Computational Molecular Bioscience
Vol.06 No.01(2016), Article ID:65284,22 pages
10.4236/cmb.2016.61001
Antibody-Like Phosphorylation Sites in Focus of Statistically Based Bilingual Approach
Jaroslav Kubrycht1*, Karel Sigler2, Pavel Souček3, Jiří Hudeček4
1Department of Physiology, Second Faculty of Medicine, Charles University, Prague, Czech Republic
2Laboratory of Cellular Biology, Institute of Microbiology, Academy of Sciences of the Czech Republic, Prague, Czech Republic
3Toxicogenomics Unit, National Institute of Public Health, Prague, Czech Republic
4Department of Biochemistry, Charles University, Prague, Czech Republic
Copyright © 2016 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 4 February 2016; accepted 28 March 2016; published 31 March 2016
ABSTRACT
In accordance with previous reports, the sequences related to phosporylated protein segments occur in conserved variable domains of immunoglobulins including first of all certain N-terminally located segments. Consequently, we look here for the sequences 1) composing human and mouse proteins different from antigen receptors, 2) identical with or highly similar to nucleotide sequence representatives of conserved variable immunoglobulin segments and 3) identical with or closely related to phosphorylation sites. More precisely, we searched for the corresponding actual pairs of DNA and protein sequence segments using five-step bilingual approach employing among others a) different types of BLAST searches, b) two in-principle-different machine-learning methods predicting phosphorylated sites and c) two large databases recording existing phosphorylation sites. The approach identified seven existing phosphorylation sites and thirty-seven related human and mouse segments achieving limits for several predictions or phylogenic parameters. Mostly serines phosporylated with ataxia-telangiectasia-related kinase (involved in regulation of DNA-double-strand-break repair) were indicated or predicted in this study. Hypermutation motifs, located in effective positions of the selected sequence segments, occurred significantly less frequently in transcribed than non-transcribed DNA strands suggesting thus the incidence of mutation events. In addition, marked differences between the numbers and proportions of human and mouse cancer-related sequence items were found in different steps of selection process. The possible role of hypermutation changes within the selected segments and the observed structural relationships are discussed here with respect to DNA damage, carcinogenesis, cancer vaccination, ageing and evolution. Taken together, our data represent additional and sometimes perhaps complementary information to the existing databases of empirically proven phosphorylation sites or pathogenically important spots.
Keywords:
Ataxia Telangiectasia-Mutated-Protein (i.e. Kinase ATM, Whose Pathogenic Mutation Is Responsible for Early Death of People), Complementarity Determining Region 1 (of Immunoglobulins, i.e. CDR1 or Hypervariable Region 1), Database (of Functional Structures), Hypermutation (i.e. Mutation of DNA Sequences Mediated by Enzymes), Immunoglobulin (i.e. Ig or Antibody), Phosphorylation (Enzyme Mediated Modification Concerns Here Mostly Protein Sequences)

1. Introduction
Protein kinase (PK) mediated phosporylation of proteins occurs specifically on phosporylated protein segments (PPS), whereas down-regulation of phosphorylated sites is specifically mediated by phosphatases. PPS can markedly influence the reactivity or interactivity of distinctly located functional sites of the same molecule or molecular complex via allosteric effects [1] - [4] . In addition, PPS represent functionally important structural elements of regulatory cascades and complex interconnected networks due to their presence in PK and phosphatases or molecules involved in triggering or modulation of the same enzymes [5] . Though the lengths of PPS mostly do not exceed ten amino acid residues (aa), these segments are specifically recognized by different PK even in cases of small sequence differences between PPS (cf. [6] [7] ).
Comparable diversified specificity of short chains is also necessary for interactions of complementary determining (hypervariable) regions (CDR) of variable immunoglobulin domains (IgV) of immunoglobulins. In accordance with this fact, it is interesting that N-terminal parts of conserved IgV (called here PPSIg) containing hypervariable region CDR1 (and also related parts of certain conserved constant immunoglobulin domains) exhibited sequence relationship to commercially accessible peptidic substrates or inhibitors of protein kinases (cf. PKSI in previous papers [8] - [10] ). The described sequence relationships were further supported by the result of successful prediction of PPS within various BLAST-accessible conserved IgV of antigenic receptors (AR). The superior prediction scores were mostly observed in two sites of these IgV. One site was located in PPSIg (the corresponding paper [11] contains also short recent summary of the corresponding investigation).
Due to their phylogeny, IgV domains of AR are adapted to effects of somatic sequence changes including hypermutation [12] [13] . In addition, these domains contain at least several types of structures or candidate structures supporting these somatic changes (cf. following paragraph). This raises the question whether at least some of the two strands of translated NS (in fact NS denotes here nucleotide sequences of the corresponding cDNA) closely similar to NS-representatives of (conserved) PPSIg but encoding PPS composing molecules distinct from AR can perform IgV-related somatic changes with functional consequences. To gradually answer to this question, newly focused bilingual approach searching for PPSIg-related NS encoding peptidic PPS was proposed. More precisely, we look here for NS 1) encoding PPS in proteins distinct from AR, 2) forming sufficiently high and dense similarities when compared with representative mRNA segments highly similar to PPSIg- related segments and 3) containing sites necessary for functionally important hypermutation.
Hypermutation mentioned above represents in fact an enzyme-mediated processes causing alteration of DNA sequences. First of all, APOBEC family member AID (activation-induced cytidine deaminase) has been extensively investigated in studies of Ig hypermutation [14] . AID exhibits a tumorigenic effect, when it is transfected and constitutively expressed and it can be found in current somatic cells different from lymphocytes in response to certain activating signals [15] - [18] . In accordance with its tumorigenic effect, AID hypermutates also genes encoding proteins different from AR [19] [20] . AID-mediated hypermutation occurs selectively at sites of certain primary and secondary DNA structures [21] [22] . First of all, this concerns well-known hypermutation motifs (HM) of sequence WRCH [21] [23] and specifically distant WRCH-pairs (W-pairs) which were frequently found in NS encoding immunoglobulins [24] [25] . In addition to this restriction of substrate specificity, initiating selective alignment between AID and DNA is also necessary for AID activity. Hence, this event does not occur at sites with sole HM, but only in G-loops [20] , and perhaps also in the other secondary DNA structures [22] or W-pairs proposed to trigger bi-bi-random mechanism of deamination [25] . Besides AID effects, mutations mediated by other APOBEC family members were observed in various non-lymphoid tissues currently not expressing AID. The research determined HM with the TCW sequence as an important target of APOBEC1, APOBEC3A, APOBEC3B, APOBEC3F and APOBEC3H reactions accompanying carcinogenesis [26] - [30] .
In summary, the goals of this paper consisted in 1) description of structurally and functionally interesting PPSIg-related PPS or PPSIg-related a posteriori reselected sequences predicted as PPS, 2) record of substantial common and extreme features of these molecules and 3) statistical evaluation of their group-related interrelationships. The corresponding search for topical PPSIg-related items occurred in five main steps (Figure 1).

Figure 1. Short methodological description of the employed heuristic bilingual approach. Five main procedural steps including both serial and parallel selection procedures determined here human, mouse and FFAS-scan-derived or NITR-related MPL. *, ** in the same box-two immediately subsequent steps (scheme simplification); repeating * in the same box-parallel procedures or common comments to these procedures; combined presence # and ∩-simultaneous occurrence of the selected items in records primarily obtained with the adjacent: 1) query sequences and 2) adjustments of BLAST searches; AR-antigen receptor(s); HM*-hypermutation motif(s) located at NS position critical with respect to possible aa alteration; NS-nucleotide sequence(s) of the corresponding both strands of cDNA. For CSB1, CSB2, Erα, MNSQ, MPL, MRNS, MSA, MSEP and SEM see inner part of this table. For additional details or abbreviations see supplementary files SF1 (cf. WF- and WP-related references), SF2 (dictionary of abbreviations) and SF3 (supplements to Figure 1). All three supplementary files are accessible on the web page address www.papersatellitesjk.com or via email jkub@post.cz.
