CellBio
Vol.04 No.03(2015), Article ID:62110,9 pages
10.4236/cellbio.2015.43005
The Cell Biology of Systemic Hyperinflammation Resulting from Failed Cytolytic Target Cell Killing
Bor Luen Tang
Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, Singapore

Copyright © 2015 by author and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 2 November 2015; accepted 19 December 2015; published 22 December 2015
ABSTRACT
Mutations in genes encoding a key component of cytotoxic granules, or the machinery for their release, underlie the systemic hyperiflammatory symptoms of familial hemophagocytic lymphohistiocytosis (FHL), a typically pediatric onset autosomal recessive disorder with five known genetic subtypes (FHL1 - 5). FHL1 mutations have been mapped to chromosome 9, while the respective genes mutated in FHL2 (PRF1), FHL3 (UNC13D/Munc13-4), FHL4 (STX11) and FHL5 (STXBP2/ Munc18b/Munc18-2) have been identified. Perforin gene mutation directly affected the cytolytic activity of the cytotoxic granules. All the other FHL mutations appear to affect some aspect of cytotoxic granule exocytosis, resulting in impaired target cell killing by cytolytic T lymphocytes (CTLs) and/or natural killer (NK) cells. Recent findings suggest that failure to kill and detach from target cells, and prolonged synapse connection time, promote cytokine hypersecretion by the defective CTLs and NKs, which in turn result in systemic inflammation. Deciphering the genetics of FHL has contributed towards our understanding of the cell biology of hyperinflammatory responses and hemophagocytic lymphohistiocytosis accompanying pathological conditions such as cancer and viral infections.
Keywords:
Cytokine Storms, Cytolytic T Lymphocytes (CTLs), Familial Hemophagocytic Lymphohistiocytosis (FHL), Natural Killer (NK) Cells, Hyperinflammation

1. Introduction
Diseased cells, particularly those that express tumor markers and viral antigens, are constantly picked up by immunological surveillance and targeted for destruction by the immune system. Target cell killing in the vertebrate adaptive and innate immune responses to cancerous or infected cells is mediated by the action of cytolytic T lymphocytes (CTLs) and natural killer (NK) cells [1] . CTLs harbor T-cell receptors with CD8 co-receptors, which recognize tumor or viral antigens presented by major histocompatibility complex (MHC) class 1 molecules, and are activated as such. NK cells are circulating lymphocytes which cytotoxic activity is determined by a balance of stimulation of their activating or inhibitory receptors, and is inhibited by self MHC class I molecules. NK cells could also be activated by antibodies that bind to disease associated antigens via its FcϒRIII (CD16) receptors. Both CTLs and NK cells kill diseased target cells using cytotoxic or lytic granules containing the complement component 9 (C9)-like pore-forming protein perforin [2] - [4] , programmed cell death inducing molecules such as Fas Ligand (FasL) and serine proteases known as granzymes [5] [6] . These granules are secretory lysosomes [7] that are generated and secreted via a specialized mode of regulated secretion in cytotoxic killer cells [8] . In recognizing and engaging target cells, the activated killer cells form a transient cell-cell junctional contact―called the immunological synapse [9] . Their cellular microtubule network re-orientates in order to facilitate vesicle movement towards the synapse. The cytotoxic granules are then delivered by motor proteins along the microtubules to the synaptic junction. Docking and fusion of the granules at the junctional membranes releases the pore-forming perforin, which facilitates the entry of granzymes into the target cells [10] . Target cell death could potentially occur by lysis, but largely exhibit variants of programmed cell death or apoptosis due to FasL and granzymes [11] , which in the case of viral infected cells would limit the systemic spread of the pathogen.
Several disease conditions could precipitate conditions of hyperinflammation resulting from hypercytokinemia, or cytokine storm [12] . The cytokine storm-type syndrome hemophagocytic lymphohistiocytosis is a rare and life-threatening condition with severe systemic hyperinflammation resulting from hypersecretion of cytokines, uncontrolled proliferations of activated lymphocytes and macrophages, as well as signs of hemophagocytosis [13] -[15] . The condition could either be primary/hereditary or secondary/acquired [15] . The former stems from mutations in a set of genes that affects CTL/NK function [16] [17] , and symptoms appear early in life upon viral infection. The latter is principally precipitated by viral infections (such as hepatitis A virus, Epstein-Barr virus and Dengue virus) [18] , lymphoid cancers [19] , or rheumatologic autoimmune diseases [20] .
Recent findings are beginning to reveal how mutations in familial hemophagocytic lymphohistiocytosis (FHL) subtypes affect disease severity and progression. Interestingly, studies on these mutations have shed light on the cell biology of lytic granule exocytosis. Recent work has also provided clues as to how defects in lytic granule activity could lead to a cytokine storm. These advances are described and discussed in the paragraphs below.
2. FHL and Its Mutations
FHL is an autosomal recessive disorder, with symptoms usually evident in early postnatal life, often triggered by viral infections [21] - [23] . There are five different subtypes (see Table 1). The locus of FHL1 has been mapped to chromosome 9q21.3-q22 [24] . For the other subtypes, biallelic loss-of-function mutations have been identified in genes encoding perforin itself, as well as variants of known components of the membrane trafficking machinery. The latter turns out to be essential for cytotoxic granule exocytosis, and genetic analysis of FHL patients have contributed much to our understanding of this fundamental cellular process.

Table 1. A tabulated summary of FHL subtypes and related hereditary diseases, the genes mutated and the nature of the encoding proteins.
