector using FuGene6 at DNA to reagent ratio of 1:3, then placing the cells in conditioned media for neural differentiation. We found that some cells did show positive staining for both markers, and most cells were negative for GFP (Figure 5 iii). Immunostaining shows expression of early neuronal (bIII-tubulin) and astrocytic (GFAP) markers at three days in neural differentiation culture. Additionally, we conducted transdifferentiation studies in vitro and in vivo with naive HMSCs, however we could not find significant production of GFAP positive cells nor b–III tubulin cells.
Transplantation results: We tested the cell fate and migration of non-viral and lentiviral dedifferentiated HMSCs in vivo three weeks post-transplantation. Following sacrifice, the brains were sectioned and examined. None of the animals displayed evidence of tumors. Immunohistochemical staining for human bIII-tubulin and GFAP did reveal evidence of both in vivo early neuronal and glial differentiation. Moreover, the presence of transplanted cells, marked by the expression of human neural markers or GFP in the CA1 regions of the hippocampus proper and dentate gyrus regions, is encouraging given the role of these structures in learning and memory (Figure 6).
4. Discussion
This study demonstrates a novel method to increase the transdifferentiation ability of adult stem cells by overexpressing genes regulating pluripotency, with the end goal of facilitating neural transdifferentiation of HMSCs, which may allow us to perform autologous cell therapies for individuals with neurodegenerative diseases and other neurological disorders. Nanog transfection of HMSCs produced proliferative cells with morphological and gene expressions resembling ES cells, though their pluripotency has not been confirmed.
We previously demonstrated that treatment with the nucleotide derivative BrdU allows for transdifferentiation [14], and other groups have demonstrated that fusion of stem cells and somatic cells can alter cell properties [13,17,50]. In the current study, we show that transdiffe-
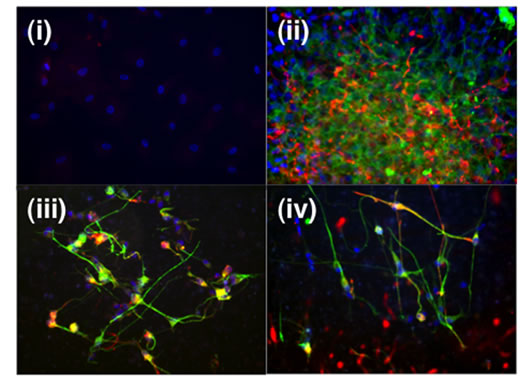
Figure 5. After co-culture with differentiated human neural stem cells, naive HMSCs showed few GFAP (red) but no bIII-tubulin (green) immunoreactivites (i). While Nanogtransfected HMSCs forming clusters of cells co-cultured with differentiated human neural stem cells attached to the culture insert membrane and migrated outward. They were positive for GFAP (red) and bIII-tubulin (green) immunoreactivites indicating neural differentiation of the cells, (low magnification in ii, high magnification in iii). Differentiated Nanog-transfected HMSCs are also stained positive for MAP2 (green) and S100 (red) at two weeks, indicating differentiation into mature neurons and astrocytes, respectively.
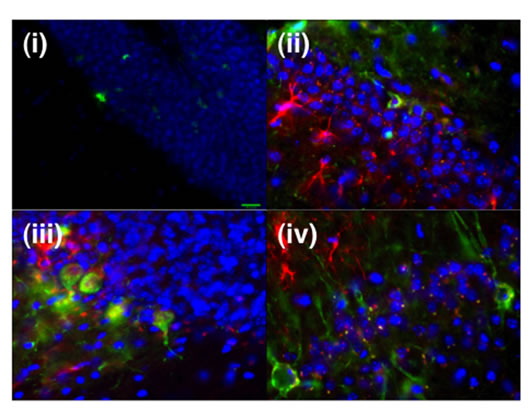
Figure 6. After transplantation into mice, Nanog-GFP transfected HMSCs HMSCs are capable of migration into hippocampus dentate gyrus (i). Immunostaining specific to human bIII-tubulin (green) and GFAP (red) indicated that the transplanted Nanog-transfected HMSCs after Cre recombinase treatment differentiated into neurons and astrocytes in the dentate gyrus (ii, iii) and CA1 regions (iv) of the hippocampus, respectively.
rentiation ability of HMSCs can be increased by gene delivery of only Nanog, although other factors already present in the cells may contribute to forming a cluster of cells whose morphology resembles that of an ESC cluster. Once the environment inside the cells have been changed by Nanog over-expression, ESC genes including Nanog, Sox2, and Oct4 is increased and maintained for longer periods due to each of the genes ability to influence each other. Recently, delivery of four factors induced pluripotency in somatic adult, fibroblasts. Their use of transcription factors Oct-4, Sox-2, and KLF4 along with the oncogenec Myc was sufficient to induce pluripotent transformation [51-54].These results have been independently achieved by different labs using human cells with the same set of genes [55-57] or with a combination of Nanog, Oct-4, Sox-2, and Lin28 [58].