About eighty percent of the predicted or existing PPSIg-related PPS found here comprised phosphorylation sites with serine processed by Ataxia telangiectasia mutated molecule (ATM), a key regulatory kinase of the DNA double-strand-break response [31] - [33] . All displayed PPSIg-related oligonucleotides contained HM WRCH and TCW at positions critical with respect to aa alterations. These critically located HM mostly occurred in non- transcribed DNA strands. The list of displayed segments contained molecules involved in carcinogenesis, cell division or specific regulatory functions.
2. Methods
2.1 Bioinformatic Tools
Programs of BLAST family [34] including conserved domain searches [35] were mostly accessed through publicly available NIH gate. Multiple-sequence alignment (MSA) was performed with Clustal W 2.1 present in the server of European Bioinformatic Institute [36] (for older version of the MSA record displayed here, see paper [11] ). The server with FFAS03 program [37] [38] was used for scanning based on the shortest possible profile- profile alignments enabling identification of conserved fold regions (FFAS-scan; cf. WP5.1). Hypermutation motifs were searched in the preformed multi-segment constructs using EMBOSS program Fuzznuc present on the web page of Pasteur Institute [25] [39] . Only PPS candidates simultaneously achieving the score 0.800 in two different machine learning programs (neural-network-based NetPhos2.0 and support-vector-machine-related KinasePhos2.0 [6] [7] ) were selected before the final reselection (cf. Figure 1 and sections WP2.4.1-2). On the other hand, alternative usage of databases Phospho.ELM a Phosida enabled us to find experimentally confirmed PPS [40] - [42] .
2.1.1. Typical Sequences
1) Conserved domain sequences (CDS; cf. [35] ), 2) phylogenically interesting actual sequence exhibiting superior similarities with CDS (IgW of clone AAB03680; igw; Figure 2; [11] [13] ) or 3) actual sequence achieving repeating superior similarities in initial searches for MNSQ units (consensus-like sequence, i.e. cls, of heavy chain with clonal names AF273898.1 and AAK20241.1; Figure 2) were all denoted here as typical sequences. Besides cls, derived in addition, the typical sequences assembled initial MSA record including two important conserved sequence blocks (CSB1 and CSB2; see Figure 2, sections 2.3 and WP2.2.2).
2.1.2. Statistical Enumerations
These enumerations were performed in accordance with the textbook of Zvárová [43] . In accordance with basic Bayesian approximation [44] , all groups of values with zero observations presented in 2 × 2 tables were increased by one and then the modified odds ratio value was enumerated as OR*[0]. Fisher exact probability of 2 × 2 tables was enumerated using Active web page [45] . This web page was used to statistically evaluate the validity of 1) currently enumerated odds ratios (OR) and 2) original 2 × 2 tables determining OR*[0]. Current search limits for long sequence similarities, i.e. 40 bits (a bit score limit for middle range BLAST similarities present in all headings of BLAST records) and p < 0.005 (indeed 5 × 10−3; cf. [34] ) restricted the validity of a) conserved segments of initial MSA record (Figure 2; cf. BS# and E# in Section 2.2.3) and b) searches for NS representatives of AR (Figure 1; sections 2.3 and WP2.4.2-4). We have to note that it holds if p < 0.05, then p < E and p ≈ E including E = E# (cf. a formula of Dembo [46] ). Specific statistically derived bit score restrictions of searches for short conserve-domain-related and MNSQ-derived similarities (MNSQ denotes multi-nucleotide sequence queries; see below) of lengths comparable with subsequently searched PPS are described in Figure 1 and section WP2.3.2.
2.1.3. Overall Scheme of Employed Procedures
For the scheme including all main procedures described in this chapter see Figure 1 and SF3.
2.2. Two Types of Consensus Sequences
We distinguish here two types of consensi (see also section WP2.2.1): 1) statistically important σ-consensus containing aa achieving the highest mean column score and 2) LE-consensus composed of aa determining length equivalents (LE) related to individual sequence block column (SBC). Both these types of consensi were differently

Figure 2. Initial multiple sequence alignment, its analysis and sequence derivatives. aThe set of sequences present in this table comprises variable domain of shark IgW sequence AAB03680 (pre-selected as representative of Ig-ancestor- related sequences of Elasmobranchii origin [11] [13] ). The included conserved Ig domains:_AL,_B,_G,_D-TCR related domains, i.e. alpha-like, beta, gamma and delta, respectively; K_L, L_L, H-kappa or lambda light chain and heavy chain domains, respectively. For further comments see sections 3.1 and 4.1. bTwo main conserved segments of sequence blocks (CSB1 and CSB2) are restricted here using bold characters of the corresponding consensus segments. The positions of the MSEP block segment (related to MNSQ1, MNSQ2) are indicated by gray background. Greek alphabets in the consensus denote aa differences in LE-/σ- related consensi determined here: α-P/S; β-N/S; γ-Y/E; δ-Y/A; ε-I/T, ζ-S/T; η-I/A; θ-W/S, λ-R/K. As follows, CSB1 related part of MSA-record determines unique consensus (consensus 1). cfin, fip-fuzzy related intervals in the ranges of negative or positive values, respectively. Numbers 1 - 9 in fip-/fin-related rows-values denoting hierarchy of fuzzy-related intervals, which represent the degrees of SBC similarities/dissimilarities (cf. Figure 3 and Section 2.2.3); n, p-negative and positive F-values described in Figure 3, respectively; plus and minus present in the fin row under fip value F = 3-presence or absence of the LE-value at least 1.5 necessary for candidate CBS edge, respectively (cf. Sections 2.2.4). For significance levels of SBC see sections WP4.1. dThe consensus pm3 is described in Sections 3.1 and WP5.1. Gray, dark gray proofs of pm3 segments localize positions corresponding specifically to CDR1 of light chains (CDR1light) or generally to all CDR1 (CDR1all), respectively. eFor details concerning the displayed structures see Sections 2.3, 3.1 and WP2.2.2.
used in enumerations described below and performed with minicomputer Casio Algebra 2 PLUS (cf. section WP2.1.4).
2.2.1. BLAST Derived Enumeration of LE
Each aa species present in evaluated SBC determined a non-invasive 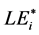 value, i.e. i-th aa-related length equivalent candidate (cf. sections WP2.1.1 and WP5.2).
value, i.e. i-th aa-related length equivalent candidate (cf. sections WP2.1.1 and WP5.2). 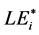 was defined as integer or non-integer height of artificial SBC composed only of i-th aa, provided that the probabilities of this artificial SBC and actual SBC (evaluated with respect to the selected i-th aa) were the same [9] . In score-related BLAST-derived evaluation of LE, we used conditioned Expect values (to include compared blank values) instead of probabilities (for details see section WP2.1.1). This evaluation finally determined the consistent formula:
was defined as integer or non-integer height of artificial SBC composed only of i-th aa, provided that the probabilities of this artificial SBC and actual SBC (evaluated with respect to the selected i-th aa) were the same [9] . In score-related BLAST-derived evaluation of LE, we used conditioned Expect values (to include compared blank values) instead of probabilities (for details see section WP2.1.1). This evaluation finally determined the consistent formula:
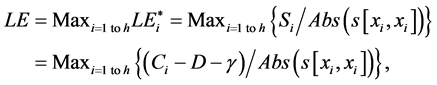 (1)
(1)
where Abs() enumerates absolute value; D is coefficient of column diversity; h is SBC height equal to the number of chains in MSA record; Si represents i-th aa related score in evaluated SBC; 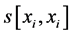 is score of i-th aa identity; xi is the number of pointing column and row positions of scores in the employed substitution matrix (PAM30 in our case, cf. limiting SBC heights of ten in Figure 2 and section WP2.1.1); γ denotes column related gap penalty. The following formulas further characterize the Ci, D and γ:
is score of i-th aa identity; xi is the number of pointing column and row positions of scores in the employed substitution matrix (PAM30 in our case, cf. limiting SBC heights of ten in Figure 2 and section WP2.1.1); γ denotes column related gap penalty. The following formulas further characterize the Ci, D and γ:
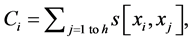 (2)
(2)
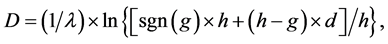 (3)
(3)
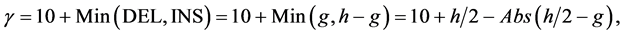 (4)
(4)
where j denotes positions of the compared aa in SBC; λ = 0.294 is BLAST constant; g is the number of gaps; d is the number of different aa in the same column; DEL and INS are the numbers of deletions and insertions deduced from the number of gaps, respectively; Min selects the minimum enumerated value in agreement with parsimony-related attempts of our approach. For explanation of 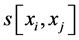 see
see 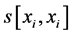 and j-values.
and j-values.