FHL2 is due to mutations of the PRF1 gene at chromosome 10q21-22, which encodes the core forming perforin [25] . Perforin gene mutations constitute the largest percentage of FHL cases (up to 50%) [26] . Perforin has structural and functional similarities to C9 [27] , and it could nonspecifically perforate target cells. Its mode of action in target cell killing is beginning to be understood. Perforation appears to induce a transient calcium flux in the target cell, which induces a quick repair of the damaged cell membrane, with both perforin and granzymes being endocytosed into enlarged endosomes. Perforin formed pores in the endosomal membrane which allow the proteases to be gradually released [4] .
While perforin gene mutation directly affected the cytolytic activity of the cytotoxic granules, the other three gene products are involved in cytotoxic granule exocytosis. FHL3 is caused by mutations mapped to UNC13D/ Munc13-4 at chromosome 17q25, which encodes Munc13-4 [28] [29] . Pathogenic, non-coding mutations of the UNC13D locus that impair gene transcription have also been described [30] [31] . Munc13-4 is a member of the Unc13 family of Ca2+-binding vesicle priming protein that regulates vesicle fusion. Munc13-4 localizes with cytotoxic granules at the immunological synapses, and a prominent feature of Munc13-4 deficiency is defective cytolytic granule exocytosis [28] . Interestingly, Munc13-4 is an effector of the small Rab GTPase, Rab27A, and is also essential for dense core granule secretion in platelets [32] .
FHL4 is caused by mutations in the gene encoding the atypical syntaxin 11, STX11, located at chromosome 6q24 [33] . Syntaxin 11 (Stx11) is a member of the syntaxin subfamily [34] of the SNARE family that mediates vesicle fusion in eukaryotes. Unlike other syntaxins, however, it does not have a C-terminal hydrophilic membrane anchor [35] . Although known to be enriched in the hematopoietic system [36] [37] , the exact function of Stx11 in lymphocytes has been unclear until its discovery as a FHL gene. NK cells from FHL4 patients fail to degranulate and lyse target cells [38] . Silencing of Stx11 did not affect proximal signaling events of cytotoxic lymphocyte activation, but diminished NK cell cytotoxicity, while over-expression increased cytotoxic killing [39] . It is now clear that Stx11 serves as a target (t)-SNARE mediating the final fusion step in cytotoxic granule exocytosis, as discussed in the section below. A new mutation found recently for Stx11 in the context of FHL occurs at its N-terminus, which abolishes its binding to Munc18-2 [40] . The latter is the causative gene of FHL5.
FHL5 mutations affect STXBP2/Munc18b/Munc18-2, located at chromosome 19p, which encodes Munc18-2, also known as Syntaxin-binding protein-2 (STXBP2) [41] [42] . CTLs and NK cells from FHL5 subjects have reduced Munc18-2 levels, and impaired cytotoxic granule exocytosis that could be alleviated by ectopic expression of Munc18-2 [42] . Munc18-2 is a member of the Sec1/Munc18 (SM) family [43] with apparently divergent roles in regulating Soluble N-ethylmaleimide sensitive factor Attachment Protein receptor (SNARE) complex formation and the vesicle priming process prior to fusion [44] in regulated exocytosis. In this case Munc18-2 is likely to be regulating the function and activity of a Stx11-containing SNARE complex [45] .
There are several inherited disorders that present clinical syndromes that have overlapping symptoms with FLH. Prominent amongst these are the Chédiak-Higashi syndrome (CHS) and Griscelli syndrome type 2 (GS-2). CHS is an autosomal recessive disorder that stems from mutations in the Lysosome Trafficking Regulator (LYST) gene [46] , and patients suffer from albinism, platelet disorders, recurrent infections and hemophagocytic lymphohistiocytosis during infections. GS-2 is also characterized by partial oculocutaneous albinism and hemophagocytic symptoms, and is caused by mutations in Rab27A [47] , a small GTPase which is important for the biogenesis and dynamics of secretory lysosome-like organelles. These associated gene mutations with clinically relevant predisposition to hyperinflammatory syndrome strongly indicate that the underlying etiology of FLH has to do with defects in cytotoxic granule exocytosis, resulting in impaired CTL/NK cell killing function.
3. Advances in Our Understanding of the Molecular Machinery for Cytotoxic Granule Secretion
Identification of FHL mutations and their analysis has been instrumental in deepening our understanding on particular aspects of cytotoxic granule secretion. Munc13-4, mutated in FHL3, is an effector of Rab27A [48] . Rab27A mutations in mice [49] and human (GS-2) [47] both exhibit impaired lytic granule exocytosis and hemophagocytic syndrome. Rab27A is involved in the biogenesis of specialized lysosome-like organelles, such as melanosomes in melanocytes [50] [51] , and has important trafficking functions in insulin secreting cells [52] . In CTLs, Rab27A is involved in the cytotoxic granule transport to the immunological synapses [53] , via its effect on synaptotagmin-like protein 3 [54] and kinesin 1 motor. The FHL5 gene, Munc18-2, on the other hand encodes an SM protein that presumably aids SNARE complex formation.
The identities of the SNAREs involve in cytotoxic granule exocytosis, particularly with regards to the final step of plasma membrane fusion, have been uncertain. There is evidence for both VAMP2 [55] and VAMP8 [56] serving as vesicle (v)-SNAREs in this regard. As for the corresponding target (t)-SNAREs, the ubiquitous surface SNARE SNAP23 is very likely to be involved, as its association with Stx11 and role in granule exocytosis have been demonstrated [57] [58] . This leaves the identity of the other t-SNARE, likely a member of the syntaxin family, outstanding. The Golgi localized Stx6 [59] and the plasma membrane localized Stx3 [60] and Stx4 [58] have all been implicated in neutrophil exocytosis. Stx11’s identification as the FHL4 gene consolidated its essential role in cytotoxic granule exocytosis, and suggests that it is likely the t-SNARE required for the final fusion step with the plasma membrane. One reason in support of this is that Munc18-2 binds Stx11 with 20 fold higher affinity compared to Stx3 [61] .
