Advances in Microbiology
Vol.4 No.7(2014), Article ID:46021,9 pages DOI:10.4236/aim.2014.47040
The Skin Microbiome of Gambusia affinis Is Defined and Selective
Annie B. Leonard1, Jeanette M. Carlson1, Dayna E. Bishoff1, Sarah I. Sendelbach1, Sonja B. Yung1, Sonya Ramzanali1, Ananda B. W. Manage2, Embriette R. Hyde3,4, Joseph F. Petrosino3, Todd P. Primm1
1Department of Biological Sciences, Sam Houston State University, Huntsville, USA
2Department of Mathematics and Statistics, Sam Houston State University, Huntsville, USA
3Alkek Center for Metagenomics and Microbiome Research, Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, USA
4Integrative Molecular and Biomedical Sciences Training Program, Baylor College of Medicine, Houston, USA
Email: tprimm@shsu.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 21 January 2014; revised 11 March 2014; accepted 10 April 2014
ABSTRACT
Metagenomics and bacterial culture were used to determine the normal skin microbiome of the Western mosquitofish (Gambusia affinis). This is the first study of G. affinis, and the most in-depth study of any fish skin, utilizing a combination of 16S profile pyrosequencing and culture analysis. Over 1800 sequences obtained from three individuals reveal that over half of all sequences come from five invariant genera, Acinetobacter, Sphingomonas, Acidovorax, Enhydrobacter, and Aquabacterium. The microbiome is diverse but has low equitability, with a total of 81 genera detected. Challenge studies suggest that non-native bacteria cannot colonize the skin. This definition of the normal skin microbiome lays the foundation for future studies with this model system.
Keywords:Fish, Microbiome, Acinetobacter, Sphingomonas, Metagenomics, Mucosal

1. Introduction
The collection of all microorganisms in and on an animal (the microbiome or microbiota) plays a central role in animal health. The revolution in DNA sequencing technology has allowed us to begin analyzing the microbiome of humans [1] and other animals in tremendous detail. Though not a dominant model for microbiome studies in relation to human health, fishes are highly tractable model systems for development, immunity, and infection studies. Fishes are evolutionarily important because piscine species compose half of all vertebrate life, and are the most ancient branch on the tree of life. Fishes are ecologically important as aquatic animals, with the majority of the Earth’s surface covered by water.
Fish gut microbiome studies have been summarized in an excellent recent review [2] . Sixteen studies examining the gut microbiome of 18 fish species were discussed, with the largest study including 3748 16S sequences and the smallest with 15 sequences. It is no surprise that the model organism zebrafish (Danio rerio) has been the best study. All samples were dominated at the phylum level by Proteobacteria, with Firmicutes and Bacteroidetes the next most common. A diverse pattern without a clear consensus was found when the bacteria were examined at the order level. The strongest factor affecting the gut population was the aquatic environment (saltwater vs freshwater).
Like humans and other animal models, the skin microbiome of fish has not been as well studied as the gut microbiome. A few studies have embarked onto this new territory; for example, the study of Landeira-Dabarca et al., in which bacteria were cultured from the cutaneous mucus of hatchery-reared Atlantic salmon (Salmo salar), and both number of colony counts and colony morphology changed between diets [3] . However, a drawback to this study was that specific bacteria were not identified. In contrast, a 2011 study identified Mycoplasma, Psychrobacter, Proteus, Photobacterium, Shewanella, Staphylococcus, Vagococcus, and Vibrio in the skin, gut, and gills of four Atlantic mackerel (Scomber scombrus) caught in the Norwegian Sea [4] . However, determination of abundance was not possible with the analysis methods used by the authors. A study of the skin of gibel carp (Carassius auratus gibelio) and bluntnose black bream (Megalobrama amblycephala Yih) cultured together in a freshwater pond found that the dominant genera on the skin were Acinetobacter, Anoxybacillus, and Enterobacter, and in this study, abundance was also estimated [5] . Finally, the skin microbiota of 102 fishes representing six species from the Gulf of Mexico was assessed [6] . The most common genera detected across all fish were Aeribacillus and Pseudomonas, with Janthinobacterium, Delftia, and Acinetobacter dominant in certain fish species only. Not surprisingly, the species of fishes was the most significant factor affecting the microbiome population.
