Computational Molecular Bioscience
Vol.07 No.02(2017), Article ID:77301,13 pages
10.4236/cmb.2017.72002
Quantitative Structure Anti-Cancer Activity Relationship (QSAR) of a Series of Ruthenium Complex Azopyridine by the Density Functional Theory (DFT) Method
Kouakou Nobel N’guessan, Mamadou Guy-Richard Koné, Kafoumba Bamba*, Ouattara Wawohinlin Patrice, Nahossé Ziao
Laboratoire de Thermodynamique et de Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, Abidjan, Côte-d’Ivoire

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: May 20, 2017; Accepted: June 26, 2017; Published: June 29, 2017
ABSTRACT
A series of ruthenium azopyridine complexes have recently been investigated due to their potential cytotoxic activities against renal cancer (A498), lung cancer (H226), ovarian cancer (IGROV), breast cancer (MCF-7) and colon cancer (WIDR). Thus, in order to predict the cytotoxic potentials of these compounds, quantitative structure-activity relationship studies were carried out using the methods of quantum chemistry. Five Quantitative Structure Activity Relationship (QSAR) models were obtained from the determined quantum descriptors and the different activities. The models present the following statistical indicators: regression correlation coefficient R2 = 0.986 - 0.905, standard deviation S = 0.516 - 0.153, Fischer test F = 106.718 - 14.220, correlation coefficient of cross-validation 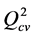 = 0.985- 0.895 and
= 0.985- 0.895 and 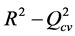 = 0.010 - 0.001. The statistical characteristics of the established QSAR models satisfy the acceptance and external validation criteria, thereby accrediting their good performance. The models developed show that the variation of the free enthalpy of reaction ΔG˚, the dipole moment μ and the charge of the ligand in the complex Ql, are the explanatory and predictive quantum descriptors correlated with the values of the anti-cancer activity of the studied complexes. Moreover, the charge of the ligand is the priority descriptor for the prediction of the cytotoxicity of the compounds studied. Furthermore, QSAR models developed are statistically significant and predictive, and could be used for the design and synthesis of new anti-cancer molecules.
= 0.010 - 0.001. The statistical characteristics of the established QSAR models satisfy the acceptance and external validation criteria, thereby accrediting their good performance. The models developed show that the variation of the free enthalpy of reaction ΔG˚, the dipole moment μ and the charge of the ligand in the complex Ql, are the explanatory and predictive quantum descriptors correlated with the values of the anti-cancer activity of the studied complexes. Moreover, the charge of the ligand is the priority descriptor for the prediction of the cytotoxicity of the compounds studied. Furthermore, QSAR models developed are statistically significant and predictive, and could be used for the design and synthesis of new anti-cancer molecules.
Keywords:
Ruthenium Azopyridine Complex, Anti-Cancer, QSAR, DFT Method

1. Introduction
The satisfaction obtained in the use of cisplatin for the treatment of tumours stimulated research on transition metal complexes. Many organometallic compounds have been synthesized and tested [1] [2] . The results obtained were promising for different metals. Especially, only ruthenium seems to be an alternative to platinum since its compounds are found to be more active and less toxic than those of platinum [3] . Thus, interest in the anticancer activity of ruthenium complexes has increased. The biological activities of ruthenium complexes are strongly influenced by the ligand structure. Indeed, several studies have shown that changing the aromatic ligand or the conformation of the complexes can have a significant influence on their anticancer activities [4] . The azopyridine ligand shown in Figure 1 and Table 1 are actually formed from the combination of a pyridine ring with an azo group. These bidentate ligands can bind to Ru ion through RuCl3, 3H2O reactive by only the lone electron pairs of the nitrogen atoms of the pyridine ring and the azo group, thereby forming a 5-membered stable ring of chelation. Thus, this reaction provides metal with excellent stability. In addition, the complexation of ruthenium (Reaction 1) by the asymmetric bidentate ligands leads to five isomers named α-Cl, β-Cl, γ-Cl, δ-Cl and ε-Cl [5] . The difference between them comes mainly from the position of both chlorine atoms (Cis or Trans configuration) and both azopyridine ligands as shown in Figure 2.