Recent reports have further delineated the role of Stx11 and other SNAREs in the process, which occurs via multiple membrane trafficking steps. Prior to perforin-containing cytotoxic granule fusion, Stx11 appears to be targeted first to the immune synapse through the recycling endosome in a VAMP8-dependent manner [45] [62] . Stx11 clustered at the immune synapse then serve as a t-SNARE for the plasma membrane fusion of cytotoxic granules [45] [62] . In other words, the plasma membrane of the CTL/NK at the immunological synapse is prepared for granule fusion by prior Stx11 accumulation. Munc13-4 may function as a Ca2+ sensor at the regulated and rate-limiting priming steps in granule exocytosis (Boswell et al. 2012).
4. Hyperinflammation Resulting from Failed Target Cell Killing
It would appear that all FHL mutations impairs killing of target cells by CTLs and NK cells, but how does this impairment lead to hyperinflammation? Logically, it is plausible that sustained presence of unkilled diseased cells induces a further degree of immune response. FHL CTLs/NKs, though defective in their killing capacity, are not handicapped in their ability to proliferate and secrete cytokines. Impaired killing of target cells could thus result in positive feedbacks that collectively heighten the inflammatory response, with immune cell hyperproliferation resulting from increase cytokine secretion reinforcing each other.
A very recent report aptly illustrated the above notion (see Figure 1). Using antigen restricted CTLs and
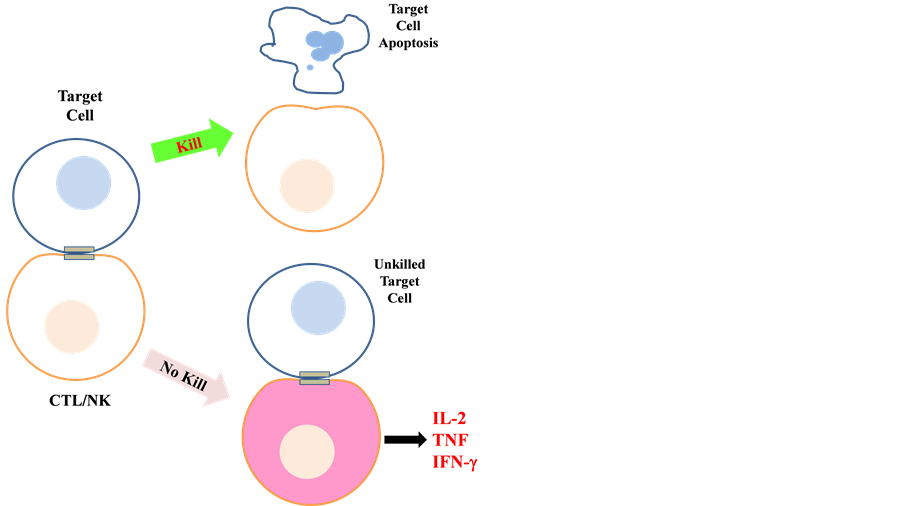
Figure 1. Schematic diagram illustrating the consequence of failure of target cell killing by CTLs/NK cells. Failure to kill target cells and eventual disengagement (i.e. prolonged synapsing time) resulted in hypersecretion of proinflammatory cytokines by the CTLs (see text for details).
NKs from mice lacking perforin or granzymes A and B, Jenkins and colleagues [63] [64] showed that these cells overproduced cytokines (such as IL-2, TNF, IFN-γ) and various chemokines when co-incubated with target cells. Amongst these, IFN-γ appears to be directly responsible for the activation and further production of proinflammatory IL-6 from macrophages. Live-cell imaging analysis of immunological synapses formed between wild- type and mutant CTLs/NK cells with the target cells revealed that one likely reason for cytokine hypersecretion in the latter is sustained Ca2+ signaling, and this is related to the failure of the cells to disengage from their unkilled targets, with a 5-fold increase in mean synapsing time. The authors also found that the signal for disengagement came from the dying target cell itself, and that this signal is caspase-dependent. These findings clearly indicate that failed killing by CTLs/NK cells and prolonged engagement with the unkilled targets causes cytokine hypersecretion, which in turn results in macrophage activation.
It is notable that the FHL subtypes differ in terms of age of onset and trigger. FHL mutations severely reducing cytotoxic lymphocyte function are more likely to eventually present full-fledge Hemophagocytic lymphohistiocytosis [65] [66] . Patients with nonsense mutations in PRF1 or UNC13D are invariably symptomatic within their two first years of life [66] , but nonsense mutations in Munc18-2 and STX11 may have a later onset and less severe phenotype. Investigations with Stx11-deficient mice challenged with lymphocytic choriomeningitis virus showed that unlike perforin-deficient mice, the hemophagocytic lymphohistiocytosis progression is not fatal [67] , apparently due to antigen specific T-cell exhaustion. Some hypomorphic mutations in FLH genes have also been associated with a late disease presentation in adulthood [68] [69] . Therefore despite the rather simplified notion of hyperinflammation resulting from failed target cell killing as the basis for the progression of hemophagocytic lymphohistiocytosis, the severity and onset is clearly influenced and complicated by other factors.
5. Epilogue
In this short review, how recent studies on FHL mutations have further our understanding of the basics of lytic granule exocytosis, and how mutant CTLs/NK cells could become a source of cytokine over-production when they engage but could not kill diseased cells, are discussed. This is an example of how genetic analysis of rare disease syndromes could aid our understanding of basic cell biological processes. Furthermore, the information obtained is critical if we are to further understand the etiology of hemophagocytic lymphohistiocytosis as the syndrome emerges in patients with comorbidities like viral infection, hematopoietic disease, neoplasm [70] [71] , autoimmunity [72] or when it is manifested in less obvious conditions such as presentation of neurological symptoms [73] . Knowing the basic cell biology of FLH would also aid the design of efficacious therapeutic interventions.