2.2.2. Enumeration of Numbers Determining the Degree of Fuzzy-Related Intervals (F or F-Values)
Each SBC-related F value was determined based on the fuzzy-related system (shown in Figure 3, see also WP5.3), implicating a two-step enumeration of F value based on υ value:
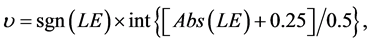 (5)
(5)
if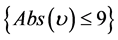 , then
, then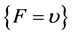 , else
, else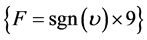 . (6)
. (6)
where int denotes an integer value of the enumerated number.
2.2.3. Restriction and Double-Sequence-Related Evaluation of the Conserved Segments of Sequence Blocks (CSB) Proposed for IgV-Related Sequences
The presence of a sequence pattern (briefly pattern determined by SBC containing unique aa species) was required in each proposed CSB including otherwise 16 - 50 neighbor SBC and achieving LE ≥1.5 in both its edge SBC. In the proposed approximating fuzzy-related approach, six rules concerned F values: 1)-3) at least 50% and 20% or at most 30% of SBC kept F ≥ 4, F ≥ 6, and F < 0, respectively. Among the state-of-the-art conditions, we assume that 4)-5) each list including F-values related to ten or five neighbor SBC had to contain at least three F ≥ 3 or at least two F ≥ 1, respectively, and 6) at least one of the three following alternative rules holds for the set of SBC composing evaluated CSB: mean(υi) ≥ 3.5, mean(Fi) ≥ 3.5 or mean(LEi) ≥ 1.75 (cf. Figure 3 or formulas 5 and 6). The selected candidates of CSB were then proved using current BLAST statistics [46] [47] . Hence, the values of double sequence similarity equivalents of bit score (BS#) and Expect value (E#) were enumerated, when assuming a simplified equal validity of all model chains in the evaluated reference MSA

Figure 3. Fuzzy-related system based on length equivalents (LE). Nineteen integer F-values from −9 to 9 are displayed here in the middle part of the graph. The following fuzzy-related description holds for the displayed positive F-values: 0―absence of similarity; 1―rampart of randomness; 2―area of “promising” noise; 3―quasi-similar (minimum but weak column similarities); 4―similar (close to “deterministic” double sequence aa identity); 5―quasi-cohesive; 6―cohesive (corresponds to aa identity in three compared chains); 7―quasi-rigid; 8―rigid (aa identity in four compared chains); 9―improved-rigid, i.e. maximum F-value in our single character system. Negative F-values approximate here more than random extent of diversity/variability (cf. sections the first section of Results and WP5.3). For MSA-record-compatible two-row entry of LE-derived fuzzy-related intervals indicating extents of SBC similarities see Figure 2.
record (more general CSB-chain-related linear combination can be also used, when employing “empirical”, i.e. Monte Carlo-related and database comparison-derived coefficients). The topical corresponding formulas follow:
 (7)
(7)
 (8)
(8)
 (9)
(9)
where ρCSB(S) is score density per single chain; K, λ are BLAST constants (K = 0.11 and λ = 0.294 in given case); BSm is equal to the mean bit score between consensus and chains forming CSB (in contrast to BS#, mentioned above, BSm disregards the negative effects of column diversity, i.e. BSm ≥ BS#; for comparison of BSm and BS# see section 3.1); Dn denotes the coefficient of n-th column diversity; L ´ h is the length of chains present in MSA record including gaps; N is the number of SBC determining the proposed CSB;  -is σ-consensus- related sum of scores concerning n-th SBC;
-is σ-consensus- related sum of scores concerning n-th SBC;  denoted minimized gap penalty with respect to the three considered models (cf. WP2.1.2). In case of our enumeration, we used the simplest alternative formula:
denoted minimized gap penalty with respect to the three considered models (cf. WP2.1.2). In case of our enumeration, we used the simplest alternative formula:
 (10)
(10)
where r is the number of chains with gaps and G denotes the number of all gaps in evaluated CSB-related segment of MSA record. The evaluation of gaps is the unique procedure in which differ evaluations of block-con- sensus and block-chain comparisons based on this section. For limits restricting valid BS# and E# see Section 2.1.2.
2.3. Generation of Two Initial Sequence Supersets Leading Separately to the Construction of Two Different Multi-Sequence Queries (MNSQ1 or MNSQ2) Representing NS of AR
In principle, two types of non-redundant different sequence sets (initial supersets of NS items) were generated, when using two types of TBLASTN records obtained with two different sets of query sequences (QS), each associated with different taxonomical restriction of searches. The first initial superset contained superior similarities with all “double conserved” sequences of CSB1 and consensus 1 (cf. Figure 1(d) in SF3), whereas the second superset comprised only the similarities found with consensus 1, three CSB1 sequences successfully examined in the previous paper [13] (i.e. cd00099, smart00406 and AAB03680_igv), their consensus-1-related derivatives and cls sequence found in addition during the formation of the second superset (see Sections 2.1.1, WP2.2.2 and Figure 2). The former and latter supersets contained sequences of Elasmobranchii or vertebrate origin, respectively. An iterative process of species- and query-related randomization consisted in 1) maximum number of five distinct species representatives as well as maximum overall numbers of selected items including non-redundant sequence samples in each set separately derived with individual QS and 2) existence of unlimited SIMULT set and hierarchy of QS-records both important for set-related rearrangement of promiscuously similar sequences (for details see WP2.2.3 or SF3). Initial TBLASTN searches employed BLOSUM62 matrix, whereas the following cumulative revision steps used two substitution matrices (BLOSUM62 and PAM30). For restriction of Expect and bit score values see Section 2.1.2.
2.4. Memory Problems in Some Searches with MNSQ1 and MNSQ2
Working memory problems were observed in differently advanced customer computers, when completing too extensive BLAST records obtained with initial steps of TCA searches and negatively Entrez-restricted two-step BLASTN searches limited by 35 bits (BNsup35; cf. Figure 1 and WP2.3.1). Consequently, special compromising processing of the corresponding records was necessary when downloading only well accessible but less extended records of items without reports of selected sequence alignments. These procedures comprised 1) a simplified total-bit-score-limited approach approximately substituting inaccessible frequencies of MUSAS (MNSQ- unit derived similarities with almost the same subject sequence positions; Figure 1) in the case of ternary combined approach (TCA) and 2) partial elimination of item names, immediately recognized as those determining AR, during the first step of BN35sup. For additional information see Altschul [34] and sections WP2.3.1-4, WP5.6 and WP5.7.
2.5. Final Selection and a Posteriori Reselection of the Displayed Items
Both final selection and reselection described in Figure 1 restricted non-redundant terminal list of items displayed in Figure 5 (for details see Sections 2.1, WP2.4.1, WP2.4.2, WP3.2 and WP3.3). Due to an unknown extent of losses following from inefficient assessing of dynamic (allosterically or proteolytically mediated) accessibility of MPL-related peptides, final selection did not include prediction of accessibility.
3. Results
3.1. Analysis of the Initiating MSA Record and Its Segments
Occurrence of the two conserved block segments (CSB1 and CSB2) was confirmed based on Section 2.2.3, when analyzing MSA record assembled by Clustal W 2.1 (Figure 2). CSB1 determined the same σ- and LE- consensi (consensus 1) containing pattern CX(10,13)WXXQXP. CSB1 achieved 1) F ≥ 6 and F < 0 in 51.3% and 12.8% SBC, respectively, 2) double sequence similarity related bit score BS# = 48.2 bits and consensus 1 related mean score BSm = 54.9 bits and 3) significant Expect value E# = 1.441 × 10−10 (for the almost equal p value see Sections 2.1.2 or WP4.1). CSB2 was characterized by 54.5% and 18.2% of columns with F ≥ 6 and F < 0, respectively, and contained the pattern LX(8)DX(3)YXC. CSB2 achieved also valid values of BS# = 44.6 bits, E# = 1.461 × 10−9 and BSm = 50.0 bits. 16 of the displayed 120 SBC (Figure 2) were classified as “at most anti- cohesive” (AAC; F ≤ −6; cf. Figure 3). CDR1-, CDR2- and CDR3-related block segments contained one, four and four of sixteen AAC SBC in Figure 2, respectively. This means that CDR1 substantially differed from CDR2 and CDR3, but not from the surrounding conservative framework regions (CDR1: positions 23 - 35, RCD = 0.89; CDR2: positions 51 - 62, RCD = 3.86, CDR3: positions 97 - 110, RCD = 3.31, where RCD denotes ratio between column densities of AAC occurrence determined in a) individual hypervariable and b) all accessible segments of framework regions).