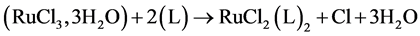 (Reaction 1)
(Reaction 1)
The recent discovery of anticancer activity azopyridine complex ruthenium [6] [7] has increased interest of researchers to find out the origin of their cytotoxicity and mechanism of their reactivity on cancer cells so as to enhance it. Thus, α-, β-, γ- [RuCl2L2] where L stands for 2-phenylazopyridine, o-tolylazopyridine and 4-methyl-2-phenylazopyridine isomers were synthesized and tested in a series of cells line of renal cancer (A498), lung cancer (H226), ovarian cancer

Figure 1. Skeleton of the azopyridine ligands with different substituents R and R1. The bidentate state of the ligand consists of the ligand binding to ruthenium or central metal by Npy and N2.

Table 1. Various ligands and substituents.

Figure 2. The five isomers of RuCl2L2 complexes. L stands for all azopyridine ligands. The arc represents azopyridine ligands highlighting their bidentate state. All these isomers have the C2 symmetry except for the β-Cl isomer. t and c represent respectively trans and cis geometries. Therefore, the three letters codes show the geometry in order of the chlorides (Cl), the pyridine ring (Npy) and the azo nitrogen N2.
(IGROV), breast cancer (MCF-7) and colon cancer (WIDR) [8] . These molecules have shown promising anticancer activity. Besides, it is admitted that two modes of binding of the complex to the cell lines were up today supposed to characterize the process: While the first trend indicates that the binding was due to the hydrolysis of both chloride atoms, thereby allowing covalent bonding between ruthenium ion and the DNA, the second indicates however that the bonding is performed between the azopyridine ligand and the DNA base-pairs of the cell lines through a π-π stacking interaction [9] . Here, the improvement of the cytotoxicity of the azopyirdine complexes requires one to know of the physicochemical properties that govern it. This would help to efficiently orient the synthesis of the ruthenium azopyridine complexes.
The Quantitative Structure Activity Relationship (QSAR) study is one of the most widely used methods to design new therapeutic agents [10] [11] [12] . It allows quantitative correlation with a mathematical model of the structure or properties of the compounds with their biological activities. It is increasingly used to reduce the excessive number of experiments, sometimes long, expensive and harmful for environment protection [13] [14] . In this work, the objective is to carry out a descriptive and predictive study of the anticancer activity of a series of nine (9) isomers of ruthenium complexes. Using the methods of quantum chemistry, this work aims to modelize the observed anticancer activities. The molecular descriptors have been calculated only from the molecular structure of the compounds and predicting the anticancer activities of analogous molecules.
2. Experimental Section
2.1. Material and Method
The six molecules of the training set and the three other external validation set molecules used in this study have IC50 ranging from 0.045 to 74 μM. Here, the term IC50 means the median concentration of molecules determined experimentally to inhibit 50% of cancer cells in a population of cancer cells. This range of concentrations makes it possible to define a quantitative relationship between the anticancer activity and the theoretical descriptors. According to Aldrik et al. the experimentation of the cytotoxicity of the aforementioned human tumor cell lines was made in vitro using the microculture sulforhodamine B test (SRB) for the estimation of the cell viability [15] . Biological data are generally expressed as the opposite of the log 10 base of activity 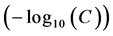 in order to obtain higher mathematical values when the structures are biologically very efficient [16] [17] . The anticancer activity is expressed by the anticancer potential pIC50 that is defined in Equation (1):
in order to obtain higher mathematical values when the structures are biologically very efficient [16] [17] . The anticancer activity is expressed by the anticancer potential pIC50 that is defined in Equation (1):
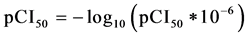 (1)
(1)
2.1.1. Calculation Level
DFT methods are generally known to generate a variety of molecular properties [18] [19] in QSAR studies that increase predictability, reduce computation time and the cost of the design of new drugs [20] [21] . Thus, all calculations were performed with the DFT methods using Becke’s three-parameter hybrid functional B3LYP [22] and the double-zeta pseudo-potential LanL2DZ [23] . All geometry optimizations of the molecules were carried out beforehand in order to obtain the structure in its ground state. Then this stable configuration was confirmed by the frequency analysis which has to reveal the absence of imaginary frequency. Furthermore, the analysis of the natural orbital population NPA was carried out at the same theoretical level. All these calculations were carried out using the software Gaussian 03 [24] . The modelling was done using the multi-linear regression method implemented in Excel [25] and XLSTAT spreadsheets [26] .
