Advances in Microbiology
Vol.06 No.12(2016), Article ID:71241,19 pages
10.4236/aim.2016.612083
Phylogenetic Analysis of Anaerobic Co-Digestion of Animal Manure and Corn Stover Reveals Linkages between Bacterial Communities and Digestion Performance
Fan Yang1, Rui Chen2, Zhengbo Yue2,3, Wei Liao2, Terence L. Marsh4*
1Department of Agricultural and Biosystems Engineering, Iowa State University, Ames, IA, USA
2Department of Biosystems and Agricultural Engineering, Michigan State University, East Lansing, MI, USA
3School of Resources and Environmental Engineering, Hefei University of Technology, Hefei, China
4 Department of Microbiology and Molecular Genetics, Michigan State University, East Lansing, MI, USA

Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: September 14, 2016; Accepted: October 14, 2016; Published: October 17, 2016
ABSTRACT
Over 3 million tons of manures are produced annually in the United States and pose environmental and health risks if not remediated. Anaerobic digestion is an effective method in treating organic wastes to reduce environmental impacts and produce methane as an alternative energy. Previous studies suggested that optimization of feed composition, hydraulic retention time, and other operational conditions can greatly improve total solids removal and increase methane productivity. These environmental factors improve functionality by altering the microbial community structure but explicit details of how the bacterial community shifts are poorly understood. Our investigations were conducted to investigate the relationship between environmental factors, microbial community structure and bioreactor efficiency by using metagenomic analysis of the microbial communities. Our results indicated that the bioreactor with the greatest methane production, digestion efficiency and reduced levels of E. coli/Shigella had a distinctive community structure at the genus level with unique and abundant uncultivated strains of Bacteroidetes. Moreover the same bioreactor was enriched in Aminomonas paucivorans and Clostridia populations that can utilize secondary metabolites produced during cellulose/hemicellulose degradation to generate hydrogen and acetate. Hence specific digestion conditions that enrich for these populations may provide a route to the optimization of co-digestion systems and control the variability in reactor performance.
Keywords:
Anaerobic Co-Digestion, Bacteroidetes, Cellulose Degrading Bacterial Community

1. Introduction
Anaerobic digestion can effectively degrade farm wastes and concomitantly produce methane, thereby reducing the health and environmental risks while producing energy [1] [2] . Methane is produced in anaerobic digestion when organic matter is in high concentration [3] . It is a highly cooperative process carried out by different groups of Bacteria and Archaea with distinct responsibilities [3] [4] . Schink and Stams [3] concluded that methanogenic degradation of organic matter was executed primarily by three groups of microorganisms. During anaerobic digestion of farm waste, cellulosic material is degraded by several bacterial groups providing smaller organic substrates that are further degraded by fermentative bacteria. The fermentation by-product hydrogen is used by hydrogen-oxidizing methanogens to produce methane, while acetate-cleaving methanogens produce methane from acetate.
A large number of studies focused on optimizing anaerobic digestions via manipulating the system’s operational parameters, such as the substrate C:N ratio [5] - [10] . However, the microbial communities within the anaerobic digestion have not been comprehensively explored. As pointed out by Schink and Stams [3] , these microbial communities are the primary force behind methane production and community shifts could affect overall performance. Studies have shown that several Clostridium species can decompose and partially hydrolyzed cellulose. Clostridia populations were also repeatedly reported in hydrogen and methane producing anaerobic bioreactors [8] [11] - [14] . The Clostridia group is well-known for their ability to produce hydrogen via [FeFe]-hydrogenase (hydA) [15] and the genes for this enzyme are found only in anaerobic bacteria and eukaryotes [16] [17] . The [FeFe]-hydrogenase energetically prefers to catalyze the reduction of protons to produce hydrogen [18] [19] . Therefore, we hypothesized that Clostridia populations are the most abundant cellulose degrading bacteria in the methane producing microbial community. Herein, a pilot-scale bioreactors study was carried on to describe microbial communities of anaerobic bioreactors fed with dairy manure (DM) and corn stover (CS). We report on phylogenetic diversity based on 16S rRNA and hydA genes of the communities detected under different operational conditions and conclude that a distinctive microbial community was associated with optimal solid degradation and methane production.
2. Materials and Methods
2.1. Anaerobic Bioreactors
Feedstock composition and the small pilot-scale anaerobic bioreactors (10 L) were set up and described in a companion study published by Yue et al. [20] . Briefly, six continuously stirred tank reactors (CSTR) were operated at 35˚C with hydraulic retention times (HRTs) of 30 days, 40 days, and 50 days. Biogas production was used to evaluate the stability of the bioreactors. Three HRTs were run for each reactors. The daily gas production became constant for all six reactors during the 2nd HRT. Due to the scope and scale of the pilot operation, replication of bioreactors was impossible. The feed for diary manure (DM) bioreactors (reactor 1, reactor 3, and reactor 5) was 5% dry matter and 95% water. The feed for manure and corn stover (DM + CS) bioreactors (reactor 2, reactor 4, and reactor 6) was a mixture of dairy manure and corn stover in a ratio of 4:1 (wt:wt) and diluted to 5% DM using water. Detailed bioreactor performance and analytical analyses were reported in the companion study [20] .
2.2. DNA Extraction and Targeted Gene Amplifications
Digestate samples were then taken from each bioreactor during the third HRTs. The samples were centrifuged at 10,000 RPM for 20 minutes in a Sorvall SS-34 rotor and the pellet was stored at −20˚C until extraction of community DNA. DNA was extracted by using Powersoil DNA Extraction Kit (Mo Bio Laboratory, Carlsbad, CA) with vigorous bead beating in a Biospec Mini-Beadbeater-8 (BioSpec Products, Inc., Bartlesville, OK).