Paralogues of AR were frequently similar to N-terminal parts of CSB1-related segments of cd00099, cd07706 and cd04980, when using FFAS-scan (Sections 2.1.1 and WP5.1). The similarities between these three selected domain segments then determined the consensus pm3, which was also N-terminally located in CSB1 (Figure 2). Since pm3-derived QS only rarely achieved the required bit score limit of 40 bits (cf. Section 2.2.3) in TBLASTN searches, we do not use pm3 for MNSQ generation. Nevertheless, a pm3-related segment of MSA record was suitable as an important fold- and sequence-related conserved block segment restricting the extension of PPSIg to C-terminus (cf. section WP5.5 and PPSIg enveloping segments of MSA record, i.e. MSEP described in Figure 2).
3.2. Selection of MNSQ1 and MNSQ2
Initial selection of sequence items restricted 121 and 169 items forming starting supersets of MNSQ1 (sequences of Elasmobranchii origin) and MNSQ2 (various vertebrate sequences), respectively. Subsequently, 41 and 108 sequence items were passed through reselection with MPQ and anti-redundant procedures assembling finally MNSQ1 and MNSQ2, respectively. Although the Ig items had not any preference in our searches, sequences of T-cell receptors (TCR) composed rarely MNSQ1 (two TCR sequences) and were not present in MNSQ2.
3.3. Paths Selecting MPL
The main paths of MPL selection are described in Table 1. OR-mediated analysis of these paths revealed interesting linkages within BLASTN-derived sets (Figure 4). Each employed OR represented ratios between cancer- related-set-derived R ratio (ratio between the numbers of human and mouse items present in the corresponding elements of Table 1) and the similarly derived Q-ratios by two species-related pairs of reference sets (BN35sup and KPO; Figure 4). Eleven and one OR values indicated strong (OR ≥ 2.0) and weak (1.4 ≤ OR < 2.0) associations of cancer-related NS items with their human origin, respectively (Figure 4). These OR values appeared to form significantly increased set of values (p < 0.05) even when skeptically considering equal distribution of OR values (as discrete values) above and under the value OR = 2.0 as random and employing model constant distribution (cf. section WP3.2) or Dirichlet statistics. In accordance with Figure 4, some of the described individual

Table 1. Most effective paths of MPL selection.
aStarting procedures included BLASTN searches or combined BLAST searches (cf. footnote d). NS―nucleotide sequences of the corresponding both strands of cDNA; hu, mu, both―sequences of human, mouse and both species origin, respectively. bAbout half of MNSQ-related mRNA segments encoded peptide segments, whereas about one seventh of these mRNA segments encoded existing or predicted PPS. cAll enumerated NS segments with predicted or database-confirmed PPS relationship (denoted here as MPL) fulfilled two conditions: 1) existence of MPL-derived similarities with chains of initial MSA record overlapping CSB1, 2) presence of at least single HM* in MPL. For details see Figure 1. dIndependent paths of MPL selection differed in their initial BLAST-derived procedures. Numbers 1 and 2 in strategy names-MNSQ1 and MNSQ2 were used as query sequences, respectively; gray and white background in left column of table elements-paths of prevailing selection of mouse or human sequences, respectively. Selections using sole BLASTN: BN1sup35, BN2sup35-molecules different from antigen receptors achieving top similarities (limit 35 bits) in set without positive Entrez restriction; C1, C2―cancer related Entrez restriction (limit 30 bits); KPO1, KPO2-Entrez restriction concerning proteins involved in phosphorylation (limit 30 bits). Combined selections: TCA1TX, TCA2TX-MNSQ1- and MNSQ2-query-derived TBLASTX variants of ternary combined approach (TCA), respectively. Each of these TCA variants used a) four searches using two different Entrez restrictions in two “subpaths” composed of two cumulative TBLASTX searches differently adjusted with respect to the matrices BLOSUM62 or PAM30 and word sizes three or two, respectively, b) limit for score maxima 25 bits, c) score limit for subject-sequence-related co-localized similarities (denoted as MUSAS) 22 bits when simultaneously requiring occurrence of at least five MUSAS in addition to segment with maximum score (i.e. limit six for all MUSAS). For additional information see Figure 1, sections 2.4, WP2.3.2-4 and WP5.6-7. eDue to independent selection paths (forming non-redundant sets), some items sometimes repeat in different sets (cf. also strategy records in Figure 5, sections WP2.3.3 and WP4.2.3).

Figure 4. Odds ratio―(OR-) mediated linkage analyses. Achievement of the three limits (i.e. sufficient sample size of at least 40 NS items, significant difference p < 0.05 and strong association indicated by OR ≥ 2) is recorded here by different column fillings: black, gray―three or two limits were attained, respectively; gradient of gray―sole sufficient sample size (important in cases of low or no associations); empty columns-other cases of OR; gray in flanks―reference R ratios compared with OR values (see above). (a) (b) Selected results of OR-mediated analysis concern Table 1 (cf. also the corresponding abbreviations) and are commented in section 3.3. Numbers 1 and 2 in abbreviation-NS items found with MNSQ1 and MNSQ2, respectively; suffixes o1, o2 and o3―selection steps 1, 2 and 3 mentioned in Table 1, respectively; B, K-BNsup35 and KPO sets, respectively; C1, C2―sets of cancer-related BLASTN-records containing sequences similar to MNSQ1 (part (a)) or MNSQ2 (part (b)), respectively; R-ratios between numbers of human and mouse NS items present in C1 and C2 sets. (c) Overall (robust) evaluation of strand-related HM* occurrences in MPL displayed in Figure 5. Strong and significant associations of HM* with non-transcribed DNA strands were found. Wp―W-pairs. For details see Abbreviations and Section 3.4.
strong associations were also significant (p < 0.05) and/or robust (sample size s ≥ 40 NS). Higher or more valid R and OR values mostly formed upper MNSQ1-related graph indicating certain phylogenic context of this evaluation due to exclusive Elasmobranchii origin of MNSQ1 units (cf. section b in Figure 4). For additional comments see Section 4.3.
Predicted phosphorylated serines (Ser) achieved scores higher than 0.990, when using both NetPhos 2.0 and KinasePhos2.0. Similarly, NetPhos-related prediction yielded score maxima for threonine (Thr) and tyrosine (Tyr) higher than 0.980, but scores lower than 0.950 were determined during the corresponding KinasePhos- mediated predictions. In accordance with this difference in score maxima, only predicted phosphorylated Ser but not Thr and Tyr were present in set of double-predicted PPS encoded by MPL. This result led us to complete MPL set with four new MPL 1) different from originally found MPL, 2) encoding predicted Thr- and Tyr-re- lated PPS, and 3) achieving minimum score of 0.950, when using NetPhos 2.0. For details concerning the results of the parallel database search for experimentally confirmed MPL see the first section of Figure 5.