2.1.2. Quantum Descriptors Used
For the development of QSAR models, some theoretical descriptors related to the conceptual DFT were determined. In particular, the variation of the formation free enthalpy ΔG˚, the natural ligand charge in the complex QL and the dipole moment μ. These descriptors are all determined from the optimized molecules. Here, the variation of free enthalpy of the reaction indicates the spontaneity of the reaction. It was calculated according to Equation (2).
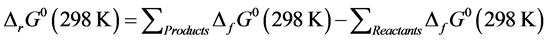 (2)
(2)
The ligand charge, which corresponds to the sum of the ligand’s natural atomic charges within the complex obtained by the NPA calculation, reflects the electrophilic or nucleophilic character of this entity. The dipole moment (μ) indicates the stability of a molecule in water. Thus, a high dipole moment will result in poor solubility in organic solvents and high solubility in water. Moreover, the interdependence of descriptors is evaluated by a linear correlation coefficient R between the pairs of the set of descriptors. Here, two descriptors are said to be independent when R < 0.95 [27] [28] .
2.1.3. Estimation of the Predictive Ability of a QSAR Model
A QSAR model is developed on the basis of statistical indicators. The quality of a model is determined on the basis of these various analysis statistical indicators, including the correlation coefficient R2, the standard deviation S, the correlation coefficients of cross validation 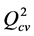 and Fischer F. R2, S and F relate to the adjustment of the calculated and experimental values: they describe the predictive capacity within the limits of the model and allow to estimate the precision of the values calculated on the learning set [29] [30] . As for the correlation coefficient of the cross-validation
and Fischer F. R2, S and F relate to the adjustment of the calculated and experimental values: they describe the predictive capacity within the limits of the model and allow to estimate the precision of the values calculated on the learning set [29] [30] . As for the correlation coefficient of the cross-validation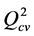 , it gives information on the predictive power of the model. This predictive power is called “internal” because it is calculated from the structures used to build this model.
, it gives information on the predictive power of the model. This predictive power is called “internal” because it is calculated from the structures used to build this model.
The squared correlation coefficient R² gives an evaluation of the dispersion of theoretical values around the experimental data. The quality of the modelling is improved when the points are close to the fitting line [31] . The adjustment of the points to this line can be evaluated by the correlation coefficient. The correlation coefficient R2 was given by the following Equation (3):
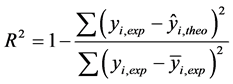 (3)
(3)
where:
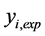 : The experimental value of the anticancer activity;
: The experimental value of the anticancer activity;
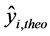 : The theoretical value of the anticancer activity;
: The theoretical value of the anticancer activity;
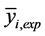 : The average value of the experimental values of the anticancer activity.
: The average value of the experimental values of the anticancer activity.