Bacterial 16S rRNA gene (16S) V3-5 regions were amplified for 454 pyrosequencing according to the Human Microbiome Project protocol (http://www.hmpdacc.org/doc/16S_Sequencing_SOP_4.2.2.pdf). For each polymerase chain reaction (PCR), 10 ng DNA template were used. Bacterial hydA were amplified according to Boyd et al. [17] using primer set FeFe-272F/FeFe-427R. For each hydA PCR, 20 ng DNA template were used.
2.3. Preparation for Pyrosequencing and Construction of Clone Libraries
The 16S rRNA gene PCR products were visualized on 2% Tris-Borate-EDTA (TBE) agarose gel and DNA fragments of the expected size (~569 bp) were excised and extracted from the gels by using Qiagen Gel Extraction Kit and further purified with QiaQuick PCR Product Purification Kit (Qiagen, Valencia, CA). All purified DNA products were quantitated with Qubit HS Double-stranded DNA Kit (Invitrogen, Carlsbad, CA) then composited at equal mass for a final DNA concentration of 0.5 ng∙μl−1. Pyrosequencing was performed at Michigan State University Research Technologies Support Facility (RTSF) by using a Roche 454 GS FLX Sequencer. Quality filtered sequences were submitted to National Center for Biotechnology Information Sequence Read Archive (BioSample accession number: SAMN04215003).
Amplified hydA genes were confirmed via electrophoresis (1% Tris-Acetate-EDTA agarose gel). Expected gene fragments (~500 bp) were cloned using Invitrogen Topo PCR2.1 Cloning Kit (Invitrogen, Carlsbad, CA) following the vendor’s protocol. The cloned fragments were purified and sequenced at RTSF on an ABI 3730 Genetic Analyzer. Quality filtered sequences are being submitted to National Center for Biotechnology Information.
2.4. Sequence Processes and Community Analyses
Bacterial 16S rRNA gene sequences obtained from pyrosequencing were processed and analyzed by using the Ribosomal Database Project (RDP) Pyrosequencing Pipeline [21] . Sequences that were shorter than 250 bp, containing any N’s in sequencing region, or with an average quality score (Phred score) less than 20 were discarded. Chimeras were removed from RDP processed sequences by using USEARCH (UCHIME reference mode with Silva gold alignment database as reference) [22] . RDP Multi-classifier was used to identify each sequence to bacterial genus level (threshold 80). Bacterial 16S rRNA sequences were also aligned using RDP Aligner and then clustered by RDP Complete Linkage Clustering at 97% sequence identity. To eliminate the clustering artifacts, sequences were also trimmed to the same length (250 bp) based on the alignment profiles prior to clustering. RDP SeqMatch was used for further identification of bacterial clusters of interest.
[FeFe]-hydrogenase gene sequences were identified using Basic Local Alignment Search Tool (BLAST) against the NCBI non-redundant amino acid database (BLASTX). The closest phylogenetic affiliate was identified as the annotated match with the best Expected Value (E < 10−5).
Operational taxonomy unit (OTU), phylotype-based community analyses and clustering were performed by using statistical software R [23] . Ecological indices were estimated for each bioreactor sample based on clustered sequences by using the R package Vegan [24] . Specifically, community richness (Chao I), diversity indices (Shannon H’ and Simpson’s D) and community evenness (E) were calculated based on equations previously described [25] . The sampling coverage (C) of each community was calculated based on Good’s method, C = 1 − (n1∙N−1), where n1 represents the number of OTUs that is a singleton and N is the total number of sequences in the sample [26] . The R package Vegan was also used to perform non-metric multidimensional scaling analysis (NMDS) to correlate the dissimilarities among bioreactor samples and the shifts in phylotype abundance [24] .
3. Results
3.1. Bacterial Community Comparisons
Bacterial 16S rRNA gene targeted sequencing revealed that we sampled over 98% of each anaerobic bioreactor community (Table 1 and Figure 1). The bacterial community evenness decreased as the HRT increased. The diversity indices followed the similar pattern with DM + CS bioreactor operated at HRT 40 days as the exception (Table 1).
Out of 33,006 quality-trimmed 16S rRNA gene sequences, approximately 5% were categorized as unclassified bacteria. A total of 16 bacterial phyla and 137 genera (6 groups unclassified at genus level) were identified in the anaerobic bioreactor samples. Firmicutes, Bacteroidetes, and Proteobacteria were the most abundant phyla in all six anaerobic bioreactor samples accounting for between 85% - 91% of community composition (data not shown). In the DM reactors, the abundance of Proteobacteria increased (15.4% > 34.2% > 44.5%) as the HRT increased with a concomitant decrease in Firmicutes. The noteworthy exception to this trend was in the corn stover-supplemented reactors where a precipitous decrease in Proteobacteria and E. coli/Shigella was detected at the mid range of HRT (40 days). In this reactor with the most efficient conversion to methane, the Proteobacteria dropped to 7% of the community and E. coli/Shigella

Table 1. Measurements of sampling size and estimation of ecological indices of bacterial community in six anaerobic bioreactors. Calculations were based on the complete linkage clustering of chimera removed and trimmed bacterial 16S rRNA gene sequence alignments at 97% similarity. N represents the total number of non-chimera sequences that passed quality trimming. Shannon index (H’) and Simpson’s index (D) were expressed in eH’ and 1-D, respectively to emphasize the differences. E represents the community evenness. C represents the sampling coverage via gene-targeted pyrosequencing.