Acknowledgements
The author declares no conflict of interest.
Cite this paper
Bor LuenTang, (2015) The Cell Biology of Systemic Hyperinflammation Resulting from Failed Cytolytic Target Cell Killing. CellBio,04,37-45. doi: 10.4236/cellbio.2015.43005
References
- 1. Trambas, C.M. and Griffiths, G.M. (2003) Delivering the Kiss of Death. Nature Immunology, 4, 399-403.
http://dx.doi.org/10.1038/ni0503-399 - 2. Pipkin, M.E. and Lieberman, J. (2007) Delivering the Kiss of Death: Progress on Understanding How Perforin Works. Current Opinion in Immunology, 19, 301-308.
http://dx.doi.org/10.1016/j.coi.2007.04.011 - 3. Voskoboinik, I., Dunstone, M.A., Baran, K., Whisstock, J.C. and Trapani, J.A. (2010) Perforin: Structure, Function, and Role in Human Immunopathology. Immunological Reviews, 235, 35-54.
http://dx.doi.org/10.1111/j.0105-2896.2010.00896.x - 4. Thiery, J., Keefe, D., Boulant, S., Boucrot, E., Walch, M., Martinvalet, D., Goping, I.S., Bleackley, R.C., Kirchhausen, T. and Lieberman, J. (2011) Perforin Pores in the Endosomal Membrane Trigger the Release of Endocytosed Granzyme B into the Cytosol of Target Cells. Nature Immunology, 12, 770-777.
http://dx.doi.org/10.1038/ni.2050 - 5. Bots, M. and Medema, J.P. (2006) Granzymes at a Glance. Journal of Cell Science, 119, 5011-5014.
http://dx.doi.org/10.1242/jcs.03239 - 6. Voskoboinik, I., Whisstock, J.C. and Trapani, J.A. (2015) Perforin and Granzymes: Function, Dysfunction and Human Pathology. Nature Reviews Immunology, 15, 388-400.
http://dx.doi.org/10.1038/nri3839 - 7. Peters, P.J., Borst, J., Oorschot, V., Fukuda, M., Krähenbühl, O., Tschopp, J., Slot, J.W. and Geuze, H.J. (1991) Cytotoxic T Lymphocyte Granules Are Secretory Lysosomes, Containing Both Perforin and Granzymes. The Journal of Experimental Medicine, 173, 1099-1109.
http://dx.doi.org/10.1084/jem.173.5.1099 - 8. de Saint Basile, G., Ménasché, G. and Fischer, A. (2010) Molecular Mechanisms of Biogenesis and Exocytosis of Cytotoxic Granules. Nature Reviews Immunology, 10, 568-579.
http://dx.doi.org/10.1038/nri2803 - 9. Yokosuka, T. and Saito, T. (2010) The Immunological Synapse, TCR Microclusters, and T Cell Activation. Current Topics in Microbiology and Immunology, 340, 81-107.
http://dx.doi.org/10.1007/978-3-642-03858-7_5 - 10. Lopez, J.A., Susanto, O., Jenkins, M.R., Lukoyanova, N., Sutton, V.R., Law, R.H.P., Johnston, A., Bird, C.H., Bird, P.I., Whisstock, J.C., Trapani, J.A., Saibil, H.R. and Voskoboinik, I. (2013) Perforin Forms Transient Pores on the Target Cell Plasma Membrane to Facilitate Rapid Access of Granzymes during Killer Cell Attack. Blood, 121, 2659- 2668.
http://dx.doi.org/10.1182/blood-2012-07-446146 - 11. Susanto, O., Stewart, S.E., Voskoboinik, I., Brasacchio, D., Hagn, M., Ellis, S., Asquith, S., Sedelies, K.A., Bird, P.I., Waterhouse, N.J. and Trapani, J.A. (2013) Mouse Granzyme A Induces a Novel Death with Writhing Morphology That Is Mechanistically Distinct from Granzyme B-Induced Apoptosis. Cell Death & Differentiation, 20, 1183-1193.
http://dx.doi.org/10.1038/cdd.2013.59 - 12. Canna, S.W. and Behrens, E.M. (2012) Making Sense of the Cytokine Storm: A Conceptual Framework for Understanding, Diagnosing, and Treating Hemophagocytic Syndromes. Pediatric Clinics of North America, 59, 329-344.
http://dx.doi.org/10.1016/j.pcl.2012.03.002 - 13. Chandrakasan, S. and Filipovich, A.H. (2013) Hemophagocytic Lymphohistiocytosis: Advances in Pathophysiology, Diagnosis, and Treatment. Journal of Pediatrics, 163, 1253-1259.
http://dx.doi.org/10.1016/j.jpeds.2013.06.053 - 14. Mehta, R.S. and Smith, R.E. (2013) Hemophagocytic Lymphohistiocytosis (HLH): A Review of Literature. Medical Oncology, 30, 740.
http://dx.doi.org/10.1007/s12032-013-0740-3 - 15. Filipovich, A.H. and Chandrakasan, S. (2015) Pathogenesis of Hemophagocytic Lymphohistiocytosis. Hematology/ Oncology Clinics of North America, 29, 895-902.
http://dx.doi.org/10.1016/j.hoc.2015.06.007 - 16. Degar, B. (2015) Familial Hemophagocytic Lymphohistiocytosis. Hematology/Oncology Clinics of North America, 29, 903-913.
http://dx.doi.org/10.1016/j.hoc.2015.06.008 - 17. Tang, B.L. (2015) A Unique SNARE Machinery for Exocytosis of Cytotoxic Granules and Platelets Granules. Molecular Membrane Biology, 32, 120-126.