More the R² value will be closer to 1 more the theoretical and experimental values will be assumed to correlate. In addition, the variance 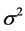 was determined by the relationship (4):
was determined by the relationship (4):
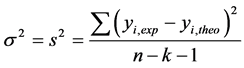 (4)
(4)
where k is the number of independent descriptors, n is the number of molecules of the training set and 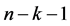 corresponds to the degree of freedom. The root mean square error s is another statistical indicator used. It allows evaluating the reliability and accuracy of a model. It is obtained from Equation (5):
corresponds to the degree of freedom. The root mean square error s is another statistical indicator used. It allows evaluating the reliability and accuracy of a model. It is obtained from Equation (5):
 (5)
(5)
The Fisher test F was also used to measure the level of statistical significance of the model, i.e. quality of the choice of descriptors constituting the model. The Fisher test F is defined from Equation (6):
 (6)
(6)
The correlation coefficient of cross-validation  to assess the accuracy of the prediction on the training set was calculated by using the following relationship:
to assess the accuracy of the prediction on the training set was calculated by using the following relationship:
 (7)
(7)
The performance of a mathematical model, for Eriksson et al. [32] , was characterized by a value of  for a satisfactory model and for an excellent model when
for a satisfactory model and for an excellent model when . According to them, for a given training set, a model will be performant if the acceptance criterion
. According to them, for a given training set, a model will be performant if the acceptance criterion  is respected.
is respected.
Moreover, the prediction power of a model can be obtained from five Tropsha’s criteria [12] [33] [34] . If at least three of the criteria were satisfied, then the model will be considered efficient in predicting the activity studied. These criteria are the following:
1) ,
,
2) ,
,
3) ,
,
4)  and
and ,
,
5)  and
and .
.
3. Results and Discussion
In this QSAR study, the training set consisting of six molecules and the three other ruthenium azopyridine complexes forming the validation set are presented in Table 2. Also, the values of the descriptor’s bivariate linear correlation coefficients R are presented in Table 3.
The calculated linear correlation coefficients R of the series of descriptors are less than 0.95 (R < 0.95). This demonstrates the non-dependence of the descriptors used to develop the models.
3.1. QSAR Model and Contribution of Descriptors
The best QSAR models obtained for the various anti-cancer activities as well as
Table 2. Quantum descriptors and experimental anti-cancer activities of the training set and the validation set. ΔG˚, Ql and µ are expressed respectively in kcal, a.u. and Debye.
a1α, 1β and 1γ correspond respectively to α-, β- and γ-RuCl2(2-phenylazopyridine)2; b2α, 2β and 2γ correspond respectively to α-, β- and γ-RuCl2(o-tolylazopyridine)2; c3α, 3β and 3γ correspond respectively to α-, β- and γ-RuCl2(4-methyl-2-phenylazopyridine)2.
Table 3. Values of the descriptor’s bivariate linear correlation coefficients.
Table 4. The most significant QSAR models for the modelling of cytotoxic activities on A498, H226, GROV, MCF-7 and WIDR cancer cells.
the statistical indicators are given in Table 4. It should be emphasized that these models were established using the same test and validation sets in Table 2.
It should be noted that the negative or positive sign of the model descriptor’s coefficient reflects the proportionality effect between the biological activity evolution of interest and this parameter of the regression equation. Thus, the negative sign indicates that when the value of the descriptor is high, the biological activity decreases while the positive sign translates the opposite effect.
The negative sign of the coefficient of the free enthalpy variation or the dipole moment indicates that the cytotoxic activity will be improved for a low value of the free enthalpy variation or the dipole moment. On the other side, the positive sign of the coefficient of the ligand charge means that the cytotoxic activity will be improved for a high value of the ligand charge. Also, the significance of the models is reflected by the Fisher coefficient F which is between 14.22 and 106.718 and the cross-validation correlation coefficient  which goes from 0.895 to 0.985. These different models are acceptable because all the values of
which goes from 0.895 to 0.985. These different models are acceptable because all the values of  are less than 0.3. External validation of these models was performed with the 1α, 1β and 1γ isomers. The different regression lines between the experimental and theoretical cytotoxic activities of the training set (blue dots) and the validation set (red dots) for each cancer cell are illustrated in Figure 3.
are less than 0.3. External validation of these models was performed with the 1α, 1β and 1γ isomers. The different regression lines between the experimental and theoretical cytotoxic activities of the training set (blue dots) and the validation set (red dots) for each cancer cell are illustrated in Figure 3.

Figure 3. Regression lines of the different models.
Table 5. Tropsha Criteria for different models.



 et
et ;
;  and
and .
.
3.2. Verification of Tropsha Criteria
The Tropsha criteria for the different models are presented in Table 5.