Figure 1. Estimation of community coverage by rarefaction analysis. Rarefaction curves of anaerobic bioreactor 16S rRNA gene sequences were constructed based on the estimation of OTUs and ChaoI indices at 97% similarity.
were reduced to ~2%. The relative abundance of phylum Bacteroidetes ranged from 21% - 40% with no apparent relationship to the HRT. Clostridia, unclassified Bacteroidetes, and E. coli/Shigella were the major groups observed within the Firmicutes, Bacteroidetes and Proteobacteria, respectively. At the phylum level a general similarity at the level of presence/absence of phyla across reactors was seen (Figure 2). However reactor 4 (corn stover supplemented at 40 d HRT) was clearly distinct in terms of the relative abundances of phyla. This difference is seen more dramatically at the genus
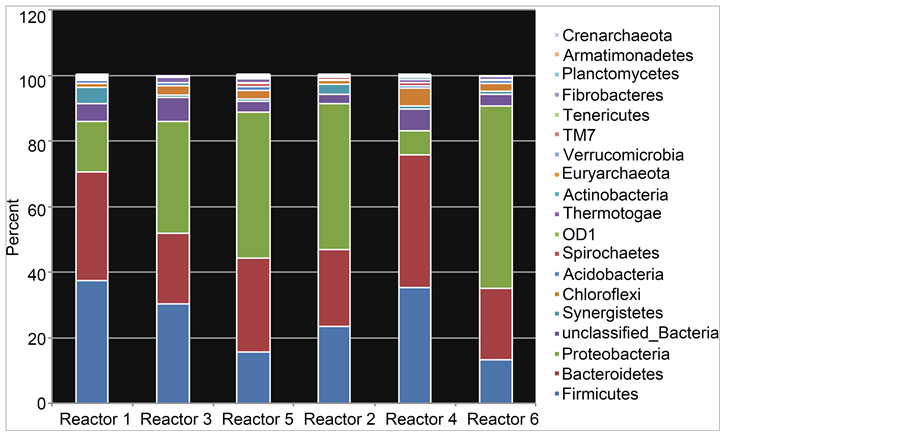
Figure 2. The phylum distribution of 16S rRNA DNA sequences in anaerobic digestors.
level (Figure 3(a)) where similarities can be easily detected between reactors 2, 3, 5 & 6 and reactor 4 is seen as distinctly different. Comparative analysis using Wards clustering of Euclidian distances (bootstrapped to 500 replicates) revealed strong similarities between reactors 2, 3, 5 & 6 (Figure 3(b)) with reactor 1 intermediate between 4 and the remainder. Clustering with Bray-Curtis dissimilarity gave similar results (data not shown). The unclassified Bacteroidetes increase from a 12% average in the other 5 reactors to 35% in reactor 4. This and small increases in other anaerobe phylotypes was accommodated by substantial reductions in the gamma-Proteobacteria populations. The combined Pseudomonas-Escherichia-Shigella percentages summed to 2.2% in reactor 4 compared to 25.8% average sum in the other 5 reactors. Reactors 5 & 6 were at the greatest HRT while reactors 2 & 3 were at different HR times. The community differences between the reactors with 30 d HRT presumably reflects the differences in the inoculum with reactor 2 supplemented with corn stover.
3.2. Shifts within Phylum Bacteroidetes
Based on a 97% cutoff, 91 different Bacteriodetes phylotypes could be detected across six reactors, many of which were singletons. These are shown in Figure 4 where each bar represents the percentage of a Bacteriodetes phylotype in the reactor. Reactors 1, 3 & 5 were dominated by phylotypes #3, #5 & #10, accounting for up to 70% of the Bacteroidetes. In sharp contrast, four unclassified Bacteroidetes phylotypes (#11, #14, #43, and #69) comprised 62% of the phylum in bioreactor 4 (DM + CS HRT40d). Phylotypes #11 & #14 were present at low levels in reactors 1, 3 & 5 and substantially more abundant in the corn stover supplemented reactors. Phylotype #43 was unique to bioreactors 4 & 6 (over 11%) while phylotype #69 was uniquely abundant in reactor 4 (DM + CS HRT40d). Over 11% and approximately 20% of the Bacteroidetes abundance was contributed by clusters #43 and #69 respectively. The insert in Figure 4 shows the Wards clustering (Euclidian distance) of the reactors based solely on the Bacteroidetes phylotypes. This analysis indicated that the Bacteroidetes populations in reactors 4 & 6 were more similar then to the remaining four reactors (bootstrap analysis highly

Figure 3. Panel (a). The top 20 most abundant genera detected in the bioreactors. Panel (b). Wards clustering (Euclidian distance) of the reactor communities. Confidence levels are indicated at the nodes and are based on bootstrapping at 500 replicates.

Figure 4. Bacteroidetes diversity and distribution in reactors. Clades within the Bacteriodetes at 97% similarity were determined through the RDP Pipeline. The insert shows Wards clustering (Euclidian distance) of the bioreactors based solely on the Bacteriodetes populations. (97% sequence similarity) between the DM + CS bioreactor with a HRT of 40 days and other bioreactor samples. Labels HRT30d, HRT40d and HRT50d represent bioreactors with a HRT of 30, 40, and 50 days, respectively. Symbol • denote populations (#11 and #14) of unclassified Bacteroidetes that are highly abundant in all DM + CS bioreactors, Symbol * denote populations in DM + CS bioreactors at long HRTs (#43) or only in reactor 4 (#69).
supported the separation).
3.3. Hydrogen Producing Population Profiles
We obtained 45 to 76 hydA gene clones from each reactor sample (352 hydA gene clones from 6 bioreactors). The coverage based on Good’s method ranged from 72% - 94% with a median of 87%. A total of 49 hydrogenase OTUs were detected of which 28 were affiliated with class Clostridia. Three major taxonomic affiliations (class Synergistia, Clostridia and Bacteroidia) were identified, accounting for 64% - 86% of the detected hydrogenases sequences. The remaining [FeFe]-hydrogenase OTUs, from more abundant to less abundant, belonged to Spirochaetales, Deltaproteobacteria. Thermotogales, Negativicutes, Candidatus Cloacamonas, Victivallales, and Gammaproteobacteria.