Another type of statistical reevaluation was performed on the subset of MPL-encoded peptides (MEP) immediately containing phosphorylated aa (i.e. uniquely limited by the same score of 0.800 in the two predictions). The constructed histogram and histogram-related distributions based on the product of the resulting two types of scores (s1 × s2) in fact approximated each result of performed double selection as unique value (Figure 6). Unexpectedly, the obtained graphs indicated bimodal-like distribution with a dominant peak located at range of superior score products. This distribution contrasted with the expected random exponential decrease assumed at least in upper half of score product values. Even the comparison with the model constant distribution (stricter evaluation than the assumed random exponential decrease) indicated strong and significant (p < 0.05) association of score products with the restricted region of dominant peak. Based on these facts and considerations concerning Figure 6, we explained the existence of the peak as the consequence of functionally conditioned structural



Figure 5. Existing and predicted antibody-like phosphorylation sites. aThe MPL subsets (subdivided according to types of reselection) were arranged in the separate table segments according to the mean positions of their feedback similarities (m(S)) between MPL and initial MSA record displayed in Figure 2 (cf. footnote i and Figure 1). BOC―brother of CDON, cell adhesion associated molecule, oncogene regulated; BP-binding protein; Copb1-coatomer protein complex, subunit beta 1; EIF4EBP1―Eukaryotic translation initiation factor 4E binding protein 1; Pnliprp1-pancreatic lipase related protein 1; regul.-regulated; Rps6ka4―ribosomal protein S6 kinase, polypeptide 4; Rps6kc1―ribosomal protein S6 kinase, polypeptide 1; SCTR―successful criteria of terminal reselection (for encoding numbers see fifth part of Figure 1(b) in SF3); VEGF― vascular endothelial growth factor. bFrS―frequencies of limited co-localizing similarities (i.e. MUSAS mentioned in Sections 2.4, WP2.3.3-4 and WP5.7); mBS―maximum bit score of the selected BLAST similarities; NS-nucleotide sequence; PS-protein sequence; *-minimum of two alternative score maxima or MUSAS numbers was recorded in cases of two co-evaluated searches. csp-species origin (current items: Hu―human, Mu―Mus musculus; reference NITR-related item: Mm-Miichthys miiuy); str-strategies yielding topical MRNS (sections WP5.6 and WP5.7). Capitals and numbers in abbreviations of Entrez restriction (section WP2.3.1): C, K-cancer- and phosphorylation-related special BLASTN searches; DX―double combined approaches comprising pairs of differently adjusted TBLASTX searches, only if they yield co-localizing similarities limited by 30 or 35 bits; S―global BLASTN searches limited by 35 bits; T―ternary combined approach (TCA, limit 25 bits); X, NX, N after T-TCA accompanying pairs of BLAST searches (selecting repeatedly co-localizing similarities; cf. WP2.3.3), i.e.: 1) two differently adjusted TBLASTX; 2) BLASTN and TBLASTX and 3) two BLASTN differently employing two MNSQ, respectively; 1, 2, 12―items selected by MNSQ1 or MNSQ2 and both MNSQ, respectively. dHaa + Waa-occurrence of effectively located hypermutation motifs (HM*) including W-pairs (for details see sections 3.4 and 4.4). Upper rows: left/right evaluation using plus or minus-WRCH/TCW are present or are not present at the critical positions, respectively. Bottom rows: #aWi, #pWi-W-pairs located at DNA strands anti-parallel or parallel with respect to the direction of transcription, respectively (i denotes the number of unclassified nucleotides inserted between the observed WRCH; if n substitutes the number i, then WRCWRCH is indicated); #aRW, #pRW-palindromic W-pairs RGYWRCY includes WRCY unit critical with respect to aa alteration in antiparallel or parallel (transcribed) DNA strands, respectively. eSEM-sequences enveloping (and including) selected MPL encoded peptides; gray background covering edges of peptide segments-chains extending central MRNS-encoded peptide sequences; underlined-predicted or empirically confirmed phosphorylated aa; CD-conserved domains; cdsBS, cdsE-conserved domain similarity-related bit scores and Expects, respectively. Lower cases (LC) in alphabets denoting peptide sequences indicate aa which could be altered during hypermutation changes via cytidines of HM (HM*-cytidines): LC and LC with―sole HM*―cytidine form WRCH and TCW, respectively; LC with *―two HM*-cytidines are present in two neighbor but antiparallelly located positions of palindromic WRCH; LC followed by dominant #―WRCH containing HM*-cytidine composes W-pair (cf. Sections 3.4 and 4.4). fpos + aa―phosphorylated aa is denoted by a single character, which is accompanied by the number of given aa position in the displayed peptide; NetP, KinP-scores obtained by NetPhos2.0 and KinasePhos2.0, respectively (maximum is equal to unity); spec―specificity of kinase―mediated phosphorylation (identified by KinasePhos2.0). gSeven experimentally proved PPS exhibited overlaps with peptides encoded by MPL (MEP), whereas phosphorylation sites of four these PPS immediately formed MEP. In addition, two proved PPS with phosphorylation sites located at position immediately neighboring to MEP were encoded by sequence containing HM* occurring within MPL. The residual (seventh) confirmed PPS (with the site outside MEP) then overlapped MEP encoded by MPL containing double-active W-pair (i.e. two HM* forming W-pair). F, P-database record of existing PPS was found using Phosida or phospho.ELM, respectively; *―indicator of empirical confirmation recorded in given databases. hOnly nine groups of protein kinases (PK) were predicted or empirically proved to phosphorylate the displayed MEP: Aur―Aurora-related kinase; ATM―Ataxia-telangiectasia mutated (kinase); CK1, CK2-casein kinases 1 and 2, respectively; GSK3-Glycogen synthetase kinase-3; MAP3K-MAPK kinase kinase (=MEKK1); MAPK-mitogen activated PK; PKB-PK B; PKC-PK C; PLK1―polo-like kinase 1, absence of abbreviation in a database-confirmed case―only phosphopeptide was observed without knowledge about PK specificity. iAlign versions of BLAST were used in feedback comparison (see WP2.4.1). O, S, m(S)―MSA-record-related positions of deduced sequence overlaps, feedback similarities, and mean aa position of these similarities (cf. footnote a), respectively. jThe segments of MSA record present in Figure 2 formed feedback (BLASTX, TBLASTN or TBLASTX; cf. section WP2.4.1) similarities at defined IgV-related positions of this “initial” MSA- record (PIM): FR1C―framework region 1 including CSB1-related overlaps (PIM from 5 to 15 - 22); CDR1L―at least 50% overlap of CDR1light (PIM: 23 - 30); CDR1a―C-terminal part of CDR1 in light chains and co-localizing CDR1 of heavy chains (PIM: 31 - 35); CDR1s-prevailing overlaps of CDR1light but short of CDR1all (PIM: 23 - 35); FR2N―N-terminus of framework region 2 (PIM: 36 - 42; the most conserved part of this framework region in Figure 2). *―different classification follows from NS differences. For additional comments see Figure 2, Section WP4.2.2 and SF2.
maintenance. Based on this statistically supported explanation, we derived the limit for score products 0.88, subsequently used in the terminal reselection. For important details see sections WP2.4.1, WP3.2, WP3.3 and file SF3.
3.4. Final Bioinformatic Analysis of the Selected Pairs of MEP and MPL
About eighty percent of regularly restricted human and mouse MEP achieved superior score related to ATM. Similarly to PPSIg, part of ATM-related sites, predicted here, achieved also co-dominant prediction of PPS

Figure 6. Distribution of score products evaluating predicted phoshorylation sites encoded by MPL. Model MPL subset of predicted sequence items was selected before overlap-, Thr-, Tyr- and database-related completions of data (cf. Figure 1 and WP2.4.1). This subset included thirty-two MPL-encoded peptide sequences containing forty predicted phosphorylation sites, which were represented here by the products of their maximum predicted scores (s1*s2). The score maxima were individually obtained with NetPhos 2.0 (s1) and KinPhos 2.0 (s2) in the searches equally restricted by the limiting minimum value 0.800 (cf. Figure 5). Axes x of the displayed graphs comprise the specific score product intervals of 0.01 extent. Upper part includes a current histogram of the score-product-related frequencies. Frequencies of the evaluated score products look like the main peak in the range of upper values. The corresponding significant prevalence to high score products (p < 0.05) perhaps consisted in the existence of phylogenic pressures keeping PPS function and enabled us to empirically assess reselection limit. In addition, we simultaneously diminished effects of possible overestimations (i.e. false negativities) frequently accompanying pre-calculated restriction of combined selection. For other comments see Sections 2.1.2, 3.3 and WP3.2. Middle part contains widely used gliding mean values of frequencies enumerated here based on three neighboring (interval-related) frequencies present in the upper histogram. Each gliding mean value is displayed specifically in the positions of central (second counted) interval yielding thus the simplest form of smoothened histogram. Bottom part contains more adequate/specific form of histogram smoothening based on linear combinations formed by central and neighbor interval related frequencies and Gaussian kernel functions which enumeration takes into consideration empirical statistics of the evaluated differences between the corresponding score pairs s1 and s2 (for details see section WP3.3).
phosphorylated with Aurora and protein kinase B. Interestingly, MPL were not always translated in the same reading frame or orientation as the compared IgV-related segments of initial MSA record. Five MEP formed chains of valid conserved domain similarity with Ig domains. Four of them achieved feedback similarity with the segment CDR1light in initial MSA record. However, feedback similarities overlapping common CDR1all segments were found only in five cases of Ig-unrelated MEP/MPL (cf. Figure 5 and section WP4.2.2; for explanation of CDR1light and CDR1all see Figure 2). Only pairs of human and mouse orthologues of HIPK2 contained equally co-localized MPL in Figure 5.