All values respect the Tropsha criteria, so these models are acceptable for predicting the ruthenium azopyridine complexes cytotoxic activity.
The study of the relative descriptors contribution in the prediction of the compounds cytotoxicity was carried out for each type of cancer cells. The different contributions are shown in Figure 4.
The charge of the ligand has a large contribution than the free enthalpy variation or the dipole moment. Thus, the charge of the ligand is revealed to be the priority descriptor in the prediction of the cytotoxic activity of the ruthenium azopyridine complexes studied.
4. Conclusion
In this work, the cytotoxic activities of six ruthenium azopyridine complexes on cancer cells that comprise renal cancer (A498), lung cancer (H226), ovarian cancer (IGROV), breast cancer (MCF-7) and colon cancer (WIDR) were correlated with the theoretical descriptors calculated by the DFT methods. The cytotoxic activities of three other ruthenium azopyridine complexes were selected to form the external validation sets for the calculated models. Multiple Linear Regression (MLR) was used to quantify the relationships between molecular descriptors and the properties of the azopyridine derivatives cytotoxic activity. A strong correlation was observed between the experimental values and the predicted values of the cytotoxic activity, indicating the validity and quality of the QSAR models obtained. The quantum descriptors of the optimized molecules, the free enthalpy variation of reaction, the dipole moment and the ligand charge, made it possible to predict the cytotoxicity of the ruthenium azopyridine complexes studied on cancer cells. Among all these descriptors the ligand charge is the descriptor which influences the cytotoxicity activ-

Figure 4. Contribution of different descriptors in different models.
ity. The QSAR models present a robustness, with good internal and external predictive capabilities.
Cite this paper
N’guessan, K.N., Guy-Richard Koné, M., Bamba, K., Patrice, O.W. and Ziao, N. (2017) Quantitative Structure Anti-Cancer Activity Relationship (QSAR) of a Series of Ruthenium Complex Azopyridine by the Density Functional Theory (DFT) Method. Computational Molecular Bioscience, 7, 19-31. https://doi.org/10.4236/cmb.2017.72002
References
- 1. Erck, A., Rainen, L., Whileyma, J., Chang, I.M., Kimball, A.P. and Bear, J. (1974) Studies of Rhodium (II) Carboxylates as Potential Antitumor Agents. Proceedings of the Society for Experimental Biology and Medicine, 145, 1278-1283.
https://doi.org/10.3181/00379727-145-37996 - 2. Higgins, J.D., Neely, L. and Fricker, S. (1993) Synthesis and Cytotoxicity of Some Cyclometallated Palladium Complexes. Journal of Inorganic Biochemistry, 49, 149.
- 3. Allardyce, C.S. and Dyson, P.J. (2001) Ruthenium in Medicine: Current Clinical Uses and Future Prospects. Platinum Metals Review, 45, 62-69.
- 4. Morris, R.E., Aird, R.E., Del Socorro Murdoch, P., Chen, H., Cummings, J., Hughes, N.D., Parsons, S., Parkin, A., Boyd, G., Jodrell, D.I. and Sadler, P.J. (2001) Inhibition of Cancer Cell Growth by Ruthenium (II) Arene Complexes. Journal of Medicinal Chemistry, 44, 3616-3621.
- 5. Velders, A.H., Van der Schilden, K., Hoste, A.C.G., Reedijk, J., Kooijman, H. and Speck, A.L. (2004) Dichlorobis (2-Phenylazopyridine) Ruthenium (II) Complexes: Characterisation, Spectroscopic and Structural Properties of Four Isomers. Dalton Transactions, 448-455.
https://doi.org/10.1039/B313182C - 6. Hotze, A.C.G., Velders, A.H., Ugozzoli, F., Biagini-Cingi, M., Manotti-Lanfredi, A.M., Haasnoot, J.G. and Reedijk, J. (2000) Synthesis, Characterization, and Crystal Structure of α-[Ru(azpy)2(NO3)2] (azpy = 2-(Phenylazo) Pyridine) and the Products of Its Reactions with Guanine Derivatives. Inorganic Chemistry, 39, 3838-3844.