The relative abundance of Synergistia-like hydA gene was more prevalent in DM + CS bioreactors then DM bioreactors (Figure 5). In contrast, Bacteroidia-like hydA

Figure 5. The relative abundance changes of the three most abundant [FeFe]-hydrogenase containing bacterial groups in anaerobic bioreactors fed with DM and DM + CS. Labels HRT30d, HRT40d and HRT50d represent bioreactors with a HRT of 30, 40, and 50 days, respectively.
genes were more abundant in the DM bioreactors. The relative abundance of Bacteroidia-like hydA gene was the least abundant in DM + CS HRT40d (reactor 4) sample. The relative abundance of Clostridia-like hydA gene decreased with increasing HRT in the DM bioreactors and increased in the DM + CS bioreactors.
At the species level, hydA gene sequences similar to those found in Aminomonas paucivorans, which comprised of 98% of Synergistetes-like hydA genes, was the most abundant. [FeFe]-hydrogenase gene sequences similar to those of Moorella thermoacetica, a member of class Clostridia, were also abundant in the DM + CS bioreactors. These two hydA gene relative abundance profiles in bioreactors were similar to the relative abundance profile for unclassified Bacteroidetes 16S rRNA genes (Figure 6).
3.4. Non-Metric Multidimensional Scaling Analysis
Non-metric multidimensional scaling (NMDS) revealed that the anaerobic bioreactor community shifts correlated strongly with changes in HRT, especially in the DM bioreactors (Figure 7). Fitting the detected bacterial genera to the bioreactor community distances revealed that the abundance of Petrimonas decreased significantly as the HRT increased and when corn stover was added. The abundance of several genera, including Caryophanon, Blastopirellula, Ketogulonicigenium, unclassified Syntrophaceae and unclassified Bacteroidetes significantly more abundant in DM + CS 40d (Figure 7 red

Figure 6. The abundance of [FeFe]-hydrogenase containing bacteria in anaerobic bioreactor samples compared to the abundance pattern of unclassified Bacteroidetes (background thick columns) determined by 16S rRNA gene sequencing. The distribution of clusters #11, #14, #43 and #69 were also plotted as part of unclassified Bacteroidetes within each sample. Labels HRT30d, HRT40d and HRT50d represent bioreactors with a HRT of 30, 40, and 50 days, respectively.

Figure 7. NMDS analysis of the bacterial community in anaerobic bioreactors (based on 16S rRNA gene sequences at 97% similarity cutoff). The green arrows indicate the increase of biogas production (Biogas), HRT (HRT) and TS removal rate (Degradation). The red and blue arrows indicate bacterial genera that shifted significantly, where P ≤ 0.001 and P ≤ 0.01, respectively. The black, yellow and purple arrows represent the [FeFe]-hydrogenase phylogenetic affiliations that shifted significantly (P ≤ 0.001, P ≤ 0.05, and P ≤ 0.01, respectively). All arrows point towards the abundant range of the gradients. Labels HRT30d, HRT40d and HRT50d represent bioreactors with a HRT of 30, 40, and 50 days, respectively.
and blue arrows). Fitting of the bacterial community based hydA gene identification showed that the abundance of those most closely affiliated with Clostridium lentocellum, and Candidatus Cloacamonas acidaminovoransa were found significantly associated with the DM + CS HRT40d (reactor 4). The abundance of several other hydA genes, such as Clostridium therocellum, and Bacteroides sp. 20_3, shifted significantly along the HRT gradient and upon the addition of corn stover (Figure 7).
4. Discussion
Anaerobic digestion is an efficient method of converting agricultural wastes into methane. The accompanied study by Yue et al. [20] demonstrated that the addition of corn stover in anaerobic digestion maximized biogas production and enhanced total solids reduction. The reported observation concluded that cellulose and hemicellulose degrading bacteria and associates contributed to the generation of hydrogen and acetate, which in turn supported the growth of methanogens.
4.1. The Significance of Clostridia and Bacteroidetes
We observe a large number of Clostridia by 16S rRNA gene profiling in this pilot scale reactor study and we anticipated greater abundance and diversity of Clostridia in the reactors supplemented with corn stover because of their cellulolytic abilities. However, Bacteroidetes were equally abundant as the Clostridia in all bioreactors. Moreover, in Bioreactor 4 where methane production and total solids reduction were greatest, unique populations of uncultivated Bacteriodetes were abundant. The shifts in the Clostridia-like hydA gene abundances indicated that unique Clostridia populations became more prominent with the addition of corn stover and the increase of HRT (Figure 5). The NMDS analysis (Figure 7) also showed that three Clostridia populations associated with the DM + CS bioreactors increased significantly (Clostridium lentocellum, Candidatus Cloacamonas acidaminovoransa, and Clostridiales bacterium 1_7_47_FAA). These observations suggested that although cellulolytic Clostridia were important, the role of specific populations of Bacteroidetes in anaerobic cellulose degradation should not be ignored. Moreover, populations of Clostridia may be crucial in both hydrogen production and cellulose degradation in our bioreactors.