While these studies have provided a good start toward analyzing the fish skin microbiome, additional, more in-depth studies utilizing the latest metagenomics analysis techniques are needed to fully assess the fish skin microbiome. Studies such as these may provide important contributions to human health, as fish skin biology has been studied as a model for human dermatology, especially related to innate antimicrobial functions [7] . Additionally, fish skin is an excellent general model for mucosal immunity, as fishes live in aquatic environments in constant exposure to high numbers and varieties of microbes.
Herein we present the first microbiological study of the Western mosquitofish (Gambusia affinis), a small (adults 0.1 to 2.0 grams) freshwater, live-bearing, Poeciliid fish [8] . It is an invasive species, having been widely introduced by humans for control of mosquito larva. It is an omnivore, primarily top-feeding on insect larva, zooplankton, and detritus. These aggressive and hardy fishes often displace native fishes and insects. It has been primarily studied scientifically for its coercive mating system [9] [10] . Ours is also the most extensive study of any fish skin microbiome to date, with respect to sequencing depth, and also includes complementary culturebased methods. Finally, this is the first study of a fish skin microbiota captured from a natural freshwater environment.
2. Materials and Methods
2.1. Collection of Skin Samples
Fishes were collected from a pristine lake in a rural neighborhood in Walker County, Texas, at +30 degrees 49 minutes 23 seconds by −95 degrees 32 minutes 35 seconds. Fishes were identified as Gambusia affinis visually. Fishes were transported back to the laboratory in a 5 L bucket of pond water, and acclimated for 24 hours. The lab temperature was 24˚C - 26˚C. The skin microbiome was extracted by placing individual fish into 2 ml of sterile PBST (140 mM NaCl, 10 mM phosphate, 3 mM KCl, 0.05% Tween-20, pH 7.4) within a sterile 15ml conical tube, and mixing using a vortexer for two minutes. This procedure has been validated in that resampling the same fish generates less than 0.1% of the number of viable bacteria compared to the first sampling, as measured by colony counts on nutrient agar (NA). NA is 5 g/L Bacto casitone (Difco), 3 g/L beef extract (Himedia), and 15 g/L Bacto agar (Difco).
2.2. DNA Extraction and Sequencing
Bacteria were collected from the PBST suspension by centrifugation at 5000 g for 5 minutes. The supernatant was decanted, and the resulting bacterial pellet was frozen at −80˚C until shipment on dry ice to Baylor College of Medicine (BCM). DNA was extracted from the thawed pellets with the MoBio PowerSoil kit following protocols developed and benchmarked during the Human Microbiome Project [1] . The V3-V5 regions of the 16 S genes were PCR amplified using barcoded universal primers (357f CCTACGGGAGGCAGCAG and 926 R CCGTCAATTCMTTTRAGT). The primers also contained adaptor oligos for 454 pyrosequencing (A adaptor, on reverse primer before the barcode sequence: CCATCTCATCCCTGCGTGTCTCCGACTCAG; B adaptor, on forward primer: CCTATCCCCTGTGTGCCTTGGCAGTCTCAG). Amplicons were sequenced on a multiplexed 454 FLX-Titanium (Roche) sequencing run at the Human Genome Sequencing Center at BCM. The resulting Standard Flowgram Format (sff) file was extracted into FASTA and qual files using mothur [11] . Sequences were then associated with each sample based on individual primer barcodes and quality trimmed to remove barcoded primers; sequence reads with more than one ambiguous base in the barcode and more than two ambiguous bases in the primer, and reads with a quality score of less than 35 over a sliding 50 bp window were removed. Reads with any ambiguous bases and with a homopolymer longer than 8bp were also removed. The FASTA files for each individual sample were then input into the CloVR 16S analysis pipeline [12] , which uses mothur and QIIME for operational taxonomic unit (OTU) picking and alpha and beta diversity analyses, UCHIME for chimera checking and removal, and RDP Bayesian classifier for taxonomic classification.