http://dx.doi.org/10.3109/09687688.2015.1079934 - 18. Parvaneh, N., Filipovich, A.H. and Borkhardt, A. (2013) Primary Immunodeficiencies Predisposed to Epstein-Barr Virus-Driven Haematological Diseases. British Journal of Haematology, 162, 573-586.
http://dx.doi.org/10.1111/bjh.12422 - 19. Yamazaki, S., Nakamura, F., Nasu, R., Nannya, Y., Ichikawa, M. and Kurokawa, M. (2011) Haemophagocytic Lymphohistiocytosis Is a Recurrent and Specific Complication of Acute Erythroid Leukaemia. British Journal of Haematology, 153, 669-672.
http://dx.doi.org/10.1111/j.1365-2141.2010.08544.x - 20. Atteritano, M., David, A., Bagnato, G., Beninati, C., Frisina, A., Iaria, C. and Cascio, A. (2012) Haemophagocytic Syndrome in Rheumatic Patients. A Systematic Review. European Review for Medical and Pharmacological Sciences, 16, 1414-1424.
- 21. Cetica, V., Pende, D., Griffiths, G.M. and Aricò, M. (2010) Molecular Basis of Familial Hemophagocytic Lymphohistiocytosis. Haematologica, 95, 538-541.
http://dx.doi.org/10.3324/haematol.2009.019562 - 22. Sieni, E., Cetica, V., Mastrodicasa, E., Pende, D., Moretta, L., Griffiths, G. and Aricò, M. (2012) Familial Hemophagocytic Lymphohistiocytosis: A Model for Understanding the Human Machinery of Cellular Cytotoxicity. Cellular and Molecular Life Sciences, 69, 29-40.
http://dx.doi.org/10.1007/s00018-011-0835-y - 23. Gholam, C., Grigoriadou, S., Gilmour, K.C. and Gaspar, H.B. (2011) Familial Haemophagocytic Lymphohistiocytosis: Advances in the Genetic Basis, Diagnosis and Management. Clinical & Experimental Immunology, 163, 271-283.
http://dx.doi.org/10.1111/j.1365-2249.2010.04302.x - 24. Ohadi, M., Lalloz, M.R., Sham, P., Zhao, J., Dearlove, A.M., Shiach, C., Kinsey, S., Rhodes, M. and Layton, D.M. (1999) Localization of a Gene for Familial Hemophagocytic Lymphohistiocytosis at Chromosome 9q21.3-22 by Homozygosity Mapping. American Journal of Human Genetics, 64, 165-171.
http://dx.doi.org/10.1086/302187 - 25. Stepp, S.E., Dufourcq-Lagelouse, R., Le Deist, F., Bhawan, S., Certain, S., Mathew, P.A., Henter, J.I., Bennett, M., Fischer, A., de Saint Basile, G. and Kumar, V. (1999) Perforin Gene Defects in Familial Hemophagocytic Lymphohistiocytosis. Science, 286, 1957-1959.
http://dx.doi.org/10.1126/science.286.5446.1957 - 26. Trizzino, A., zur Stadt, U., Ueda, I., Risma, K., Janka, G., Ishii, E., Beutel, K., Sumegi, J., Cannella, S., Pende, D., Mian, A., Henter, J.I., Griffiths, G., Santoro, A., Filipovich, A. and Aricò, M., Histiocyte Society HLH Study Group (2008) Genotype-Phenotype Study of Familial Haemophagocytic Lymphohistiocytosis Due to Perforin Mutations. Journal of Medical Genetics, 45, 15-21.
http://dx.doi.org/10.1136/jmg.2007.052670 - 27. Rosado, C.J., et al. (2007) A Common Fold Mediates Vertebrate Defense and Bacterial Attack. Science, 317, 1548-1551.
http://dx.doi.org/10.1126/science.1144706 - 28. Feldmann, J., Callebaut, I., Raposo, G., Certain, S., Bacq, D., Dumont, C., Lambert, N., Ouachée-Chardin, M., Chedeville, G., Tamary, H., Minard-Colin, V., Vilmer, E., Blanche, S., Le Deist, F., Fischer, A. and de Saint Basile, G. (2003) Munc13-4 Is Essential for Cytolytic Granules Fusion and Is Mutated in a Form of Familial Hemophagocytic Lymphohistiocytosis (FHL3). Cell, 115, 461-473.
http://dx.doi.org/10.1016/S0092-8674(03)00855-9 - 29. Yamamoto, K., Ishii, E., Sako, M., Ohga, S., Furuno, K., Suzuki, N., Ueda, I., Imayoshi, M., Yamamoto, S., Morimoto, A., Takada, H., Hara, T., Imashuku, S., Sasazuki, T. and Yasukawa, M. (2004) Identification of Novel MUNC13-4 Mutations in Familial Haemophagocytic Lymphohistiocytosis and Functional Analysis of MUNC13-4-Deficient Cytotoxic T Lymphocytes. Journal of Medical Genetics, 41, 763-767.
http://dx.doi.org/10.1136/jmg.2004.021121 - 30. Meeths, M., et al. (2011) Familial Hemophagocytic Lymphohistiocytosis Type 3 (FHL3) Caused by Deep Intronic Mutation and Inversion in UNC13D. Blood, 118, 5783-5793.
http://dx.doi.org/10.1182/blood-2011-07-369090 - 31. Entesarian, M., Chiang, S.C.C., Schlums, H., Meeths, M., Chan, M.Y., Mya, S.N., Soh, S.Y., Nordenskjöld, M., Henter, J.I. and Bryceson, Y.T. (2013) Novel Deep Intronic and Missense UNC13D Mutations in Familial Haemophagocytic Lymphohistiocytosis Type 3. British Journal of Haematology, 162, 415-418.