We found levels of Oct-4, Sox-2, and other genes related to pluripotency and self-renewal were significantly increased after Nanog over-expression. Previous reports failed to show production of cells expressing these genes using any single ESC gene when delivered to stem cells or fibroblasts [53,59], nor by combining Oct-4, Sox-2, Klf4 and cMyc in adult HMSCs [56]. The use of additional vectors encoding for the simian virus large T antigen (SV40T) and the catalytic subunit for telomerase (hTERT) produced a few colonies resembling ES cell clusters but still showed cellular loss with expansion [56]. Such discrepancy between the studies analyzing ESC genes in HMSCs could be because those cells expressing ESC genes are a very minor sub population in HMSCs and could be altered by culture conditions.
We observed that the number of colonies resembling the morphology of ES cell cluster that formed after Nanog transfection did not directly correspond to the number of cells receiving the gene. This may be likely the result of a number of critical factors. First, MSCs have a limited capacity for expansion and vary in their ability to proliferate and differentiate, qualities that decrease with age and vary among sources. They are sensitive to culturing conditions, particularly plating density, supplements and serum quality [60-63]. Thus, heterogeneity in the culture may be responsible for this existence of responder and non-responder in the culture. Gene delivery is also challenging in these cell types given their difficulty to transfect, death from toxicity [64-67], their propensity to undergo senescence after several passages [47,68,69], or toxicity associated with viral transductions [67,70]. Additionally, original heterogeneity within HMSCs and variation between cultures may account for t lesser efficiency. Previous studies are inconsistent regarding the expression of pluripotency transcription factors Nanog, Oct-4, and Sox-2 in adult stem cells. Oct-4 is present at low levels in HMSC in vitro cultures or can be induced in a subtype of cells using various culture conditions [10,60,71,72]. However, low levels of Sox2 and Nanog are detected in some, but not all HMSCs [44,60,73-75]. This inconsistency extends to telomerase activity and the ability to immortalize HMSCs.
Similar to genes associated with pluripotency, telomerase activity has been detected by some groups [76,77], but not by others [47,48,78,79]. Conflicting results are also observed when groups attempt to immortalize HMSCs through viral delivery of telomerase. Overexpression of telomerase appears to overcome early senescence and generate immortalized cell lines [32,80-83] while other groups report hTERT alone is insufficient [78,79]. Alternatively, only a subpopulation of HMSCs may increase transdifferentiation ability by Nanog. Presumably cells that endogenously express other necessary stem cell genes would be responsive to Nanog over-expression. This hypothesis is supported by the ability to convert NSCs, which already express many stem cell factors including Sox2 and cMyc, to pluripotent cells through forced expression of two factors, Oct-4 and Klf-4 [84].
In addition to the presence of critical stem cell genes, the level of expression is likely to be imperative in determining cell conversion. Since the combination of Oct-4 and Sox-2, which may up-regulates Nanog, is reported to fail increasing transdifferentiation ability of the adult cells [34,35], high levels of Nanog may be the critical factor. Other research found that selection of the cells expressing high levels of Nanog after transfection with a combination of Oct-4 and Sox-2 has yielded ESC-like colonies [52].
Cells receiving a non-viral transfection of Nanog tended to lose proliferative capability while lentiviral-transfected Nanog cells can be maintained over forty passages. HMSCs show extremely low rates of stable transfection using non-viral transfection [65]. Thus, differences between cells receiving Nanog through chemical transfection compared to those receiving it through viral delivery might explain the lack of stable expression of Nanog in the non-viral transfected cells. However, stable Nanog expression may not only increase their rate of proliferation but also prevent them from differentiating into functional cells.
The presented work demonstrates that forced expression of Nanog in HMSCs interacts with endogenous factors to induce neural transdifferentiation ability in cells committed to a mesoderm lineage and increase the developmental potential of the cells. We repeated the experiments with both Nanog pseudogene 8 and Nanog clone from Dr. Austin Smith and we found the same results. The ability to generate human neural cells from adult bone marrow derived stem cells may allow us to perform autologous regeneration therapies for neurological diseases using the patient’s own cells. This will eliminate the issues associated with use of ESCs or adult NSCs. Although we observed both early glial and neuronal differentiation in aged wild-type mice, functionality of the cells needs to be confirmed and we have to consider effects of pathological condition on the cells in each disease [85,86]. This technology may also open a door for the possible production of disease-specific functional cells from patients that can be used to create disease models for research of the disease mechanisms and the development of personalized drugs. However, further detail studies are needed to prove pluripotency by formation of teratoma and/or chimera of the Nanog-transfected HMSC-forming colonies resembling the morphology of ES cell clusters and to show functionality of neural cells derived from the Nanog-transfected HMSCs by head-to-head comparison with the primary neural cells and neural cells derived form NSCs and/or ESCs.