2.3. Culture Analysis
To measure the culturable skin bacteria, skin was extracted as above in Section 2.1. and ten-fold serial dilutions made in PBST. Most Probable Number (MPN) was estimated by adding 50 μL of each dilution to 150 μL of Nutrient Broth (NB, identical to NA without the agar) or Luria Broth (LB from Difco) or Bacto Tryptic Soy Broth (TSB from Difco), with five replicates for each dilution, using 96-well plates (Falcon). Growth was read visually after a 24 hour incubation at 25˚C. Calculations were made according to the FDA BAM [13] . Colony Forming Units (CFU) were determined by spreading 100 μL of each dilution in duplicate onto NA and Eosin Methylene Blue (EMB Levine from Difco) plates, and counting colonies after a 48 hour incubation at 25˚C.
2.4. Challenge Experiments
Challenge bacteria were grown 48 hours at 25˚C on NA, then suspended in PBST. Fishes were exposed to the challenge bacteria in Artificial Pond Water (APW, 0.11 g/L CaCl2, 0.11 g/L MgSO4, 0.04 g/L NaHCO3) for 48 hours at 25˚C, followed by replacement with fresh APW. Bacterial counts on the skin were measured as above, with challenge bacteria being identified using unique morphological characteristics not present in the normal fish flora. Micrococcus luteus (initial exposure 2.1 × 107 CFU/ml APW) was identified by appearance of nonfermentive, yellow-pigmented colonies on Mannitol Salt Agar (MSA, from Difco). No colonies were observed from the unexposed fish on MSA. E. coli strain B (initial exposure 2.7 × 108 CFU/ml) was identified by colonies with a metallic green sheen on EMB. No colonies on EMB from the unexposed fish displayed the metallic green phenotype. Serratia marcescens (initial exposure 1.4 × 109 CFU/ml) exhibited a strong red pigment on NA plates. Bacillus megaterium (initial exposure 1.5 × 106 CFU/ml) grows on MSA with large white colonies that turn the surrounding media yellow. No fish fatalities were observed in the challenge experiments.
2.5. Community Analysis
Diversity in ecosystems (biomes) can be quantified by diversity indices. A set of three common indices are α, β, and γ diversity [14] . Alpha diversity, or species richness, is total number of OTUs in one individual fish skin biome. Beta diversity is the total number of OTUs unique to each biome (subtractive) between two compared ecosystems, two fishes in this case. Gamma diversity is the toal unique OTUs among all biomes (additive) in a region, in this case, all three of the fishes. To quantify organism richness and evenness in the 16S profiles from the fish skin, the Simpson’s Diversity Index was calculated [15] .
3. Results
3.1. 16S Profile of Fish Skin
After quality filtering and normalization, 585, 798, and 807 16S rDNA sequences were obtained from the skin of three fishes. Of these, 378 (17.2%) could not be unambiguously classified at the genus level. The majority of the unclassified sequences were from one fish, being from the family Peptostrepococcaceae (179 sequences, or almost half). Thus, the current databases are very efficient (82.8%) at high-confidence genus level identification using short (~400 bp) 16S sequence reads. Within the sequence reads, the most abundant OTUs matched were in the genera Acinetobacter (16.5% of all recovered sequences classified to the genus level), Sphingomonas (15.6%), Acidovorax (8.8%), Enhydrobacter (7.2%), Aquabacterium (7.1%), and Myroides (4.1%).