http://dx.doi.org/10.1111/bjh.12371 - 32. Shirakawa, R., Higashi, T., Tabuchi, A., Yoshioka, A., Nishioka, H., Fukuda, M., Kita, T. and Horiuchi, H. (2004) Munc13-4 Is a GTP-Rab27-Binding Protein Regulating Dense Core Granule Secretion in Platelets. Journal of Biological Chemistry, 279, 10730-10737.
http://dx.doi.org/10.1074/jbc.M309426200 - 33. zur Stadt, U., Schmidt, S., Kasper, B., Beutel, K., Diler, A.S., Henter, J.I., Kabisch, H., Schneppenheim, R., Nürnberg, P., Janka, G. and Hennies, H.C. (2005) Linkage of Familial Hemophagocytic Lymphohistiocytosis (FHL) Type-4 to Chromosome 6q24 and Identification of Mutations in Syntaxin 11. Human Molecular Genetics, 14, 827-834.
http://dx.doi.org/10.1093/hmg/ddi076 - 34. Teng, F.Y., Wang, Y. and Tang, B.L. (2001) The Syntaxins. Genome Biology, 2, Reviews 3012.1-Reviews 3012.7.
- 35. Tang, B.L., Low, D.Y. and Hong, W. (1998) Syntaxin 11: A Member of the Syntaxin Family without a Carboxyl Terminal Transmembrane Domain. Biochemical and Biophysical Research Communications, 245, 627-632.
http://dx.doi.org/10.1006/bbrc.1998.8490 - 36. Prekeris, R., Klumperman, J. and Scheller, R.H. (2000) Syntaxin 11 Is an Atypical SNARE Abundant in the Immune System. European Journal of Cell Biology, 79, 771-780.
http://dx.doi.org/10.1078/0171-9335-00109 - 37. Zhang, S., Ma, D., Wang, X., Celkan, T., Nordenskjöld, M., Henter, J.I., Fadeel, B. and Zheng, C. (2008) Syntaxin-11 Is Expressed in Primary Human Monocytes/Macrophages and Acts as a Negative Regulator of Macrophage Engulfment of Apoptotic Cells and IgG-Opsonized Target Cells. British Journal of Haematology, 142, 469-479.
http://dx.doi.org/10.1111/j.1365-2141.2008.07191.x - 38. Bryceson, Y.T., Rudd, E., Zheng, C., Edner, J., Ma, D., Wood, S.M., Bechensteen, A.G., Boelens, J.J., Celkan, T., Farah, R.A., Hultenby, K., Winiarski, J., Roche, P.A., Nordenskjöld, M., Henter, J.I., Long, E.O. and Ljunggren, H.G. (2007) Defective Cytotoxic Lymphocyte Degranulation in Syntaxin-11 Deficient Familial Hemophagocytic Lymphohistiocytosis 4 (FHL4) Patients. Blood, 110, 1906-1915.
http://dx.doi.org/10.1182/blood-2007-02-074468 - 39. Arneson, L.N., Brickshawana, A., Segovis, C.M., Schoon, R.A., Dick, C.J. and Leibson, P.J. (2007) Cutting Edge: Syntaxin 11 Regulates Lymphocyte-Mediated Secretion and Cytotoxicity. Journal of Immunology, 179, 3397-3401.
http://dx.doi.org/10.4049/jimmunol.179.6.3397 - 40. Müller, M.L., Chiang, S.C.C., Meeths, M., Tesi, B., Entesarian, M., Nilsson, D., Wood, S.M., Nordenskjöld, M., Henter, J.I., Naqvi, A. and Bryceson, Y.T. (2014) An N-Terminal Missense Mutation in STX11 Causative of FHL4 Abrogates Syntaxin-11 Binding to Munc18-2. Frontiers in Immunology, 4, 515.
http://dx.doi.org/10.3389/fimmu.2013.00515 - 41. zur Stadt, U., Rohr, J., Seifert, W., Koch, F., Grieve, S., Pagel, J., Strauss, J., Kasper, B., Nürnberg, G., Becker, C., Maul-Pavicic, A., Beutel, K., Janka, G., Griffiths, G., Ehl, S. and Hennies, H.C. (2009) Familial Hemophagocytic Lymphohistiocytosis Type 5 (FHL-5) Is Caused by Mutations in Munc18-2 and Impaired Binding to Syntaxin 11. American Journal of Human Genetics, 85, 482-492.
http://dx.doi.org/10.1016/j.ajhg.2009.09.005 - 42. Côte, M., Ménager, M.M., Burgess, A., Mahlaoui, N., Picard, C., Schaffner, C., Al-Manjomi, F., Al-Harbi, M., Alangari, A., Le Deist, F., Gennery, A.R., Prince, N., Cariou, A., Nitschke, P., Blank, U., El-Ghazali, G., Ménasché, G., Latour, S., Fischer, A. and de Saint Basile, G. (2009) Munc18-2 Deficiency Causes Familial Hemophagocytic Lymphohistiocytosis Type 5 and Impairs Cytotoxic Granule Exocytosis in Patient NK Cells. Journal of Clinical Investigation, 119, 3765-3773.
http://dx.doi.org/10.1172/JCI40732 - 43. Toonen, R.F.G. and Verhage, M. (2003) Vesicle Trafficking: Pleasure and Pain from SM Genes. Trends in Cell Biology, 13, 177-186.
http://dx.doi.org/10.1016/S0962-8924(03)00031-X - 44. Bonifacino, J.S. and Glick, B.S. (2004) The Mechanisms of Vesicle Budding and Fusion. Cell, 116, 153-166.