The DNA-strand-related occurrence of well-known hypermutation motifs (HM; [21] [23] [24] [30] ) was observed, when selecting HM subset (HM*) located at MPL sequence positions critical with respect to aa alteration. Interestingly, the number of HM* was significantly higher (p < 0.05) in the non-transcribed DNA strands of MPL than in transcribed ones when comparing the strand-related occurrences of HM* with those of HM incapable to alter aa (non-HM*). The corresponding significant increase concerned also certain WRCH-related pairs (W-pairs; OR*[0] = 11.6 but an extremely low set of 22 items) determined by the patterns: RGY-WRCY, WRCWRCH, WRCHNWRCH and WRCHN(9)WRCH (see section 4.4 and previous papers [24] [25] ). For the graphs of HM*-related OR see Figure 4. The occurrences of HM*-related W-pairs in framework-associated and CDR1-associated regions of MPL (determined by feedback comparison) also differed, determining 3.85 times higher density of such W-pairs in CDR1-associated regions. In addition, we found three types of MPL relationships interesting with respect to HM* occurrence. This concerned: 1) knocking-out alteration of predicted phosphorylated serine in cases of twelve human and mouse MPL and one reference MPL of bony fish origin, 2) distinct relationship of the two HM* (WRCH and TCW) to transcribed DNA strand in site encoding phosphorylated serine following from genetic code and 3) maximum number of HM* per single MPL (two different MPL triads distinctly contained four times such WRCH or such TCW). For additional details and comments see Figure 5, Sections 4.4, WP2.4.1 and WP4.2.1-3.
4. Discussion
4.1. Structures Found in Initial MSA-Record
Both sequences of patterns displayed in Figure 2 exhibit certain relationship to the published data. The first two aa of CSB1-related pattern CX(10,13)WXXQXP include well known positional markers restricting CDR1. Cysteine is the current C-terminal aa of FR1 composing light chains of IgV, whereas tryptophan mostly occurs in N-terminus of FR2 in both light and heavy chains of IgV [48] [49] . Trans-cis isomerization of C-terminal aa of the same pattern, i.e. proline, is probably important for local flexibility of IgV [8] [50] . Another CSB2-related pattern identified here, LX(8)DX(3)YXC, contains the sequence DX(3)YXC identical with the pattern found in non-vertebrate molecules closely related to AR [13] .
The marked differences between the occurrence of at least anti-cohesive columns in CDR1 and other two hypervariable regions (cf. Section 3.1) indicate higher conservativeness of usually effectively hypermutating CDR1. If we also consider location of CDR1 in CSB1 and PKSIg [8] [9] [11] (cf. also Figure 2), the indicated conservativeness supports as an additional fact the model importance of N-terminally located PPSIg region for studies of antibody-like PPS.
4.2. Methodological Aspects
The bilingual approach employed here was in fact based on simultaneous evaluation of 1) occurrence of HM motifs present in MPL composing IgV-related mRNA or cDNAs segments (the first formal language; Figure 1), 2) existing or predicted phosphorylation of MPL encoded peptides extended by their immediately neighboring chains (the second formal language) and 3) possible alteration of aa composing the selected PPS via HM (site specific translation). Due to various actual reading frames of MPL, the bilingual approach determined a larger set of MPL than would simply follow from the searches for protein sequence similarities.
To unify the selection of MRNS (cf. Figure 1), we supplemented here a simplified BLAST derived fuzzy- system associated restriction of CSB (Section 2.2.3) consistent with ELEMS principles [9] [25] . The unified usage of BLAST-related evaluation in the three starting main steps (cf. Figure 1) does not mean refusing the possible prediction by means of machine-learning methods such as SVM or neural networks [51] - [55] . More likely, we assume that some results obtained in our searches represent important starting information for future machine learning. This concerns among others 1) feedback similarities and 2) reevaluation similarity extents and cooperative effects related to different elementary MNSQ units participating in or associated with MPL selection process.
4.3. Statistical and Phylogenic Contexts of Employed Search Procedures
In fact, we did not observe here NS mutations in cell clones, but only potential NS mutation. Consequently, we considered here only inherited changes of DNA, when looking for an explanation of unexpected prevailing occurrence of cancer-related NS items in the human set described in section 3.3. This consideration focused our attention first of all on 1) several events occurring in germ-line cells (GLC) or 2) parameters of these events, i.e.: a) prevention of DNA attacks by oxygen radicals in GLC, b) extents or specificity of meiotic hypermutation (comprising also the described changes in Ig-related HM [56] ) and its possible effects on genome plasticity, c) percentage of mutated descendants bred by old individuals d) immune surveillance on GLC. Better understanding of the observed and potential relationships, including phylogenically interesting (Elasmobranchii-related) MNSQ1 linkage (cf. Sections 2.3, 3.3 and Figure 4), needs also more detailed analysis on a larger set of PPSIg- related molecules and MPL in future.
In accordance with our working hypothesis [13] , at least some MPL displayed here (encoding most probably ATM-phosphorylated MEP; cf. section 3.4) and PPSIg segments originate in common ancestral oligonucleotide segments of possible increased mutability. This hypothesis appears to be in agreement with the displayed strong and significant association of score products with superior score values (Figure 6; Sections 3.3 and WP3.2) and existence of four molecules containing MPL/MPL-like segments most frequently (i.e. four times) selected in terminal reselection (see the first column of Figure 5 and section WP4.3). The latter possibility concerns: 1) NITR2 of close phylogenic relationship to AR [57] otherwise only near the limits of PPS prediction (cf. WP4.3), 2) two molecules containing MPL with Ig domain context molecules (KDR of superior PPS-related scores, oncogene regulated adhesion molecule BOC); 3) MPL of glioma tumor suppressor candidate region gene 1 (Gltscr1). In contrast to other molecules, Gltscr1 did not achieve any conserved Ig domain similarity, the corresponding CDART-indicated relationship to Ig superfamily (IgSF) representatives or local PPSIg-related fold similarity in records obtained with FFAS03 program (cf. section 3.1). To explain this anomalous sequence behavior of Gltscr1, we assumed participation of well-known mechanisms such as a) transposition of short repeats, b) gene conversion and/or c) perhaps also functionally conditioned convergence of DNA segments (cf. [5] ).
4.4. MPL in the Light of Hypermutation Events
Collisions of transcription apparatus (synthesizing mRNA copy of transcribed strand of DNA) with APOBEC enzymes interacting with HM present in transcribed DNA strand (CTAE) are able to enlarge hypermutation effects of these deaminases. This occurs due to 1) extended low-fidelity repair of generated double strand breaks with error-prone DNA polymerases and 2) accompanying insertion deletion changes [58] - [60] . In accordance with this mechanism the observed significant differences in distributions of HM* in transcribed and non-tran- scribed DNA strands (Section 3.4; Figure 4) can be explained by similarly selective CTAE-triggered events. Since the comparison of DNA strands does not concern altered nucleotide sequences, inherited but not somatic changes have to be considered, which looks like a contradiction when dealing with somatic mutation changes. Nevertheless, HM WRCH participates also in inherited meiotic hypermutation via a similar though less frequent mechanism than in case of Ig gene hypermutation [56] . Consequently (and in accordance with less frequent incidence of meiotic-mutations), the statistics of HM* occurrence in non-altered DNA strands (see Figure 4) suggests parallel somatic mutation changes via CTAE events in at least some transcribed DNA strands encoding MEP-peptides and containing HM*. In accordance with this parallelism, HM* located in their transcribed DNA appear to be more perilous than others, indicating thus MEP of probably increased hypermutation risk in molecules such as human RhoGEF kinase, MAGEE1, KDR, zinc finger protein 687, rhabdomyo-sarcoma FBO1A, mouse anaplastic lymphoma kinase and zinc finger protein 619 and both reference AR-related NITR molecules of bony fish origin present in Figure 5 or Table WPT2 (see section WP4.3).