For comparing and interpreting Figure 1 and Figure 2, the taxonomic classifications and relatedness of the nine most abundant organisms from Figure 1 are shown in Table 1.
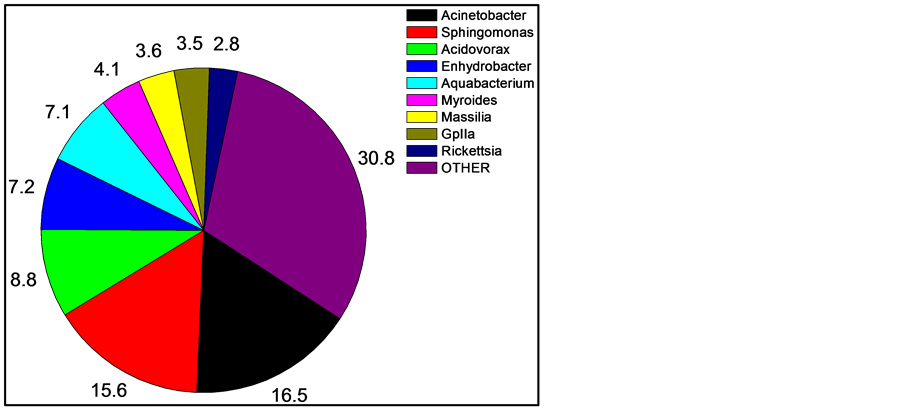
Figure 1. Mean skin abundance of sequences at the genus level of three sampled fishes. Numbers are percent abundance, with OTHER being a combination of all sequences in genera with less than two percent abundance and sequences unclassified at the genus level. Shown are the nine most abundant genera.
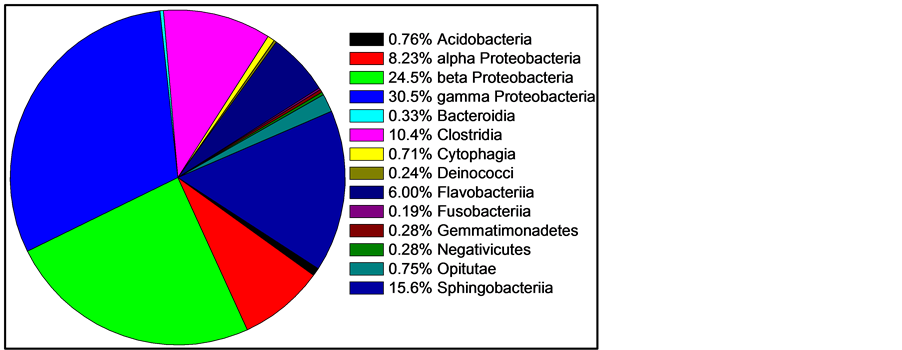
Figure 2. Mean skin abundance of sequences at the class level of three fishes. Two classes containing only one sequence are not shown: Chloroflexi and Vurrucomicrobiae. Two classes represented by only two recovered sequences are also not shown: Bacilli and Mollicutes. All other classes are shown.

Table 1. Relatedness of dominant organisms.
*Incertasedis means uncertain placement. Adjacencies in the table are according to relatedness. GpIIa is a genus of uncertain placement, in the phylum Cyanobacteria.
3.2. Culture Analysis
MPN using NB gave an estimate of 9.5 × 105 bacteria per gram of fish weight, with a 95% CI of 3.0 to 23 × 105, or about one million culturable organisms per gram. MPN using LB and TSB gave highly similar results (data not shown). NA plates yielded a count of 4.8 ± 2.9 × 105 CFU/g, while EMB 6.8 ± 0.4 × 105 CFU/g. The higher counts on EMB are consistent with the sequencing data, as EMB is designed to grow enterics, and the skin is dominated by Proteobacteria (Table 1).