5. Acknowledgements
This research was supported by NIH grant R01 AG 23472-01. We would like to thank Dr. Austin Smith, University of Cambridge, England, for Nanog clone and Drs. Didier Trono and Patrick Salmon, LVPU, Centre MédicalUniversitaire, Switzerland, for pLox lentivirus vector. We would like to thank Ms. Stephanie Merchant for providing lab assistance and the manuscript preparation.
6. Author Disclosure Statement
No competing financial interests exist.
REFERENCES
- D. C. Hess and C. V. Borlongan, “Stem Cells and Neurological Diseases,” Cell Proliferation, Vol. 41, Suppl. 1, 2008, pp. 94-114.
- E. Juengst and M. Fossel, “The Ethics of Embryonic Stem Cells-Now and Forever, Cells without End,” The Journal Of the American Medical Association, Vol. 284, No. 24, 2000, pp. 3180-3184.
- A. McLaren, “Important Differences between Sources of Embryonic Stem Cells,” Nature, Vol. 408, 2000, p. 513.
- A. McLaren, “Ethical and Social Considerations of Stem Cell Research,” Nature, Vol. 414, No. 8859, 2001, pp. 129-131.
- R. A. Barker and H. Widner, “Immune Problems in Central Nervous System Cell Therapy,” NeuroRx, Vol. 1, No.4, 2004, pp. 472-481.
- J. A. Bradley, E. M. Bolton and R. A. Pedersen, “Stem cell Medicine Encounters the Immune System,” Nature Reviews Immunology, Vol. 2, No. 11, 2002, pp. 859-871.
- S. Arnhold, H. Klein, I. Semkova, K. Addicks and U. Schraermeyer, “Neurally Selected Embryonic Stem Cells Induce Tumor Formation after Long-Term Survival Following Engraftment into the Subretinal Space,” Investigative Ophthalmology & Visual Science, Vol. 45, No. 12, 2004, pp. 4251-4255.
- E. Bieberich, J. Silva, G. Wang, K. Krishnamurthy and B. G. Condie, “Selective Apoptosis of Pluripotent Mouse and Human Stem Cells by Novel Ceramide Analogues Prevents Teratoma Formation and Enriches for Neural Precursors in Es Cell-Derived Neural Transplants,” The Journal of Cell Biology, Vol. 167, No. 4, 2004, pp. 723-734.
- D. Rubio, J. Garcia-Castro, M. C. Martin, R. de la Fuente, J. C. Cigudosa, A. C. Lloyd and A. Bernad, “Spontaneous Human Adult Stem Cell Transformation,” Cancer Research, Vol. 65, No. 8, 2005, pp. 3035-3039.
- Y. Jiang, B. N. Jahagirdar, R. L. Reinhardt, R. E. Schwartz, C. D. Keene, X. R. Ortiz-Gonzalez, M. Reyes, T. Lenvik, T. Lund, M. Blackstad, J. Du, S. Aldrich, A. Lisberg, W. C. Low, D. A. Largaespada and C. M. Verfaillie, “Pluripotency of Mesenchymal Stem Cells Derived from Adult Marrow,” Nature, Vol. 418, No. 6893, 2000, pp. 41-49.
- Y. Jiang, B. Vaessen, T. Lenvik, M. Blackstad, M. Reyes and C. M. Verfaillie, “Multipotent Progenitor Cells Can be Isolated from Postnatal Murine Bone Marrow, Muscle, and Brain,” Experimental Hematology, Vol. 30, No. 8, 2002, pp. 896-904.
- M. Alvarez-Dolado, R. Pardal, J. M. Garcia-Verdugo, J. R. Fike, H. O. Lee, K. Pfeffer, C. Lois, S. J. Morrison and A. Alvarez-Buylla, “Fusion of Bone-Marrow-Derived Cells with Purkinje Neurons, Cardiomyocytes and Hepatocytes,” Nature, Vol. 425, 2003, pp. 968-973.
- N. Terada, T. Hamazaki, M. Oka, M. Hoki, D. M. Mastalerz, Y. Nakano, E. M. Meyer, L. Morel, B. E. Petersen and E. W. Scott, “Bone Marrow Cells Adopt the Phenotype of Other Cells by Spontaneous Cell Fusion,” Nature, Vol. 416, No. 6880, 2002, pp. 542-545.