http://dx.doi.org/10.1016/S0092-8674(03)01079-1 - 45. Halimani, M., Pattu, V., Marshall, M.R., Chang, H.F., Matti, U., Jung, M., Becherer, U., Krause, E., Hoth, M., Schwarz, E.C. and Rettig, J. (2014) Syntaxin11 Serves as a t-SNARE for the Fusion of Lytic Granules in Human Cytotoxic T Lymphocytes. European Journal of Immunology, 44, 573-584.
http://dx.doi.org/10.1002/eji.201344011 - 46. Ji, X., Chang, B., Naggert, J.K. and Nishina, P.M. (2016) Lysosomal Trafficking Regulator (LYST). Advances in Experimental Medicine and Biology, 854, 745-750.
http://dx.doi.org/10.1007/978-3-319-17121-0_99 - 47. Ménasché, G., Pastural, E., Feldmann, J., Certain, S., Ersoy, F., Dupuis, S., Wulffraat, N., Bianchi, D., Fischer, A., Le Deist, F. and de Saint Basile, G. (2000) Mutations in RAB27A Cause Griscelli Syndrome Associated with Haemophagocytic Syndrome. Nature Genetics, 25, 173-176.
http://dx.doi.org/10.1038/76024 - 48. Neeft, M., Wieffer, M., de Jong, A.S., Negroiu, G., Metz, C.H.G., van Loon, A., Griffith, J., Krijgsveld, J., Wulffraat, N., Koch, H., Heck, A.J.R., Brose, N., Kleijmeer, M. and van der Sluijs, P. (2005) Munc13-4 Is an Effector of rab27a and Controls Secretion of Lysosomes in Hematopoietic Cells. Molecular Biology of the Cell, 16, 731-741.
http://dx.doi.org/10.1091/mbc.E04-10-0923 - 49. Haddad, E.K., Wu, X., Hammer, J.A. and Henkart, P.A. (2001) Defective Granule Exocytosis in Rab27a-Deficient Lymphocytes from Ashen Mice. Journal of Cell Biology, 152, 835-842.
http://dx.doi.org/10.1083/jcb.152.4.835 - 50. Hume, A.N. and Seabra, M.C. (2011) Melanosomes on the Move: A Model to Understand Organelle Dynamics. Biochemical Society Transactions, 39, 1191-1196.
http://dx.doi.org/10.1042/BST0391191 - 51. Booth, A.E.G., Seabra, M.C. and Hume, A.N. (2012) Rab27a and Melanosomes: A Model to Investigate the Membrane Targeting of Rabs. Biochemical Society Transactions, 40, 1383-1388.
http://dx.doi.org/10.1042/BST20120200 - 52. Kimura, T., Kaneko, Y., Yamada, S., Ishihara, H., Senda, T., Iwamatsu, A. and Niki, I. (2008) The GDP-Dependent Rab27a Effector Coronin 3 Controls Endocytosis of Secretory Membrane in Insulin-Secreting Cell Lines. Journal of Cell Science, 121, 3092-3098.
http://dx.doi.org/10.1242/jcs.030544 - 53. Kurowska, M., Goudin, N., Nehme, N.T., Court, M., Garin, J., Fischer, A., de Saint Basile, G. and Ménasché, G. (2012) Terminal Transport of Lytic Granules to the Immune Synapse Is Mediated by the Kinesin-1/Slp3/Rab27a Complex. Blood, 119, 3879-3889.
http://dx.doi.org/10.1182/blood-2011-09-382556 - 54. Fukuda, M. (2002) The C2A Domain of Synaptotagmin-Like Protein 3 (Slp3) Is an Atypical Calcium-Dependent Phospholipid-Binding Machine: Comparison with the C2A Domain of Synaptotagmin I. Biochemical Journal, 366, 681-687.
http://dx.doi.org/10.1042/bj20020484 - 55. Matti, U., Pattu, V., Halimani, M., Schirra, C., Krause, E., Liu, Y., Weins, L., Chang, H.F., Guzman, R., Olausson, J., Freichel, M., Schmitz, F., Pasche, M., Becherer, U., Bruns, D. and Rettig, J. (2013) Synaptobrevin2 Is the v-SNARE Required for Cytotoxic T-Lymphocyte Lytic Granule Fusion. Nature Communications, 4, 1439.
http://dx.doi.org/10.1038/ncomms2467 - 56. Loo, L.S., Hwang, L.A., Ong, Y.M., Tay, H.S., Wang, C.C. and Hong, W. (2009) A Role for Endobrevin/VAMP8 in CTL Lytic Granule Exocytosis. European Journal of Immunology, 39, 3520-3528.
http://dx.doi.org/10.1002/eji.200939378 - 57. Valdez, A.C., Cabaniols, J.P., Brown, M.J. and Roche, P.A. (1999) Syntaxin 11 Is Associated with SNAP-23 on Late Endosomes and the Trans-Golgi Network. Journal of Cell Science, 112, 845-854.
- 58. Mollinedo, F., Calafat, J., Janssen, H., Martín-Martín, B., Canchado, J., Nabokina, S.M. and Gajate, C. (2006) Combinatorial SNARE Complexes Modulate the Secretion of Cytoplasmic Granules in Human Neutrophils. Journal of Immunology, 177, 2831-2841.
http://dx.doi.org/10.4049/jimmunol.177.5.2831 - 59. Martín-Martín, B., Nabokina, S.M., Blasi, J., Lazo, P.A. and Mollinedo, F. (2000) Involvement of SNAP-23 and Syntaxin 6 in Human Neutrophil Exocytosis. Blood, 96, 2574-2583.