3.3. Challenge Experiments
To explore the selectivity of the fish skin microbiome, fishes were bath exposed to several non-pathogenic and non-native bacterial species. The pattern observed was initial colonization of the skin, followed by rapid decline and loss of the challenge bacteria. With the M. luteus (Gram positive cocci) exposure, 7.7 ± 5.8 × 104 CFU/g of fish weight was observed on MSA plates immediately following the 48 hours exposure, with 2.2 ± 0.3 × 105 CFU/g on NA plates. Three days after the water change, no colonies were seen on MSA, with 1.2 ± 0.3 × 105 CFU/g on NA plates. With the E. coli (Gram negative gamma Proteobacteria), before exposure the fish skin generated 6.1 ± 1.3 × 105 CFU/g on EMB plates (none being metallic green), and 5.3 ± 4.4 × 106 CFU/g on NA plates. Immediately after the 48 hour exposure, 1.6 ± 0.6 x 107 CFU/g was observed on EMB plates (0.03% being metallic green) and 6.9 ± 1.4 × 108 CFU/g on NA plates. Two days after the water change, 8.0 ± 3.3 × 104 CFU/g was observed on EMB plates (1.2% being metallic green) and 3.8 ± 2.8 × 107 CFU/g on NA plates. Three days after the water change, 2.6 ± 0.4 × 104 CFU/g was observed on EMB plates (none being metallic green) and the NA plates were too numerous to count. With S. marcescens (Gram negative gamma Proteobacteria), no red colonies were observed from the normal fish microbiota. Following the 48 hour exposure, 1.6 ± 0.6 × 107 CFU/g was observed on NA, with 15.8% of colonies being red. Three days after the water change, 3.8 ± 1.8 × 104 CFU/g was observed on NA plates (1.2% being red), and four days after 6.0 ± 2.7 × 104 CFU/g was observed on NA plates (none being red). With B. megaterium (Gram positive rod), no colonies appeared on MSA plates during the experiment (counts from NA after the 48 hour exposure of 1.6 ± 0.5 × 105 CFU/g, and three days after of 1.6 ± 0.1 × 105 CFU/g).
3.4. Community Analysis
Alpha, beta, and gamma diversity indices were calculated separately for both the dominant and the remaining OTUs from the fish skin.
The organism richness number in the Simpson’s Index, or the S value, is the combination of the dominant and rare alpha numbers from Table 2, or 53 from fish A, 69 from fish B, and 68 from fish C. The diversity index D from fish A is 0.109, from fish B is 0.164, and from fish C is 0.070. The equitability (organism evenness), or ED, is 0.204 for fish A, 0.111 from fish B, and 0.268 from fish C. Removing the unclassified genera had a negligible effect on these numbers, for example, the D for all three fishes together including the unclassified is 0.094 and after removal of unclassified is 0.093.
Table 2. Diversity among dominant and rare oganisms.
Dominant organisms are the most abundant seven genera of Acinetobacter, Sphingomonas, Acidovorax, Enhydrobacter, Aquabacterium, Myroides, and Massilia, collectively 58.8% of all sequences. Rare organisms are all the other OTUs, none of which are more than 2% abundance in any of the three fishes.
4. Discussion
We have generated the most detailed examination to date of any fish skin microbiome, utilizing DNA pyrosequencing to determine 16S rRNA gene profiles of three mosquitofishes. The microbiome was dominated by a few organisms (Figure 1), with the top five genera comprising 55.2% of all sequences, and the top seven being 58.8%. This dominant group was very stable and consistent across fish (Figure 3), while the more rare organisms were much less stable and consistent (Table 2). However, while the number of times a 16S sequence is recovered which is recovered is a good measurement of abundance, it does not directly correlate to quantity of the organism, primarily because bacteria can contain a wide range, from one to 15 copies, of the 16S gene in their genomes [16] . Typically these copies are all identical or highly similar (1 - 3 bp difference). Since the organisms in the skin microbiome can only be identified to the genus level, the exact number of 16S copies in the bacteria cannot be known for certain. Using the Ribosomal RNA Database [17] , the number can be estimated. The database contains 11 species of Acinetobacter, which contain four to seven 16S gene copies, with an average of six. Sphingomonas is only represented by the S. wittichii species, which contains two copies, while two species are described for Acidovorax, which both have three copies. The genera Enhydrobacter, Aquabacterium, Myroides, and Massila have no entries. Myroides is in the family Flavobacteriaceae, which has ten entries, with an average of three copies each, but unfortunately a range of one to six. Massilia is in the family Oxalobacteraceae, which has four entries in the database, which have two or three copies, and an average of 2.5. This suggests that the truly most abundant genus on the skin is Sphingomonas.