- T. Y. Qu, X. J. Dong, I. Sugaya, A. Vaghani, J. Pulido and K. Sugaya, “Bromodeoxyuridine Increases Multipotency of Human Bone Marrow-Derived Stem Cells,” Restor Neurol Neurosci, Vol. 22, No. 6, 2004, pp. 459-468.
- J. T. Do and H. R. Scholer, “Cell-Cell Fusion as a Means to Establish Pluripotency,” Ernst Schering Research Foundation Workshop, Vol. 60, 2006, pp. 35-45.
- M. Tada and T. Tada, “Nuclear Reprogramming Of Somatic Nucleus Hybridized with Embryonic Stem Cells by Electrofusion,” Methods in Molecular Biology, Vol. 329, 2006, pp. 411-420.
- M. Tada, Y. Takahama, K. Abe, N. Nakatsuji and T. Tada, “Nuclear Reprogramming of Somatic Cells by in Vitro Hybridization with Es Cells,” Current Biology, Vol. 11, No. 19, 2001, pp. 1553-1558.
- J. T. Do and H. R. Scholer, “Comparison of Neurosphere Cells with Cumulus Cells after Fusion with Embryonic Stem Cells: Reprogramming Potential,” Reproduction, Fertility and Development, Vol. 17, No. 1-2, 2005, pp. 143-149.
- I. Chambers, D. Colby, M. Robertson, J. Nichols, S. Lee, S. Tweedie and A. Smith, “Functional Expression Cloning of Nanog, a Pluripotency Sustaining Factor in Embryonic Stem Cells,” Cell, Vol. 113, No. 5, 2003, pp. 643-655.
- K. Mitsui, Y. Tokuzawa, H. Itoh, K. Segawa, M. Murakami, K. Takahashi, M. Maruyama, M. Maeda and S. Yamanaka, “The Homeoprotein Nanog is Required for Maintenance of Pluripotency in Mouse Epiblast and Es Cells,” Cell, Vol. 113, No. 5, 2003, pp. 631-642.
- Y. Fan, M. F. Melhem and J. R. Chaillet, “Forced Expression of the Homeobox-Containing Gene Pem Blocks Differentiation of Embryonic Stem Cells,” Developmental Biology, Vol. 210, No.2, 1999, pp. 481-496.
- R. Eiges, M. Schuldiner, M. Drukker, O. Yanuka, J. Itskovitz-Eldor and N. Benvenisty, “Establishment of Human Embryonic Stem Cell-Transfected Clones Carrying a Marker for Undifferentiated Cells,” Current Biology, Vol. 11, 2001, pp. 514-518.
- H. Niwa, J. Miyazaki and A. G. Smith, “Quantitative Expression of Oct-3/4 Defines Differentiation, Dedifferentiation or Self-Renewal of Es Cells,” Nature Genetics, Vol. 24, No. 4, 2000, pp. 372-376.
- N. al Yacoub, M. Romanowska, N. Haritonova and J. Foerster, “Optimized Production and Concentration of Lentiviral Vectors Containing Large Inserts,” The Journal of Gene Medicine, Vol. 9, 2007, pp. 579-584.
- B. Mitta, M. Rimann and M. Fussenegger, “Detailed Design and Comparative Analysis of Protocols for Optimized Production of High-Performance Hiv-1-Derived Lentiviral Particles,” Metabolic Engineering, Vol. 7, No. 5-6, 2005, pp. 426-436.
- Y. Le, S. Gagneten, D. Tombaccini, B. Bethke and B. Sauer, “Nuclear Targeting Determinants of the Phage P1 Cre DNA Recombinase,” Nucleic Acids Research, Vol. 27, No. 24, 1999, pp. 4703-4709.
- Y. Le, J. L. Miller and B. Sauer, “Gfpcre Fusion Vectors with Enhanced Expression,” Analytical Biochemistry, Vol. 270, 1999, pp. 334-336.
- K. J. Livak and T. D. Schmittgen, “Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-delta delta c (t)) Method,” Methods, Vol. 25, 2001, pp. 402-408.
- J. S. Yuan, A. Reed, F. Chen and C. N. Stewart, Jr., “Statistical Analysis of Real-Time Pcr Data,” BMC Bioinformatics, Vol. 7, No. 85, 2006, pp. 1471-2105.
- H. A. Booth and P. W. Holland, “Eleven Daughters of Nanog,” Genomics, Vol. 84, No. 2, 2004, pp. 229-238.
- A. H. Hart, L. Hartley, M. Ibrahim and L. Robb, “Identification, Cloning and Expression Analysis of the Pluripotency Promoting Nanog Genes in Mouse and Human,” Developmental dynamics, Vol. 230, 2004, pp. 187-198.