In accordance with the theories of ageing, abnormal phosphorylation (or loss of phosphorylation) is assumed as an alteration possibly important during ageing [61] . Abnormal phosphorylation can be among others caused via mutation or hypermutation changes of PPS generating knocked-out, alternatively or weakly reacting PPS. If we assume that such PPS changes frequently occur in different PPS phosphorylated by the same PK, then their change can imitate the loss or decrease of the involved PK activity. In accordance with the given assumption, about eighty percent of MEP appear to be dominant substrates of ATM (section 3.4), which is the reason to consider possible imitation of functional loss of ATM via mutation of these PPS. As well known ATM is involved in regulation of double-strand-break response (cf. Introduction). Functional loss of mutated ATM causes autosomal recessive disease (ataxia-telangiectasia; AT) characterized by median survival 19 - 25 years (a wide range) and death due to cancer and respiratory failure [62] . In addition, AT is also accompanied by neurodegeneration and immunodeficiency or worsened immune response otherwise frequently appearing in elderly people [63] . Consequently, a substantial question arises: Can changes in sequences of PPSIg/MPL-related PPS influence important regulatory or even ageing-related effects of ATM?
Some MPL are not translated in the same reading frame as PPSIg (cf. Section 3.4). The corresponding peptides (M_out) thus can avoid cross-reactivity with Ig epitopes, which mostly contain only “in frame” insertions and deletions [58] [64] . This raises a question of possible usage of at least some mutants of M_out as components of future complex/multicomponent anticancer vaccines. On the other hand, it is a question whether at least some MPL-derived peptides encoded in the same reading frame as PPSIg (M_in) or their mutants can cross- react with rheumatoid autoantibodies. The positive answer could be among others important for protein engineering improving autoantibody specificities with respect to M_in mutants.
5. Conclusions
Though the databases of empirically proved phosphorylation sites are considerably incomplete, seven MEP contain such database-confirmed sites. The nonrandom main peak of MPL-derived products of phosphorylation- related prediction scores then suggests the phosphorylation of at least some additional MEP (Section 3.3; Figure 6). The significantly prevailing occurrence of HM* and the corresponding W-pairs in non-transcribed DNA strands of MPL indicates certain mutation events in the corresponding gene segments including parallel somatic changes (Section 4.4; Figure 4). In accordance with these parallel changes, MPL segments with HM* present in transcribed DNA strand appear to be most critical (for list see Section 4.4). The occurrence of HM* in MPL meets also three important functional aspects. First of all, most MPL (about 80%) encode peptides identical with or predicted as phosphorylated by ATM molecules, known for their relationship to aging and regulation of DNA double-strand-break response (Sections 3.4 and 4.4). Secondly, cancer-related MRNS are significantly more frequent in humans than in mice (Figure 4; Sections 3.3 and 4.3). Thirdly, feedback similarities of MPL containing W-pairs co-localized mainly with (hypervariable) CDR1 segments of IgV chains forming both MNSQ (Table 1, Figure 2 and Figure 5, Section 3.4). These three functional aspects moreover concern several existing groups of MPL with extreme properties, e.g. 1) MPL with HM* encoding directly predicted phosphorylated amino acid, 2) CDR1-related MPL containing superior numbers of HM* and 3) MPL/MEP pairs frequently selected in the alternative steps of terminal reselection (Figure 5; Sections 4.3, 4.4 and WP4.2.1-3).
Though the actual carcinogenic effect of mutation changes in phosphorylation sites is known for a long time [65] - [67] , the set of the corresponding investigated sequences is not sufficiently large. In accordance with this fact, we have not yet found experimental confirmation of mutation changes in the seven existing PPS segments (displayed in Figure 5) in the literature. Consequently, these PPS, as well as the displayed predicted segments, represent an inspiration and challenge for the subsequent experimental researches in specialized laboratories and perhaps also subjects interested in certain bioinformatic trends (cf. [68] [69] , Sections 4.2 and 4.4; see also below). In our opinion, future experimental research should comprise 1) comparative sequence studies of ATM- related MPL composing not only DNA of cancer patients (for the corresponding methods see also our papers [70] - [72] ) but also DNA of old people and 2) usage of phage displayed libraries or protein engineering in case of the considered immunological relationships ( [73] - [75] and Section 4.4). Theoretically based bioinformatic investigation of the immunological relationships could moreover efficiently select the subset of possible candidates for vaccine-related promiscuous epitopes specifically recognized by cytotoxic T cells ( [76] - [78] and Section 4.4) even in case of more extended HM*-related set of MEP mutants than is that displayed in Figure 5 (for other corresponding bioinformatic aims and trends see section WP5.8). In addition to the preceding medicinal aspects, an interesting question refers to a possible phylogenic relationship between IgV domains and certain NS encoding peptide segments a) containing several near ATM-related PPS and b) composing for instance immunoglobulin-like archaeal surface layer proteins or related metazoan cell surface proteins [13] [79] .
Cite this paper
Jaroslav Kubrycht,Karel Sigler,Pavel Souček,Jiří Hudeček, (2016) Antibody-Like Phosphorylation Sites in Focus of Statistically Based Bilingual Approach. Computational Molecular Bioscience,06,1-22. doi: 10.4236/cmb.2016.61001
References
- 1. Kornev, A.P., Haste, N.M., Taylor, S.S. and Ten Eyck, L.F. (2006) Surface Comparison of Active and Inactive Protein Kinases Identifies a Conserved Activation Mechanism. Proceedings of National Academy of Sciences of the United States of America, 103, 17783-17788.
http://dx.doi.org/10.1073/pnas.0607656103 - 2. Kornev, A.P., Taylor, S.S. and Ten Eyck, L.F. (2008) A Helix Scaffold for the Assembly of Active Protein Kinases. Proceedings of National Academy of Sciences of the United States of America, 105, 14377-14382.
http://dx.doi.org/10.1073/pnas.0807988105 - 3. Joseph, R.E., Xie, Q. and Andreotti, A.H. (2010) Identification of an Allosteric Signaling Network within Tec Family Kinases. Journal of Molecular Biology, 403, 231-242.
http://dx.doi.org/10.1016/j.jmb.2010.08.035 - 4. Seco, J., Ferrer-Costa, C., Campanera, J.M., Soliva, R. and Barril, X. (2012) Allosteric Regulation of PKCq: Understanding Multistep Phosphorylation and Priming by Ligands in AGC Kinases. Proteins, 80, 269-280.
http://dx.doi.org/10.1002/prot.23205 - 5. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. (2008) Molecular Biology of the Cell. 5th Edition, Garland Science, New York.
- 6. Blom, N., Gammeltoft, S. and Brunak, S. (1999) Sequence- and Structure-Based Prediction of Eukaryotic Protein Phosphorylation Sites. Journal of Molecular Biology, 294, 1351-1362.
http://dx.doi.org/10.1006/jmbi.1999.3310 - 7. Wong, Y.H., Lee, T.Y., Liang, H.K., Huang, C.M., Wang, T.Y., Yang, Y.H., Chu, C.H., Huang, H.D., Ko, M.T. and Hwang, J.K. (2007) KinasePhos 2.0: A Web Server for Identifying Protein Kinase-Specific Phosphorylation Sites Based on Sequences and Coupling Patterns. Nucleic Acids Research, 35, W588-W594.
http://dx.doi.org/10.1093/nar/gkm322 - 8. Kubrycht, J. and Sigler, K. (1997) Animal Membrane Receptors and Adhesive Molecules. Critical Reviews on Biotechnology, 17, 123-147.
http://dx.doi.org/10.3109/07388559709146610 - 9. Kubrycht, J., Borecky, J. and Sigler, K. (2002) Sequence Similarities of Protein Kinase Peptide Substrates and Inhibitors: Comparison of Their Primary Structures with Immunoglobulin Repeats. Folia Microbiologica, 47, 319-358.