Like class Clostridia, Bacteroidetes populations were also commonly reported as cellulose/hemicellulose degraders [27] - [29] and observed in anaerobic digestion at low abundance (average 17%) [30] - [34] . There were 21% - 40% of Bacteroidetes populations in our pilot bioreactors and we observed a shift from the dominance of identifiable Bacteroidetes to unclassified Bacteroidetes, which correlated with the overall improved system performance reported in the companion study [20] . The four unclassified Bacteroidetes clusters that were uniquely abundant in corn stover supplemented bioreactors were highly similar to bacterial 16S rRNA gene clones found in methanogenic environments, specifically, domestic wastewater enriched microbial fuel cells (#43, SeqMatch score = 0.998), biogas slurry (#11, SeqMatch score = 0.966), Kinneret lake sediments (#14, SeqMatch score = 0.983), methanogenic zone of a hydrocarbon- and chlorinated-solvent contaminated aquifer (#69, SeqMatch score = 0.988) and an anaerobic bioreactor fed with butyrate and sulfate enriched paper mill wasterwater (#69, SeqMatch score = 0.988, unpublished work) [35] - [38] . Further, a polysaccharide utilization locus in uncultured Bacteroidetes phylotype (SRM-1) was recently identified, which allows SRM-1 to target different major components of plant biomass [39] . Thus, unclassified Bacteroidetes may be more crucial in anaerobic degradation of recalcitrant carbons than we have previously perceived. While the 16S rRNA gene based survey suggested that members of Bacteroidetes played an important role in the anaerobic digestion, the hydA clone library profile revealed that Bacteroidetes-like hydA genes were not as abundant as would be expected if they were linked to these important Bacteriodetes populations. Bacteroidetes-like hydA genes were also less abundant in the DM + CS than the DM bioreactors (Figure 5). The 16S rRNA and hydA gene profiling together suggests that unclassified Bacteroidetes might play a substantial role in cellulose/hemicellulose degradation and total solid removal but not as direct providers of substrates for methanogens in biogas production.
4.2. The Hydrogen Producing Populations
Besides Clostridia-and Bacteroidetes-like hydA genes, we observed a high abundance of Synergistia-like hydrogenase genes (at least 35%) in all of the DM + CS bioreactors. This observation suggested that Synergistia were active in hydrogen production in cellulose/hemicellulose rich environments. At species level, Synergistia was solely contributed by Aminomonas paucivorans, an anaerobic bacterium that was previously identified as an amino-acid-degrader. Baena et al. [40] described an A. paucivorans isolate that was able to grow on arginine, histidine and glutamate when cultivated syntrophically with methanogen Methanobacterium formicicum. The end products of the co-culture were propionate, acetate, CO2 and methane [40] . There are no previous studies indicating that Aminomonas paucivorans is capable of cellulose degradation. Hence, concluding from the available information and our observations, A. paucivorans might be contributing largely to hydrogen production by utilizing the secondary fermentation products of degraded dairy manure [40] .
4.3. The Anaerobic Co-Digesting Ecosystem
Anaerobic bioreactor is a complex ecosystem and frequently referred to as a black box. This pilot study was designed to correlate the microbial community structure with operating conditions. From our results, we postulate that a unique community structure including specific uncultivated Bacteriodetes populations and syntrophic populations in anaerobic bioreactors fed with agricultural wastes led to optimal methane production, total solids reduction, and reduced E. coli/Shigella abundance (Figure 8).
NMDS analysis revealed a complex network among bacterial communities in the methane producing anaerobic bioreactors. Correlating 16S rRNA gene with hydA gene diversities, we observed that the high abundance of unclassified Bacteroidetes was significantly associated with the DM + CS bioreactor with a HRT of 40 days that produced the most methane. Besides unclassified Bacteroidetes, the abundance of Caryophanon, Blastopirellula, Ketogulonicigenium and unclassified Syntrophaceae increased significantly in the DM + CS bioreactor with a HRT of 40 days. Little has been discovered regarding the metabolic capabilities of Caryophanon and Blastopirellula. Members of Ketogulonicigenium and Syntrophaceae were previously identified as fermenters with versatile metabolisms [41] - [44] . Further, the syntrophic importance of members of Syntrophaceae was linked to methane production systems using hydrocarbons as substrate [45] - [47] . It is important to note that hydrocarbons are secondary metabolites from plant material degradation. Hence, unclassified Bacteroidetes might play a substantial role in primary cellulose/hemicellulose degradation and that genera Caryophanon, Blastopirellula, Ketogulonicigenium and unclassified Syntrophaceae could potentially participate in secondary fermentation (Figure 8). NMDS analysis also showed that different members of Clostridia and Bacteroidetes participated in different activities among all six anaerobic bioreactors. For example, Bacteroides sp. 1_1_14 appeared to be affiliated with hydrogen production during dairy manure digestion at a low HRT, while Bacteroides sp. 20_3 was found significantly associated with DM + CS digestion

Figure 8. The potential biogas production model in anaerobic bioreactors. Thick lines indicate potential important processes (figure adapted from Schink & Stams [3] ).
at an elevated HRT. Similarly, C. thermocellum-like hydA genes were found to be significantly correlated with the low HRT bioreactors and Clostridiales bacterium and Odoribacter splanchnicus might play a significant role during methane production in DM + CS digestion at elevated HRT. Although A. paucivorans-(a member of family Synergistaceae) and M. thermoacetica-(a member of class Clostridia) like hydA genes were highly abundant and varied in anaerobic bioreactors, NMDS analysis indicated that the shifts of their abundance were not significant across all bioreactors (Figure 7). However, we observed that the abundance of these two groups of hydA genes followed the abundance pattern of unclassified Bacteroidetes determined based on 16S rRNA gene sequences (Figure 6). From these observations, we hypothesized that hydrogen-producing population A. paucivorans responded to the growth of unclassified Bacteroidetes. This is consistent with our prediction that unclassified Bacteroidetes were responsible for cellulose/hemicellulose degradation but not for hydrogen production. Subsequently, cellulose/hemicellulose degradation products, such as xylose, glucose and cellulobiose, could be consumed by hydrogen producers (A. paucivorans and M. thermoacetica).
The companion study [20] described the archaeal community structures in these anaerobic bioreactors, where they observed that the communities were dominated by Methanobacterium, Methanosarcina and Methanosaeta populations (Figure 8). Members of Methanobacterium are known for producing methane via hydrogen oxidation. The presence of this group of methanogens may be metabolically link to hydrogen produced by A. paucivorans, Clostridia and Bacteroidetes. The literature indicates that many members of hydA containing bacteria generate acetate as one of the final products [17] . The presence of a large amount of Methanosarcina and Methanosaeta suggests that acetate was a substantial source for methane generation. Yue et al. [20] also concluded that the community shifts from Methanosarcina dominant to Methanosaeta dominant was due to the decrease in acetate concentration in the bioreactors as HRT increased. This also explains the decreasing trend in Methanobacterium abundance in both the DM and DM + CS bioreactors as the HRT increased from 30 to 50 days. These observations are consistent with the observation that the biogas/methane production rate approached plateau when the operational HRT was more than 40 days.