- 60. Naegelen, I., Plançon, S., Nicot, N., Kaoma, T., Muller, A., Vallar, L., Tschirhart, E.J. and Bréchard, S. (2015) An Essential Role of Syntaxin 3 Protein for Granule Exocytosis and Secretion of IL-1α, IL-1β, IL-12b, and CCL4 from Differentiated HL-60 Cells. Journal of Leukocyte Biology, 97, 557-571.
http://dx.doi.org/10.1189/jlb.3A0514-254RR - 61. Hackmann, Y., Graham, S.C., Ehl, S., Höning, S., Lehmberg, K., Aricò, M., Owen, D.J. and Griffiths, G.M. (2013) Syntaxin Binding Mechanism and Disease-Causing Mutations in Munc18-2. Proceedings of the National Academy of Sciences of the United States of America, 110, E4482-E4491.
http://dx.doi.org/10.1073/pnas.1313474110 - 62. Marshall, M.R., Pattu, V., Halimani, M., Maier-Peuschel, M., Müller, M.L., Becherer, U., Hong, W., Hoth, M., Tschernig, T., Bryceson, Y.T. and Rettig, J. (2015) VAMP8-Dependent Fusion of Recycling Endosomes with the Plasma Membrane Facilitates T Lymphocyte Cytotoxicity. Journal of Cell Biology, In Press.
- 63. Jenkins, M.R., Rudd-Schmidt, J.A., Lopez, J.A., Ramsbottom, K.M., Mannering, S.I., Andrews, D.M., Voskoboinik, I. and Trapani, J.A. (2015) Failed CTL/NK Cell Killing and Cytokine Hypersecretion Are Directly Linked through Prolonged Synapse Time. Journal of Experimental Medicine, 212, 307-317.
http://dx.doi.org/10.1084/jem.20140964 - 64. Trapani, J.A., Voskoboinik, I. and Jenkins, M.R. (2015) Perforin-Dependent Cytotoxicity: “Kiss of Death” or Prolonged Embrace with Darker Elocation-Idnseque11es? Oncoimmunology, 4, Article ID: e1036215.
http://dx.doi.org/10.1080/2162402X.2015.1036215 - 65. Jessen, B., Kögl, T., Sepulveda, F.E., de Saint Basile, G., Aichele, P. and Ehl, S. (2013) Graded Defects in Cytotoxicity Determine Severity of Hemophagocytic Lymphohistiocytosis in Humans and Mice. Frontiers in Immunology, 4, 448.
http://dx.doi.org/10.3389/fimmu.2013.00448 - 66. Sepulveda, F.E., Debeurme, F., Ménasché, G., Kurowska, M., Côte, M., Pachlopnik Schmid, J., Fischer, A. and de Saint Basile, G. (2013) Distinct Severity of HLH in Both Human and Murine Mutants with Complete Loss of Cytotoxic Effector PRF1, RAB27A, and STX11. Blood, 121, 595-603.
http://dx.doi.org/10.1182/blood-2012-07-440339 - 67. Kögl, T., Müller, J., Jessen, B., Schmitt-Graeff, A., Janka, G., Ehl, S., zur Stadt, U. and Aichele, P. (2013) Hemophagocytic Lymphohistiocytosis in Syntaxin-11-Deficient Mice: T-Cell Exhaustion Limits Fatal Disease. Blood, 121, 604- 613.
http://dx.doi.org/10.1182/blood-2012-07-441139 - 68. Zhang, K., Jordan, M.B., Marsh, R.A., Johnson, J.A., Kissell, D., Meller, J., Villanueva, J., Risma, K.A., Wei, Q., Klein, P.S. and Filipovich, A.H. (2011) Hypomorphic Mutations in PRF1, MUNC13-4, and STXBP2 Are Associated with Adult-Onset Familial HLH. Blood, 118, 5794-5798.
http://dx.doi.org/10.1182/blood-2011-07-370148 - 69. Meeths, M., Chiang, S.C.C., Löfstedt, A., Müller, M.L., Tesi, B., Henter, J.I. and Bryceson, Y.T. (2014) Pathophysiology and Spectrum of Diseases Caused by Defects in Lymphocyte Cytotoxicity. Experimental Cell Research, 325, 10-17.
http://dx.doi.org/10.1016/j.yexcr.2014.03.014 - 70. Clementi, R., Locatelli, F., Dupré, L., Garaventa, A., Emmi, L., Bregni, M., Cefalo, G., Moretta, A., Danesino, C., Comis, M., Pession, A., Ramenghi, U., Maccario, R., Aricò, M. and Roncarolo, M.G. (2005) A Proportion of Patients with Lymphoma May Harbor Mutations of the Perforin Gene. Blood, 105, 4424-4428.
http://dx.doi.org/10.1182/blood-2004-04-1477 - 71. Machaczka, M., Klimkowska, M., Chiang, S.C.C., Meeths, M., Müller, M.L., Gustafsson, B., Henter, J.I. and Bryceson, Y.T. (2013) Development of Classical Hodgkin’s Lymphoma in an Adult with Biallelic STXBP2 Mutations. Haematologica, 98, 760-764.
http://dx.doi.org/10.3324/haematol.2012.073098 - 72. Ravelli, A., Grom, A.A., Behrens, E.M. and Cron, R.Q. (2012) Macrophage Activation Syndrome as Part of Systemic Juvenile Idiopathic Arthritis: Diagnosis, Genetics, Pathophysiology and Treatment. Genes & Immunity, 13, 289-298.
http://dx.doi.org/10.1038/gene.2012.3 - 73. Deiva, K., Mahlaoui, N., Beaudonnet, F., de Saint Basile, G., Caridade, G., Moshous, D., Mikaeloff, Y., Blanche, S., Fischer, A. and Tardieu, M. (2012) CNS Involvement at the Onset of Primary Hemophagocytic Lymphohistiocytosis. Neurology, 78, 1150-1156.
http://dx.doi.org/10.1212/WNL.0b013e31824f800a