The most dominant seven genera present are also the most stable (Table 2 and Figure 3). Those seven genera were present in each of the three fishes invariably, and always composed the most abundant sequences, although not always in the same order of abundance. In contrast, high beta diversity showed that the other 74 genera varied considerably in their presence on each fish. Of those 74, 33 (44%) were recovered from only one of the three fishes sampled (from 1 to 6 sequences recovered). This suggests the rare sequences may represent transient members of the skin microbiota. Another way to quantify rarity is to count how many genera of all 81 found consist of no more than one sequence per fish, which was 31 (38%). This again supports the idea that, while a diversity of 81 different genera were recovered from the fish skin; it is likely that less than ten are stable and dominant in the microbiome. Future sampling of more fish will help confirm this.
Since the microbiome is dominated by a few organisms, we explored selectivity by challenge studies. While B. megaterium was excluded, the other three non-native species were able to temporarily establish into the fish skin biome. The initial dose of organism correlated with persistence on the skin, with higher doses being recovered for longer periods, suggesting some mechanism to actively push out invading species. Experiments are underway to determine if selectively is determined by the fishes themselves, possibly in secretions, or from the dominant bacteria in the microbiome, or some combination of both. The challenge results are consistent with the sequence analysis, in that the skin microenvironment is dominated by just a few species and selective against nonnative species.
Simpson’s diversity D values range from 0 for infinite diversity to 1 for no diversity. Thus the lower values

Figure 3. Variation among the three normal fishes of the nine most abundant sequences at the genus level. Column is average of the mean abundance, and the error bars are standard deviation.
(all > 0.2) indicate the skin as rather diverse, consistent with the presence of over 80 genera. The evenness ED also ranges from 0 to 1, with 1 being complete equitability. The lower numbers indicate the microbiomes are uneven, which reflects the domination of a complex (approximately 65 OTUs in each fish) skin community by a relatively few (about 7 OTUs).
All seven of the dominant genera are Gram negative rods. In fact, of the 1812 sequences classified at the genus level, only 13 are from Gram positive organisms (0.7%). Consistent with this, when skin extract was plated onto MSA, Chapman Stone agar (Difco), or Phenylethyl Alcohol Agar (Difco), no growth was observed (data not shown). The mechanism of this striking selective bias is under investigation. The Svanevik and Lunestad study [4] of Atlantic mackerel identified eight genera present on the skin, five of which were Gram negative, and all of those Proteobacteria. Abundance was not determined. The Wang et al. study [5] of bream and carp also identified Acinetobacter as dominant, but did recover Gram positive Firmicutes and Actinocymetes. The Larsen et al. survey of six Gulf species [6] found the Proteobacteria members Pseudomonas, Acinetobacter, Janthinobacterium, and Delftia were dominant. Thus, prior studies of fish skin that measured abundance found Proteobacteria as the major phylum, so this is a consistent point among the published surveys. However, the domination by Gram negative bacteria in Gambusia is unique so far among fishes studied. Thus our study agrees with Larsen et al. in suggesting the uniqueness of the skin microbiome of each fish species.