5. Conclusion
Our results, while descriptive, suggest that specific populations of unclassified Bacteroidetes, Clostridia and supporting metabolic guilds are required to establish optimal conditions for methane generating farm waste digesters. Establishing optimal physical and chemical conditions in an anaerobic digester may select for the optimal microbial community only if the members are present. However, knowing with certainty the structure of this microbial community provides the option of preseeding reactors with critical populations so that digestion rapidly reaches and maintains efficient operation of what is a complex community metabolism. Information regarding key populations involved in co-digestion might lead to rational selective strategies for initiating and maintaining optimal community structure. While it is not surprising that close attention to operating conditions will optimize waste processing, it might be greatly enhanced if an optimal community structure could be seeded into reactors and nurtured rather than select and hope for optimality.
Acknowledgements
This research has been supported by the Michigan Public Service Commission (PSC- 09-19), the Michigan Animal Agriculture Initiative, the Michigan State Agricultural Station, and the MSU Vice President for Research and Graduate Studies (WL) as well as additional support through a USGS grant to TLM.
Cite this paper
Yang, F., Chen, R., Yue, Z.B., Liao, W. and Marsh, T.L. (2016) Phylogenetic Analysis of Anaerobic Co-Di- gestion of Animal Manure and Corn Stover Reveals Linkages between Bacterial Communities and Digestion Performance. Advances in Microbiology, 6, 879-897. http://dx.doi.org/10.4236/aim.2016.612083
References
- 1. Hedegaard, M. and Jaensch, V. (1999) Anaerobic Co-Digestion of Urban and Rural Wastes. Renewable Energy, 16, 1064-1069.
http://dx.doi.org/10.1016/S0960-1481(98)00372-3 - 2. Banks, C.J., Salter, A.M., Heaven, S. and Riley, K. (2011) Energetic and Environmental Benefits of Co-Digestion of Food Waste and Cattle Slurry: A Preliminary Assessment. Resources, Conservation and Recycling, 56, 71-79.
http://dx.doi.org/10.1016/j.resconrec.2011.09.006 - 3. Schink, B. and Stams, A.J.M. (2006) Syntrophism among Prokaryotes. In: Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H. and Stackebrandt, E., Eds., The Prokaryotes, Springer, New York, 309-335.
http://dx.doi.org/10.1007/0-387-30742-7_11 - 4. Bryant, M.P., Wolin, E.A., Wolin, M.J. and Wolfe, R.S. (1967) Methanobacillus Omelianskii, a Symbiotic Association of Two Species of Bacteria. Archiv für Mikrobiologie, 59, 20-31.
http://dx.doi.org/10.1007/BF00406313 - 5. Frear, C., Liao, W., Ewing, T. and Chen, S. (2011) Evaluation of Co-Digestion at a Commercial Dairy Anaerobic Digester. CLEAN—Soil, Air, Water, 39, 697-704.
http://dx.doi.org/10.1002/clen.201000316 - 6. Curtis, T.P., Sloan, W.T. and Scannell, J.W. (2002) Estimating prokaryotic Diversity and Its Limits. Proceedings of the National Academy of Sciences USA, 99, 10494-10499.
http://dx.doi.org/10.1073/pnas.142680199 - 7. Lehtomaki, A., Huttunen, S. and Rintala, J.A. (2007) Laboratory Investigations on Co-Digestion of Energy Crops and Crop Residues with Cow Manure for Methane Production: Effect of Crop to Manure Ratio. Resources, Conservation and Recycling, 51, 591-609.
http://dx.doi.org/10.1016/j.resconrec.2006.11.004 - 8. Wang, H., Lehtomaki, A., Tolvanen, K., Puhakka, J. and Rintala, J. (2009) Impact of Crop Species on Bacterial Community Structure during Anaerobic Co-Digestion of Crops and Cow Manure. Bioresource Technology, 100, 2311-2315.
http://dx.doi.org/10.1016/j.biortech.2008.10.040 - 9. Fang, C., Boe, K. and Angelidaki, I. (2011) Anaerobic Co-Digestion of By-Products from Sugar Production with Cow Manure. Water Research, 45, 3473-3480.
http://dx.doi.org/10.1016/j.watres.2011.04.008 - 10. Nges, I.A., Escobar, F., Fu, X. and Bjornsson, L. (2012) Benefits of Supplementing an Industrial Waste Anaerobic Digester with Energy Crops for Increased Biogas Production. Waste Management, 32, 53-59.
http://dx.doi.org/10.1016/j.wasman.2011.09.009 - 11. Ohmiya, K. (2005) Anaerobic Bacterial Degradation for the Effective Utilization of Biomass. Biotechnology and Bioprocess Engineering, 10, 482-493.
http://dx.doi.org/10.1007/BF02932282 - 12. O’Sullivan, C.A., Burrell, P.C., Clarke, W.P. and Blackall, L.L. (2005) Structure of a Cellulose Degrading Bacterial Community during Anaerobic Digestion. Biotechnology and Bioengineering, 92, 871-878.
http://dx.doi.org/10.1002/bit.20669 - 13. Pobeheim, H., Munk, B., Müller, H., Berg, G. and Guebitz, G.M. (2010) Characterization of an Anaerobic Population Digesting a Model Substrate for Maize in the Presence of Trace Metals. Chemosphere, 80, 829-836.