https://doi.org/10.1021/ic000066v - 7. Velders, A.H., Kooijman, H., Spek, A.L., Haasnoot, J.G., De Vos, D. and Reedijk, J. (2000) Strong Differences in the in Vitro Cytotoxicity of Three Isomeric Dichlorobis (2-Phenylazopyridine) Ruthenium(II) Complexes. Inorganic Chemistry, 39, 2966-2967.
https://doi.org/10.1021/ic000167t - 8. Hotze, A.C.G., Caspers, S.E., De Vos, D., Kooijman, H., Spek, A.L., Flamigni, A., Bacac, M., Sava, G., Haasnoot, J.G. and Reedijk, J. (2004) Structure-Dependent in Vitro Cytotoxicity of the Isomeric Complexes [Ru(L)2Cl2] (L= O-Tolylazopyridine and 4-Methyl-2-phenylazopyridine) in Comparison to [Ru(azpy)2Cl2]. Journal of Biological Inorganic Chemistry, 9, 354-364.
https://doi.org/10.1007/s00775-004-0531-6 - 9. Bamba, K., Ouattara, W.P., N’Guessan, K.N. and Ziao, N. (2016) SARs Investigation of α-, β-, γ-, δ-, ε-RuCl2(Azpy)2 Complexes as Antitumor Drugs. Computational Chemistry, 4, 1-10.
https://doi.org/10.4236/cc.2016.41001 - 10. Chhabria, M.T., Mahajan, B.M. and Brahmkshatriya, P.S. (2011) QSAR Study of a Series of Acyl Coenzyme A (CoA): Cholesterol Acyltransferase Inhibitors Using Genetic Function Approximation. Medicinal Chemistry Research, 20, 1573-1580.
https://doi.org/10.1007/s00044-010-9413-3 - 11. Buha, V.M., Rana, D.N., Chhabria, M.T., Chikhalia, K.H., Mahajan, B.M., Brahmkshatriya, P.S. and Shah, N.K. (2013) Syn-thesis, Biological Evaluation and QSAR Study of a Series of Substituted Quinazolines as Antimicrobial Agents. Medicinal Chemistry Research, 22, 4096-4109.
https://doi.org/10.1007/s00044-012-0408-0 - 12. Tropsha, A. (2010) Best Practices for QSAR Model Development, Validation, and Exploitation. Molecular Informatics, 29, 476-488.
https://doi.org/10.1002/minf.201000061 - 13. Oprea, T.I. (2005) Chemoinformatics in Drug Discovery. Wiley-VCH Verlag, Allemagne.
- 14. Rekka, E.A. and Kourounakis, P.N. (2008) Chemistry and Molecular Aspects of Drug Design and Action. Taylor & Francis Group, Etats Unies.
- 15. Aldrik, H., Velders, Kooijman, H., Spek, A.L., Haasnoot, J.G., de Vos, D. and Reedijk, J. (2000) Strong Differences in Vitro Cytotoxicity of Three Isomeric Dichlorobis(2-phenylazopyridine)ruthenium(II) Complexes. Inorganic Chemistry, 39, 2966-2967.
https://doi.org/10.1021/ic000167t - 16. Chaltterjee, S., Hadi, A. and Price, B., (2000) Regression Analysis by Examples. Wiley VCH, New York.
- 17. Phuong, H.T.N. (2007) Synthèse et étude des relations structure/activité quantitatives (QSAR/2D) d’analogues Benzo [c] phénanthridiniques. Thèse de doctorat, Université d’Angers.
- 18. Chattaraj, P.K., Cedillo, A. and Parr, R.G. (1995) Variational Method for Determining the Fukui Function and Chemical Hardness of an Electronic System. The Journal of Physical Chemistry, 103, 7645.
https://doi.org/10.1063/1.470284 - 19. Parr, R.G., Donnelly, R.A., Levy, M. and Palke, W.E. (1978) Electronegativity: The Density Functional View Point. The Journal of Chemical Physics, 68, 3801.
https://doi.org/10.1063/1.436185 - 20. Hansch, C., Sammes, P.G. and Taylor, J.B. (1990) Computers and the Medicinal Chemist. In: Comprehensive Medicinal Chemistry, Vol. 4, Pergamon Press, Oxford, 33-58.