In contrast, Gram positive organisms, such as Staphylococcus, Streptococcus, Propionibacterium, and Corynebacterium, tend to dominate the skin of humans [1] and other land animals. Humans secrete antimicrobial proteins, such as psoriasin, to prevent colonization of the skin by enteric Gram negatives [18] . This study suggests that the fish skin and gut are more homogenous than in humans, which may be a major difference between aquatic and terrestrial animals, but will need further verification. Thus the skin of fish may not be very representative models for human skin biology, but instead serve as suitable models for mucosal immunity, such as in the human lung and urogenital tract. The abundance of Gram negative bacteria on fish skin is matched in the fish gut [2] . While the gut is also dominated by Proteobacteria, the Gram positive phyla of Firmicutes and Actinobacteria are also present at significant levels (from 5% to 35% combined). Unlike humans, in fish, both the skin and the gut are mucosal surfaces, although the gut is presumably far more anaerobic. The seven dominant fish skin genera identified in this study are non-fermentative strict aerobes (Acinetobacter, Sphingomonas, Myroides, and Massilia) or non-fermentative with some species able to carry out anaerobic respiration using nitrate (Acidovorax and Aquabacterium), with only one genus (Enhydrobacter) having some fermentation as a facultative anaerobe, according to Bergey’s Manual [19] . This suggests aerobic metabolism dominates in the skin microenvironment, unlike the gut.
Noteworthy was the recovery of the obligate intracellular pathogen Rickettsia from the fish skin (composed 4.1% of sequences from fish A, 2.3% from fish B, and 1.9% from fish C), suggesting either a low-level infection in this fish population or a consistent colonization. The related Neorickettsia (different family, same order) was found only in striped mullet (Mugil cephalus) among the Gulf fish [6] , but at a significant level (approximately 7% of sequences). None of the other skin or gut studies reported Rickettsiales. Rickettsia has been reported as fish pathogens, but their role is as yet unclear [20] .
The skin microbiome of Gambusia affinis is dominated by seven taxa which exhibit low variability between individual fishes, and thus can serve as an excellent future model system for studies of antibiotics, probiotics, infections, and immunity. In animal systems, skin is inherently easier to manipulate experimentally than the gut. Uncovering the mechanisms behind the impressive selectivity of fish skin may lead to breakthroughs in mucosal immunity.
References
- Human Microbiome Project Consortium (2012) Structure, Function and Diversity of the Healthy Human Microbiome. Nature, 486, 207-214. http://dx.doi.org/10.1038/nature11234
- Sullam, K.E., Essinger, S.D., Lozupone, C.A., O’Conner, M.P., Rosen, G.L., Knight, R., Kilham, S.S. and Russell, J.A. (2012) Environmental and Ecological Factors That Shape the Gut Bacterial Communities of Fish: A Meta-Analysis. Molecular Ecology, 21, 3363-3378. http://dx.doi.org/10.1111/j.1365-294X.2012.05552.x
- Landeira-Dabarca, A., Siero, C. and Alvarez, M. (2013) Change in Food Ingestion Induces Rapid Shifts in the Diversity of Microbiota Associated with Cutaneous Mucus of Atlantic Salmon Salmosalar. Journal of Fish Biology, 82, 893-906. http://dx.doi.org/10.1111/jfb.12025
- Svanevik, C.S. and Lunestad, B.T. (2011) Characterization of the Microbiota of Atlantic Mackerel (Scomberscombrus). International Journal of Food Microbiology, 151, 164-170.
- Wang, W., Zhou, Z., He, S., Liu, Y., Cao, Y., Shi, P., Yao, B. and Ringo, E. (2010) Identification of the Adherent Microbiota on the Gills and Skin of Poly-Cultured Gibel Carp (Carassiusauratusgibelio) and Bluntnose Black Bream (Megalobramaamblycephala Yih). Aquaculture Research, 41, e72-e83. http://dx.doi.org/10.1111/j.1365-2109.2009.02459.x
- Larsen, A., Tao, Z., Bullard, S.A. and Arias, C.R. (2013) Diversity of the Skin Microbiota of Fishes: Evidence for Host Species Specificity. FEMS Microbial Ecology, 85, 1-12.