http://dx.doi.org/10.1016/j.chemosphere.2010.06.011 - 14. Cheng, X.-Y., Li, Q. and Liu, C.-Z. (2012) Coproduction of Hydrogen and Methane via Anaerobic Fermentation of Cornstalk Waste in Continuous Stirred tank Reactor Integrated with Up-Flow Anaerobic Sludge Bed. Bioresource Technology, 114, 327-333.
http://dx.doi.org/10.1016/j.biortech.2012.03.038 - 15. Calusinska, M., Happe, T., Joris, B. and Wilmotte, A. (2010) The Surprising Diversity of Clostridial Hydrogenases: A Comparative Genomic Perspective. Microbiology, 156, 1575-1588.
http://dx.doi.org/10.1099/mic.0.032771-0 - 16. Meyer, J. (2007) [FeFe] Hydrogenases and Their Evolution: A Genomic Perspective. Cellular and Molecular Life Sciences, 64, 1063-1084.
http://dx.doi.org/10.1007/s00018-007-6477-4 - 17. Boyd, E.S., Spear, J.R. and Peters, J.W. (2009) [FeFe] Hydrogenase Genetic Diversity Provides Insight into Molecular Adaptation in a Saline Microbial Mat Community. Applied and Environmental Microbiology, 75, 4620-4623.
http://dx.doi.org/10.1128/AEM.00582-09 - 18. Frey, M. (2002) Hydrogenases: Hydrogen-Activating Enzymes. Chembiochem, 3, 153-160.
http://www.ncbi.nlm.nih.gov/pubmed/11921392
http://dx.doi.org/10.1002/1439-7633(20020301)3:2/3<153::AID-CBIC153>3.0.CO;2-B - 19. Xing, D., Ren, N. and Rittmann, B.E. (2008) Genetic Diversity of Hydrogen-Producing Bacteria in an Acidophilic Ethanol-H2-Coproducing System, Analyzed Using the [Fe]-Hydrogenase Gene. Applied and Environmental Microbiology, 74, 1232-1239.
http://dx.doi.org/10.1128/AEM.01946-07 - 20. Yue, Z., Chen, R., Yang, F., MacLellan, J., Marsh, T., Liu, Y., et al. (2012) Effects of Dairy Manure and Corn Stover Co-Digestion on Anaerobic Microbes and Corresponding Digestion Performance. Bioresource Technology, 128, 65-71.
http://dx.doi.org/10.1016/j.biortech.2012.10.115 - 21. Cole, J.R., Wang, Q., Fish, J., Chai, B., McGarrell, D.M., Sun, Y., et al. (2014) Ribosomal Database Project: Data and Tools for High Throughput rRNA Analysis. Nucleic Acids Research, 42, D633-D642.
http://dx.doi.org/10.1093/nar/gkt1244 - 22. Edgar, R.C., Haas, B.J., Clemente, J.C., Quince, C. and Knight, R. (2011) UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics, 27, 2194-2200.
http://dx.doi.org/10.1093/bioinformatics/btr381 - 23. R Core Team (2012) R: A Language and Environment for Statistical Computing.
http://www.r-project.org/ - 24. Oksanen, J., Blanchet, F.G., Kindt, R., Legendre, P., Minchin, P.R., O’Hara, R.B., et al. (2012) Vegan: Community Ecology Package.
http://cran.r-project.org/package=vegan - 25. Magurran, A.E. (2004) Measuring Biological Diversity. Blackwell Publishing, Malden.
http://books.google.com/books?id=tUqzLSUzXxcC - 26. Esty, W. (1986) The Efficiency of Good’s Nonparametric Coverage Estimator. Annals of Statistics, 14, 1257-1260.
http://www.jstor.org/stable/10.2307/3035574
http://dx.doi.org/10.1214/aos/1176350066 - 27. Mullings, R. and Parish, J. (1984) Mesophilic Aerobic Gram Negative Cellulose Degrading Bacteria from Aquatic Habitats and Soils. Journal of Applied Microbiology, 57, 455-468.
http://onlinelibrary.wiley.com/doi/10.1111/j.1365-2672.1984.tb01411.x/abstract
http://dx.doi.org/10.1111/j.1365-2672.1984.tb01411.x - 28. Lednicka, D., Mergaert, J., Cnockaert, M. and Swings, J. (2000) Isolation and Identification of Cellulolytic Bacteria Involved in the Degradation of Natural Cellulosic Fibres. Systematic and Applied Microbiology, 23, 292-299.
http://dx.doi.org/10.1016/S0723-2020(00)80017-X - 29. Stursová, M., Zifcáková, L., Leigh, M.B., Burgess, R. and Baldrian, P. (2012) Cellulose Utilization in Forest Litter and Soil: Identification of Bacterial and Fungal Decomposers. FEMS Microbiology Ecology, 80, 735-746.
http://dx.doi.org/10.1111/j.1574-6941.2012.01343.x - 30. Wirth, R., Kovács, E., Maróti, G., Bagi, Z., Rákhely, G. and Kovács, K.L. (2012) Characterization of a Biogas-Producing Microbial Community by Short-Read Next Generation DNA Sequencing. Biotechnology for Biofuels, 5, 41-56.
http://dx.doi.org/10.1186/1754-6834-5-41 - 31. Schlüter, A., Bekel, T., Diaz, N.N., Dondrup, M., Eichenlaub, R., Gartemann, K.-H., et al. (2008) The Metagenome of a Biogas-Producing Microbial Community of a Production-Scale Biogas Plant Fermenter Analysed by the 454-Pyrosequencing Technology. Journal of Biotechnology, 136, 77-90.
http://dx.doi.org/10.1016/j.jbiotec.2008.05.008 - 32. Wang, H., Vuorela, M., Keranen, A.-L., Lehtinen, T.M., Lensu, A., Lehtomaki, A., et al. (2010) Development of Microbial Populations in the Anaerobic Hydrolysis of Grass Silage for Methane Production. FEMS Microbiology Ecology, 72, 496-506.