- 21. Franke, R. (1984) Theoretical Drug Design Methods. Elsevier, Amsterdam.
- 22. Becke, A.D. (1993) A New Mixing of Hartree-Fock and Local Density-Functional Theories. The Journal of Chemical Physics, 98, 1372-1377.
https://doi.org/10.1063/1.464304 - 23. Foresman, J.B. and Frisch, Æ. (1996) Exploring Chemistry with Electronic Structure Methods. 2nd Edition, Gaussian Inc., Pittsburgh.
- 24. Frisch, M.J., Trucks, G.W., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Scalmani, G., Barone, V., Mennucci, B., Petersson, G.A., Nakatsuji, H., Caricato, M., Li, X., Hratchian, H.P., Izmaylov, A.F., Bloino, J., Zheng, G., Sonnenberg, J.L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery, J.A., Peralta, J.E., Ogliaro, F., Bearpark, M., Heyd, J.J., Brothers, E., Kudin, K.N., Staroverov, V.N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J.C., Iyengar, S.S., Tomasi, J., Cossi, M., Rega, N., Millam, M.J., Klene, M., Knox, J.E., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Martin, R.L., Morokuma, K., Zakrzewski, V.G., Voth, G.A., Salvador, P., Dannenberg, J.J., Dapprich, S., Daniels, A.D., Farkas, Ö., Foresman, J.B., Ortiz, J.V., Cioslowski, J. and Fox, D.J. (2004) Gaussian 03, Revision E01. Gaussian, Inc., Wallingford.
- 25. Microsoft® Excel® 2013 (15.0.4420.1017) MSO (15.0.4420.1017) 64 Bits (2013) Partie de Microsoft Office Professionnel Plus.
- 26. XLSTAT Version 2014.5.03 Copyright Addinsoft 1995-2014 (2014) XLSTAT and Addinsoft Are Registered Trademarks of Addinsoft.
https://www.xlstat.com - 27. Katritzky, A.R., Lobanov, V.S. and Karelson, M. (1994) CODESSA Comprehensive Descriptors for Structural and Statistical Analysis, Reference Manual, version 2.0. University of Florida, Gainesville.
- 28. Dagnélie, P. (1998) Statistique Théorique et Appliquée, Tomes 1: Statistique descriptive et bases de l’inférence statistique. De Boeck and S. Larcier, Bruxelle, 508.
- 29. Snedecor, G.W. and Cochran, W.G. (1967) Statistical Methods. Oxford and IBH, New Delhi, 381.
- 30. Diudea, M.V. (2000) QSPR/QSAR Studies for Molecular Descriptors. Nova Science, Huntingdon, New York.
- 31. Esposito, E.X., Hopfinger, A.J. and Madura, J.D. (2004) Methods for Applying the Quantitative Structure-Activity Relationship Paradigm. Methods in Molecular Biology, 275, 131-213.
- 32. Eriksson, L., Jaworska, J., Worth, A., Cronin, M.T.D., Mc Dowell, R.M. and Gramatica, P. (2003) Methods for Reliability and Uncertainty Assessment and for Applicability Evaluations of Classification- and Regression-Based QSARs. Environmental Health Perspectives, 111, 1361-1375.
https://doi.org/10.1289/ehp.5758 - 33. Golbraikh, A. and Tropsha, A. (2002) Beware of QSAR. Journal of Molecular Graphics and Modelling, 20, 269.
- 34. Tropsha, A., Gramatica, P. and Gombar, V.K. (2003) The Importance of Being Earnest: Validation Is the Absolute Essential for Successful Application and Interpretation of QSPR Models. QSAR and Combinatorial Science, 22, 69-77.
https://doi.org/10.1002/qsar.200390007