- Rakers, S., Gebert, M., Uppalapati, S., Meyer, W., Maderson, P., Sell, A.F., Kruse, C. and Paus, R. (2010) Fish Matters: The Relevance of Fish Skin Biology to Investigative Dermatology. Experimental Dermatology, 19, 313-324. http://dx.doi.org/10.1111/j.1600-0625.2009.01059.x
- Pawlitz, R.J. and Kayla, D.D. (2012) The National Nonindigenous Aquatic Species Program. US Geological Survey, Reston. http://nas.er.usgs.gov
- Cureton II, J.C., Martin, M.E. and Deaton, R. (2010) Short Term Changes in Sex Ratio and Density Alter Coercive Male Mating Tactics. Behavior, 11, 1431-1442. http://dx.doi.org/10.1163/000579510X519495
- Deaton, R. (2008) Factors Influencing Male Mating Behavior in Gambusia affinis (Baird & Girard) with a Coercive Mating System. Journal of Fish Biology, 72, 1607-1622. http://dx.doi.org/10.1111/j.1095-8649.2008.01827.x
- Schloss, P.D., Westcott, S.L, Ryabin, T., Hall, J.R., Hartmann, M., Hollister, E.B., Lesniewski, R.A, Oakley, B.B., Parks, D.H., Robinson, C.J., Sahl, J.W., Stres, B., Thallinger, G.G, Van Horn, D.J. and Weber, C.F. (2009) Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Applied and Environmental Microbiology, 75, 7537-7541. http://dx.doi.org/10.1128/AEM.01541-09
- Angiuoli, S.V., Matalka, M., Gussman, G., Galens, K., Vangala, M., Riley, D.R., Arze, C., White, J.R., White, O. and Fricke, W.F. (2011) CloVR: A Virtual Machine for Automated and Portable Sequence Analysis from the Desktop Using Cloud Computing. BMC Bioinformatics, 12, 356. http://dx.doi.org/10.1186/1471-2105-12-356
- Blodgett, R. (2010) Bacteriological Analytical Manual. Food and Drug Administration USA, Appendix 2.
- Whittaker, R.H. (1972) Evolution and Measurement of Species Diversity. Taxon, 21, 213-251. http://dx.doi.org/10.2307/1218190
- Beals, M., Gross, L. and Harrell, S. (1999) Diversity Indices: Simpson’s D and E. http://www.tiem.utk.edu/~gross/bioed/bealsmodules/simpsonDI.html
- Kembel, S.W., Wu, M., Eisen, J.A. and Green, J.L. (2012) Incorporating 16S Gene Copy Number Information Improves Estimates of Microbial Diversity and Abundance. PLoS Computational Biology, 8, Article ID: e1002743. http://dx.doi.org/10.1371/journal.pcbi.1002743
- Lee, Z.M., Bussema 3rd, C. and Schmidt, T.M. (2009) rrnDB: Documenting the Number of rRNA and tRNA Genes in Bacteria and Archaea. Nucleic Acids Research, 37, 489-493. http://dx.doi.org/10.1093/nar/gkn689
- Wiesner, J. and Vilcinskas, A (2010) Antimicrobial Peptides: The Ancient Arm of the Human Immune System. Virulence, 5, 440-464. http://dx.doi.org/10.4161/viru.1.5.12983
- Garrity, G., Brenner, D.J., Krieg, N.R. and Staley, J.R. (2005) Bergey’s Manual of Systematic Bacteriology. 2nd Edition, Springer, Berlin.
- Fryer, J.L. and Mauel, M.J. (1997) The Rickettsia: An Emerging Group of Pathogens in Fish. Emerging Infectious Diseases, 3, 137-144. http://dx.doi.org/10.3201/eid0302.970206