- D. Pain, G. W. Chirn, C. Strassel and D. M. Kemp, “Multiple Retropseudogenes from Pluripotent Cell-Specific Gene Expression Indicates a Potential Signature for Novel Gene Identification,” Journal of Biological Chemistry, Vol. 280, No. 2, 2005, pp. 6265-6268.
- J. L. Chew, Y. H. Loh, W. Zhang, X. Chen, W. L. Tam, L. S. Yeap, P. Li, Y. S. Ang, B. Lim, P. Robson and H. H. Ng, “Reciprocal Transcriptional Regulation of Pou5f1 and Sox2 Via the Oct4/Sox2 Complex in Embryonic Stem Cells,” Molecular and Cellular Biology, Vol. 25, No. 14, 2005, pp. 6031-6046.
- T. Kuroda, M. Tada, H. Kubota, H. Kimura, S. Y. Hatano, H. Suemori, N. Nakatsuji and T. Tada, “Octamer and sox Elements are Required for Transcriptional Cis Regulation of Nanog Gene Expression,” Moecular and Celluar Biology, Vol. 25, No. 6, 2005, pp. 2475-2485.
- D. J. Rodda, J. L. Chew, L. H. Lim, Y. H. Loh, B. Wang, H. H. Ng and P. Robson, “Transcriptional Regulation of Nanog by Oct4 and Sox2,” Journal of Biological Chemistry, Vol. 280, No. 26, 2005, pp. 24731-24737.
- P. Deb-Rinker, D. Ly, A. Jezierski, M. Sikorska and P. R. Walker, “Sequential DNA Methylation of the Nanog and Oct-4 Upstream Regions in Human Nt2 Cells during Neuronal Differentiation,” Journal of Biological Chemistry, Vol. 280, No. 8, 2005, pp. 6257-6260.
- P. Perry, S. Sauer, N. Billon, W. D. Richardson, M. Spivakov, G. Warnes, F. J. Livesey, M. Merkenschlager, A. G. Fisher and V. Azuara, “A Dynamic Switch in the Replication Timing of Key Regulator Genes in Embryonic Stem Cells upon Neural Induction,” Cell Cycle, Vol. 3, No. 12, 2004, pp. 1645-1650.
- G. J. Pan, D. Q. Pei, “Identification of Two Distinct Transactivation Domains in the Pluripotency Sustaining Factor Nanog,” Cell Research, Vol. 13, No.6, 2003, pp. 499-502.
- A. Suzuki, A. Raya, Y. Kawakami, M. Morita, T. Matsui, K. Nakashima, F. H. Gage, C. Rodriguez-Esteban and J. C. Izpisua Belmonte, “Nanog Binds to Smad1 and Blocks Bone Morphogenetic Protein-Induced Differentiation of Embryonic Stem Cells,” Proceedings of the National Academy of Sciences, Vol. 103, No. 27, 2006, pp. 10294- 10299.
- G. Pan and D. Pei, “The Stem Cell Pluripotency Factor Nanog Activates Transcription with Two Unusually Potent Subdomains at Its C Terminus,” Journal of Biological Chemistry, Vol. 280, No. 2, 2005, pp. 1401-1407.
- M. Eberhardt, P. Salmon, M. A. von Mach, J. G. Hengstler, M. Brulport, P. Linscheid, D. Seboek, J. Oberholzer, A. Barbero, I. Martin, B. Muller, D. Trono and H. Zulewski, “Multipotential Nestin and Isl-1 Positive Mesenchymal Stem Cells Isolated from Human Pancreatic Islets,” Biochemical Biophysical Research Communications, Vol. 345, No. 3, 2006, pp. 1167-1176.
- P. Salmon, J. Oberholzer, T. Occhiodoro, P. Morel, J. Lou and D. Trono, “Reversible Immortalization of Human Primary Cells by Lentivector-Mediated Transfer of Specific Genes,” Molecular Theraby, Vol. 2, No. 4, 2000, pp. 404- 414.
- M. Reyes and C. M. Verfaillie, “Characterization of Multipotent Adult Progenitor Cells, a Subpopulation of Mesenchymal Stem Cells,” Annals of the New York Academy of Sciences, Vol. 938, 2001, pp. 231-233, discussion 233- 235.
- F. Ulloa-Montoya, B. L. Kidder, K. A. Pauwelyn, L. G. Chase, A. Luttun, A. Crabbe, M. Geraerts, A. A. Sharov, Y. Piao, M. S. Ko, W. S. Hu and C. M. Verfaillie, “Comparative Transcriptome Analysis of Embryonic and Adult Stem Cells with Extended and Limited Differentiation Capacity,” Genome Biology, Vol. 8, No. 8, 2007, p. R163.