http://dx.doi.org/10.1111/j.1574-6941.2010.00850.x - 33. Nolla-Ardèvol, V., Peces, M., Strous, M., Tegetmeyer, H.E., Angelidaki, I., Ellegaard, L., et al. (2015) Metagenome from a Spirulina Digesting Biogas Reactor: Analysis via Binning of Contigs and Classification of Short Reads. BMC Microbiology, 15, 277.
http://dx.doi.org/10.1186/s12866-015-0615-1 - 34. Sun, L., Liu, T., Müller, B. and Schnürer, A. (2016) The Microbial Community Structure in Industrial Biogas Plants Influences the Degradation Rate of Straw and Cellulose in Batch Tests. Biotechnology for Biofuels, 9, 1-20.
http://dx.doi.org/10.1186/s13068-016-0543-9 - 35. Dojka, M. and Hugenholtz, P. (1998) Microbial Diversity in a Hydrocarbon- and Chlorinated-Solvent-Contaminated Aquifer Undergoing Intrinsic Bioremediation. Applied and Environmental Microbiology, 64, 3869-3877.
http://aem.asm.org/content/64/10/3869.short - 36. Roest, K., Heilig, H.G.H.J., Smidt, H., de Vos, W.M., Stams, A.J.M. and Akkermans, A.D.L. (2005) Community Analysis of a Full-Scale Anaerobic Bioreactor Treating Paper Mill Wastewater. Systematic and Applied Microbiology, 28, 175-185.
http://dx.doi.org/10.1016/j.syapm.2004.10.006 - 37. Schwarz, J.I.K., Eckert, W. and Conrad, R. (2007) Community Structure of Archaea and Bacteria in a Profundal Lake Sediment Lake Kinneret (Israel). Systematic and Applied Microbiology, 30, 239-254.
http://dx.doi.org/10.1016/j.syapm.2006.05.004 - 38. Lefebvre, O., Nguyen, T.T.H., Al-Mamun, A., Chang, I.S. and Ng, H.Y. (2010) T-RFLP Reveals High β-Proteobacteria Diversity in Microbial Fuel Cells Enriched with Domestic Wastewater. Journal of Applied Microbiology, 109, 839-850.
http://dx.doi.org/10.1111/j.1365-2672.2010.04735.x - 39. Mackenzie, A.K., Naas, A.E., Kracun, S.K., Schückel, J., Fangel, J.U., Agger, J.W., et al. (2015) A Polysaccharide Utilization Locus from an Uncultured Bacteroidetes Phylotype Suggests Ecological Adaptation and Substrate Versatility. Applied and Environmental Microbiology, 81, 187-195.
http://dx.doi.org/10.1128/AEM.02858-14 - 40. Baena, S., Fardeau, M., Ollivier, B., Labat, M., et al. (1993) Aminomonas paucivorans gen. nov., sp. nov., A Mesophilic, Anaerobic, Amino-Acid-UtiIizing Bacterium. International Journal of Systematic Bacteriology, 49, 975-982.
http://dx.doi.org/10.1099/00207713-49-3-975 - 41. Graber, J.R., Leadbetter, J.R. and Breznak, J.A. (2004) Description of Treponema azotonutricium sp. nov. and Treponema primitia sp. nov., the First Spirochetes Isolated from Termite Guts. Applied and Environmental Microbiology, 70, 1315-1320.
http://dx.doi.org/10.1128/AEM.70.3.1315-1320.2004 - 42. Magot, M., Fardeau, M.-L., Arnauld, O., Lanau, C., Ollivier, B., Thomas, P., et al. (1997) Spirochaeta smaragdinae sp. nov., a New Mesophilic Strictly Anaerobic Spirochete from an Oil Field. FEMS Microbiology Letters, 155, 185-191.
http://dx.doi.org/10.1111/j.1574-6968.1997.tb13876.x - 43. Sugisawa, T., Miyazaki, T. and Hoshino, T. (2005) Microbial Production of L-Ascorbic Acid from D-Sorbitol, L-Sorbose, L-Gulose, and L-Sorbosone by Ketogulonicigenium vulgare DSM 4025. Bioscience, Biotechnology, and Biochemistry, 69, 659-662.
http://japanlinkcenter.org/JST.JSTAGE/bbb/69.659?from=Google
http://dx.doi.org/10.1271/bbb.69.659 - 44. Jia, N., Ding, M.-Z., Du, J., Pan, C.-H., Tian, G., Lang, J.-D., et al. (2016) Insights into Mutualism Mechanism and Versatile Metabolism of Ketogulonicigenium vulgare Hbe602 Based on Comparative Genomics and Metabolomics Studies. Scientific Reports, 6, Article Number: 23068.
http://dx.doi.org/10.1038/srep23068 - 45. Gray, N.D., Sherry, A., Grant, R.J., Rowan, A.K., Hubert, C.R.J., Callbeck, C.M., et al. (2011) The Quantitative Significance of Syntrophaceae and Syntrophic Partnerships in Methanogenic Degradation of Crude Oil Alkanes. Environmental Microbiology, 13, 2957-2975.
http://dx.doi.org/10.1111/j.1462-2920.2011.02570.x - 46. Cheng, L., Ding, C., Li, Q., He, Q., Rong Dai, L. and Zhang, H. (2013) DNA-SIP Reveals That Syntrophaceae Play an Important Role in Methanogenic Hexadecane Degradation. PLoS ONE, 8, 1-11.
http://dx.doi.org/10.1371/journal.pone.0066784 - 47. Ding, C., Ma, T., Hu, A., Dai, L., He, Q., Cheng, L., et al. (2015) Enrichment and Characterization of a Psychrotolerant Consortium Degrading Crude Oil Alkanes Under Methanogenic Conditions. Microbial Ecology, 70, 433-444.
http://dx.doi.org/10.1007/s00248-015-0590-y