- Y. H. Loh, Q. Wu, J. L. Chew, V. B. Vega, W. Zhang, X. Chen, G. Bourque, J. George, B. Leong, J. Liu, K. Y. Wong, K. W. Sung, C. W. Lee, X. D. Zhao, K. P. Chiu, L. Lipovich, V. A. Kuznetsov, P. Robson, L. W. Stanton, C. L. Wei, Y. Ruan, B. Lim and H. H. Ng, “The Oct4 and Nanog Transcription Network Regulates Pluripotency in Mouse Embryonic Stem Cells,” Nature Genetics, Vol. 38, No. 4, 2006, pp. 431-440.
- G. Pan, J. Li, Y. Zhou, H. Zheng and D. Pei, “A Negative Feedback Loop of Transcription Factors That Controls Stem Cell Pluripotency and Self-Renewal,” The Federation of American Societies for Experimental Biology, Vol. 20, No. 10, 2006, pp. 1730-1732.
- M. E. Bernardo, N. Zaffaroni, F. Novara, A. M. Cometa, M. A. Avanzini, A. Moretta, D. Montagna, R. Maccario, R. Villa, M. G. Daidone, O. Zuffardi and F. Locatelli, “Human Bone Marrow Derived Mesenchymal Stem Cells Do Not Undergo Transformation after Long-Term in Vitro Culture and Do Not Exhibit Telomere Maintenance Mechanisms,” Cancer Research, Vol. 67, No. 19, 2007, pp. 9142-9149.
- S. Zimmermann, M. Voss, S. Kaiser, U. Kapp, C. F. Waller and U. M. Martens, “Lack of Telomerase Activity in Human Mesenchymal Stem Cells,” Leukemia, Vol. 17, No. 6, 2003, pp. 1146-1149.
- U. Riekstina, I. Cakstina, V. Parfejevs, M. Hoogduijn, G. Jankovskis, I. Muiznieks, R. Muceniece and J. Ancans, “Embryonic Stem Cell Marker Expression Pattern in Human Mesenchymal Stem Cells Derived from Bone Marrow, Adipose Tissue, Heart and Dermis,” Stem Cell Review, Vol. 5, No. 4, 2009, pp. 378-386.
- L. M. Eisenberg and C. A. Eisenberg, “Stem Cell Plasticity, Cell Fusion, and Transdifferentiation,” Birth Defects Research C Embryo Today, Vol. 69, No. 3, 2003, pp. 209- 218.
- A. Meissner, M. Wernig and R. Jaenisch, “Direct Reprogramming of Genetically Unmodified Fibroblasts into Pluripotent Stem Cells,” Nature Biotechnology, Vol. 25, No. 10, 2007, pp. 1177-1181.
- K. Okita, T. Ichisaka and S. Yamanaka, “Generation of Germline-Competent Induced Pluripotent Stem Cells,” Nature, Vol. 448, No. 7151, 2007, pp. 313-317.
- K. Takahashi and S. Yamanaka, “Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors,” Cell, Vol. 126, No. 4, 2006, pp. 663-676.
- S. Yamanaka, “Induction of Pluripotent Stem Cells from Mouse Fibroblasts by Four Transcription Factors,” Cell Proliferation, Vol. 41, Suppl. 1, 2008, pp. 51-56.
- W. E. Lowry, L. Richter, R. Yachechko, A. D. Pyle, J. Tchieu, R. Sridharan, A. T. Clark and K. Plath, “Generation of Human Induced Pluripotent Stem Cells from Dermal Fibroblasts,” Proceedings of the National Academy of Sciences, Vol.105, No. 8, 2008, pp. 2883-2888.
- I. H. Park, R. Zhao, J. A. West, A. Yabuuchi, H. Huo, T. A. Ince, P. H. Lerou, M. W. Lensch and G. Q. Daley, “Reprogramming of Human Somatic Cells to Pluripotency with Defined Factors,” Nature, Vol. 451, No. 7175, 2008, pp. 141-146.
- K. Takahashi, K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda and S. Yamanaka, “Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors,” Cell, Vol. 131, No. 5, 2007, pp. 861-872.
- J. Yu, M. A. Vodyanik, K. Smuga-Otto, J. Antosiewicz-Bourget, J. L. Frane, S. Tian, J. Nie, G. A. Jonsdottir, V. Ruotti, R. Stewart, I. I. Slukvin and J. A. Thomson, “Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells,” Science, Vol. 318, No. 5858, 2007, pp. 1917-1920.
- M. J. Go, C. Takenaka and H. Ohgushi, “Forced Expression of Sox2 or Nanog in Human Bone Marrow Derived Mesenchymal Stem Cells Maintains Their Expansion and Differentiation Capabilities,” Experimental Cell Research, Vol. 314, No. 5, 2008, pp. 1147-1154.
- V. L. Battula, P. M. Bareiss, S. Treml, S. Conrad, I. Albert, S. Hojak, H. Abele, B. Schewe, L. Just, T. Skutella and H. J. Buhring, “Human placenta and Bone Marrow Derived Msc Cultured in Serum-Free, B-Fgf-Containing Medium Express Cell Surface Frizzled-9 and Ssea-4 and Give Rise to Multilineage Differentiation,” Differentiation, Vol. 75, No. 4, 2007, pp. 279-291.
- I. Sekiya, B. L. Larson, J. R. Smith, R. Pochampally, J. G. Cui and D. J. Prockop, “Expansion of Human Adult Stem Cells from Bone Marrow Stroma: Conditions That Maximize the Yields of Early Progenitors and Evaluate Their Quality,” Stem Cells, Vol.20, No. 6, 2002, pp. 530-541.
- P. A. Sotiropoulou, S. A. Perez, M. Salagianni, C. N. Baxevanis and M. Papamichail, “Characterization of the Optimal Culture Conditions for Clinical Scale Production of Human Mesenchymal Stem Cells,” Stem Cells, Vol. 24, No. 2, 2006, pp. 462-471.
- M. Yokoyama, H. Miwa, S. Maeda, S. Wakitani and M. Takagi, “Influence of Fetal Calf Serum on Differentiation of Mesenchymal Stem Cells to Chondrocytes During Expansion,” Journal of Bioscience and Bioengineering, Vol. 106, No. 1, 2008, pp. 46-50.
- D. Baksh, R. Yao and R. S. Tuan, “Comparison of Proliferative and Multilineage Differentiation Potential of Human Mesenchymal Stem Cells Derived from Umbilical Cord and Bone Marrow,” Stem Cells, Vol. 25, No. 6, 2007, pp. 1384-1392.
- H. Haleem-Smith, A. Derfoul, C. Okafor, R. Tuli, D. Olsen, D. J. Hall and R. S. Tuan, “Optimization of High-Efficiency Transfection of Adult Human Mesenchymal Stem Cells in Vitro,” Molecular Biotechnology, Vol. 30, No. 1, 2005, pp. 9-20.
- A. Hamm, N. Krott, I. Breibach, R. Blindt and A. K. Bosserhoff, “Efficient Transfection Method for Primary Cells,” Tissue Engineering, Vol. 8, No. 2, 2002, pp. 235-245.
- J. M. McMahon, S. Conroy, M. Lyons, U. Greiser, C. O'Shea, P. Strappe, L. Howard, M. Murphy, F. Barry and T. O'Brien, “Gene Transfer into Rat Mesenchymal Stem Cells: A Comparative Study of Viral and Nonviral Vectors,” Stem Cells and Devlopment, Vol. 15, No. 1, 2006, pp. 87-96.
- M. M. Bonab, K. Alimoghaddam, F. Talebian, S. H. Ghaffari, A. Ghavamzadeh and B. Nikbin, “Aging of Mesenchymal Stem Cell in Vitro,” BMC Cell Bioloty, Vol. 7, 2006, pp. 7-14.
- F. Moussavi-Harami, Y. Duwayri, J. A. Martin and J. A. Buckwalter, “Oxygen Effects on Senescence in Chondrocytes and Mesenchymal Stem Cells: Consequences for Tissue Engineering,” Iowa Orthopaedic Journal, Vol. 24, 2004, pp. 15-20.
- X. Y. Zhang, V. F. La Russa and J. Reiser, “Transduction of Bone-Marrow-Derived Mesenchymal Stem Cells by Using Lentivirus Vectors Pseudotyped with Modified Rd114 Envelope Glycoproteins,” Journal Virology, Vol. 78, No. 3, 2004, pp. 1219-1229.
- S. J. Greco, K. Liu and P. Rameshwar, “Functional Similarities among Genes Regulated by Oct4 in Human Mesenchymal and Embryonic Stem Cells,” Stem Cells, Vol. 25, No. 12, 2007, pp. 3143-3154.
- S. Wislet-Gendebien, G. Hans, P. Leprince, J. M. Rigo, G. Moonen and B. Rogister, “Plasticity of Cultured Mesenchymal Stem Cells: Switch from Nestin-Positive to Excitable Neuron-Like Phenotype,” Stem Cells, Vol. 23, No. 3, 2005, pp. 392-402.
- R. Gonzalez, C. B. Maki, J. Pacchiarotti, S. Csontos, J. K. Pham, N. Slepko, A. Patel and F. Silva, “Pluripotent Marker Expression and Differentiation of Human Second Trimester Mesenchymal Stem Cells,” Biochemical Biophysical Research Communication, Vol. 362, No. 2, 2007, pp. 491-497.
- R. Izadpanah, T. Joswig, F. Tsien, J. Dufour, J. C. Kirijan and B. A. Bunnell, “Characterization of Multipotent Mesenchymal Stem Cells from the Bone Marrow of Rhesus Macaques,” Stem Cells Development, Vol. 14, No. 4, 2005, pp. 440-451.
- M. S. Tsai, S. M. Hwang, Y. L. Tsai, F. C. Cheng, J. L. Lee and Y. J. Chang, “Clonal Amniotic Fluid-Derived Stem Cells Express Characteristics of Both Mesenchymal and Neural Stem Cells,” Biology Reproduction, Vol. 74, No. 3, 2006, pp. 545-551.
- L. Liu, C. M. DiGirolamo, P. A. Navarro, M. A. Blasco and D. L. Keefe, “Telomerase Deficiency Impairs Differentiation of Mesenchymal Stem Cells,” Experimental Cell Research, Vol. 294, No. 1, 2004, pp. 1-8.
- D. Parsch, J. Fellenberg, T. H. Brummendorf, A. M. Eschlbeck and W. Richter, “Telomere Length and Telomerase Activity during Expansion and Differentiation of Human Mesenchymal Stem Cells and Chondrocytes,” Journal of Molecular Medicine, Vol. 82, No. 1 2004, pp. 49-55.
- T. Mori, T. Kiyono, H. Imabayashi, Y. Takeda, K. Tsuchiya, S. Miyoshi, H. Makino, K. Matsumoto, H. Saito, S. Ogawa, M. Sakamoto, J. Hata and A. Umezawa, “Combination of Htert and Bmi-1, E6, Or E7 Induces Prolongation of the Life Span of Bone Marrow Stromal Cells From an Elderly Donor without Affecting Their Neurogenic Potential,” Molecular and Cellular Biology, Vol. 25, No. 12, 2005, pp. 5183-5195.
- T. Okamoto, T. Aoyama, T. Nakayama, T. Nakamata, T. Hosaka, K. Nishijo, T. Nakamura, T. Kiyono and J. Toguchida, “Clonal Heterogeneity in Differentiation Potential of Immortalized Human Mesenchymal Stem Cells,” Biochemical Biophysical Research Communications, 295, 2002, pp. 354-361.
- B. M. Abdallah, M. Haack-Sorensen, J. S. Burns, B. Elsnab, F. Jakob, P. Hokland, M. Kassem, “Maintenance of Differentiation Potential of Human Bone Marrow Mesenchymal Stem Cells Immortalized by Human Telomerase Reverse Transcriptase Gene Despite [Corrected] Extensive Proliferation,” Biochemical Biophysical Research Communications, Vol. 326, 2005, pp. 527-538.
- E. S. Jun, T. H. Lee, H. H. Cho, S. Y. Suh and J. S. Jung, “Expression of Telomerase Extends Longevity and Enhances Differentiation in Human Adipose Tissue-Derived Stromal Cells,” Cellular Physiology Biochemistry, Vol. 14, 2004, pp. 261-268.
- S. K. Kang, L. Putnam, J. Dufour, J. Ylostalo, J. S. Jung and B. A. Bunnell, “Expression of Telomerase Extends the Lifespan and Enhances Osteogenic Differentiation of Adipose Tissue-Derived Stromal Cells,” Stem Cells, Vol. 22, No. 7, 2004, pp. 1356-1372.
- J. L. Simonsen, C. Rosada, N. Serakinci, J. Justesen, K. Stenderup, S. I. Rattan, T. G. Jensen and M. Kassem, “Telomerase Expression Extends the Proliferative Life-Span and Maintains the Osteogenic Potential of Human Bone Marrow Stromal Cells,” Nature Biotechnology, Vol. 20, No. 6, 2002, pp. 592-596.
- J. B. Kim, H. Zaehres, G. Wu, L. Gentile, K. Ko, V. Sebastiano, M. J. Arauzo-Bravo, D. Ruau, D. W. Han, M. Zenke, H. R. Scholer, “Pluripotent Stem Cells Induced from Adult Neural Stem Cells by Reprogramming with Two Factors,” Nature, Vol. 454, 2008, pp. 646-650.
- K. Sugaya, “Neuroreplacement Therapy and Stem Cell Biology under Disease Conditions,” Cellular and Molecular Life Sciences, Vol. 60, No. 9, 2003, pp. 1891- 1902.
- K. Sugaya, A. Alvarez, A. Marutle, Y. D. Kwak and E. Choumkina, “Stem Cell Strategies for Alzheimer’s Disease Therapy,” Panminerva Medica, Vol. 48, 2006, pp. 87-96.
NOTES
*This research was supported by NIH grant R01 AG 23472-01.