http://dx.doi.org/10.1007/BF02818689 - 10. Kubrycht, J., Borecky, J., Soucek, P. and Jezek, P. (2004) Sequence Similarities of Protein Kinase Substrates and Inhibitors with Immunoglobulins and Model Immunoglobulin Homologue: Cell Adhesion Molecule from the Living Fossil Sponge Geodia Cydonium. Mapping of Coherent Database Similarities and Implications for Evolution of CDR1 and Hypermutation. Folia Microbiologica, 49, 219-246.
http://dx.doi.org/10.1007/BF02931038 - 11. Kubrycht, J., Sigler, K., Soucek, P. and Hudecek, J. (2013) Structures Composing Protein Domains. Biochimie, 95, 1511-1524.
http://dx.doi.org/10.1016/j.biochi.2013.04.001 - 12. James, L.C., Roversi, P. and Tawfik, D.S. (2003) Antibody Multispecificity Mediated by Conformational Diversity. Science, 299, 1362-1367.
http://dx.doi.org/10.1126/science.1079731 - 13. Kubrycht, J., Sigler, K., Ruzicka, M., Soucek, P., Borecky, J. and Jezek, J. (2006) Ancient Phylogenetic Beginnings of Immunoglobulin Hypermutation. Journal of Molecular Evolution, 63, 691-706.
http://dx.doi.org/10.1007/s00239-006-0051-9 - 14. Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y. and Honjo, T. (2000) Class Switch Recombination and Hypermutation Require Activation-Induced Cytidine Deaminase (AID), a Potential RNA Editing Enzyme. Cell, 102, 553-563.
http://dx.doi.org/10.1016/S0092-8674(00)00078-7 - 15. Okazaki, I.M., Hiai, H., Kakazu, N., Yamada, S., Muramatsu, M., Kinoshita, K. and Honjo, T. (2003) Constitutive Expression of AID Leads to Tumorigenesis. The Journal of Experimental Medicine, 197, 1173-1181.
http://dx.doi.org/10.1084/jem.20030275 - 16. Kou, T., Marusawa, H., Kinoshita, K., Endo, Y., Okazaki, I., Ueda, Y., et al. (2006) Expression of Activation-Induced Cytidine Deaminase in Human Hepatocytes during Hepatocarcinogenesis. International Journal of Cancer, 120, 469-476.
http://dx.doi.org/10.1002/ijc.22292 - 17. Endo, Y., Marusawa, H., Kinoshita, K., Morisawa, T., Sakurai, T., Okazaki, I.M., Watashi, K., Shimotohno, K., Honjo, T. and Chiba, T. (2007) Expression of Activation-Induced Cytidine Deaminase in Human Hepatocytes via NF-kappaB Signaling. Oncogene, 26, 5587-5595.
http://dx.doi.org/10.1038/sj.onc.1210344 - 18. Matsumoto, Y., Marusawa, H., Kinoshita, K., Endo, Y., Kou, T., Morisawa, T., Azuma, T., Okazaki, I.M., Honjo, T. and Chiba, T. (2007) Helicobacter Pylori Infection Triggers Aberrant Expression of Activation-Induced Cytidine Deaminase in Gastric Epithelium. Nature in Medicine, 13, 470-476.
http://dx.doi.org/10.1038/nm1566 - 19. Gordon, M.S., Kanegai, C.M., Doerr, J.R. and Wall, R. (2003) Somatic Hypermutation of the B Cell Receptor Genes B29 (Igbeta, CD79b) and mb1 (Igalpha, CD79a). Proceedings of National Academy of Sciences of the United States of America, 100, 4126-4131.
http://dx.doi.org/10.1073/pnas.0735266100 - 20. Duquette, M.L., Pham, P., Goodman, M.F. and Maizels, N. (2005) AID Binds to Transcription-Induced Structures in c-MYC that Map to Regions Associated with Translocation and Hypermutation. Oncogene, 24, 5791-5798.
http://dx.doi.org/10.1038/sj.onc.1208746 - 21. Rogozin, I.B. and Kolchanov, N.A. (1992) Somatic Hypermutagenesis in Immunoglobulin Genes. II. Influence of Neighbouring Base Sequences on Mutagenesis. Biochimica et Biophysica Acta, 1171, 11-18.
http://dx.doi.org/10.1016/0167-4781(92)90134-L - 22. Wright, B.E., Schmidt, K.H., Davis, N., Hunt, A.T. and Minnick, M.F. (2008) II. Correlations between Secondary Structure Stability and Mutation Frequency during Somatic Hypermutation. Molecular Immunology, 45, 3600-3608.
http://dx.doi.org/10.1016/j.molimm.2008.05.012 - 23. Rogozin, I.B. and Diaz, M. (2004) Cutting Edge: DGYW/WRCH Is a Better Predictor of Mutability at G:C Bases in Ig Hypermutation than the Widely Accepted RGYW/WRCY Motif and Probably Reflects a Two-Step Activation-Induced Cytidine Deaminase-Triggered Process. The Journal of Immunology, 172, 3382-3384.
http://dx.doi.org/10.4049/jimmunol.172.6.3382 - 24. Dorner, T., Foster, S.J., Brezinschek, H.P. and Lipsky, P.E. (1998) Analysis of the Targeting of the Hypermutational Machinery and the Impact of Subsequent Selection on the Distribution of Nucleotide Changes in VHDJH Rearrangements. Immunology Reviews, 162, 161-171.
http://dx.doi.org/10.1111/j.1600-065X.1998.tb01439.x - 25. Kubrycht, J. and Sigler, K. (2008) Length of the Hypermutation Motif DGYW/WRCH in the Focus of Statistical Limits. Implications for a Double-Motif or Extended Motif Recognition Models. Journal of Theoretical Biology, 255, 8-15.
http://dx.doi.org/10.1016/j.jtbi.2008.07.039 - 26. Beale, R.C.L., Petersen-Mahrt, S.K., Watt, I.N., Harris, R.S., Rada, C. and Neuberger, M.S. (2004) Comparison of the Differential Context-Dependence of DNA Deamination by APOBEC Enzymes: Correlation with Mutation Spectra in Vivo. Journal of Molecular Biology, 337, 585-596.
http://dx.doi.org/10.1016/j.jmb.2004.01.046 - 27. Bishop, K.N., Holmes, R.K., Sheehy, A.M., Davidson, N.O., Cho, S.J. and Malim, M.H. (2004) Cytidine Deamination of Retroviral DNA by Diverse APOBEC Proteins. Current Biology, 14, 1392-1396.
http://dx.doi.org/10.1016/j.cub.2004.06.057 - 28. Henry, M., Guetard, D., Suspene, R., Rusniok, C., Wain-Hobson, S. and Vartanian, J.P. (2009) Genetic Editing of HBV DNA by Monodomain Human APOBEC3 Cytidine Deaminases and the Recombinant Nature of APOBEC3G. PLoS ONE, 4, e4277.
http://dx.doi.org/10.1371/journal.pone.0004277 - 29. Thielen, B.K., McNevin, J.P., McElrath, M.J., Hunt, B.V., Klein, K.C. and Lingappa, J.R. (2010) Innate Immune Signaling Induces High Levels of TC-Specific Deaminase Activity in Primary Monocyte-Derived Cells through Expression of APOBEC3A Isoforms. The Journal of Biological Chemistry, 285, 27753-27766.
http://dx.doi.org/10.1074/jbc.M110.102822 - 30. Roberts, S.A., Sterling, J., Thompson, C., Harris, S., Mav, D., Shah, R., et al. (2012) Clustered Mutations in Yeast and in Human Cancers Can Arise from Damaged Long Single-Strand DNA Regions. Molecular Cell, 46, 424-435.
http://dx.doi.org/10.1016/j.molcel.2012.03.030 - 31. Nakagawa, K., Taya, Y., Tamai, K. and Yamaizumi, M. (1999) Requirement of ATM in Phosphorylation of the Human p53 Protein at Serine 15 Following DNA Double-Strand Breaks. Molecular and Cellular Biology, 19, 2828-2834.
http://dx.doi.org/10.1128/MCB.19.4.2828 - 32. Lavin, M.F. (2007) ATM and the Mre11 Complex Combine to Recognize and Signal DNA Double-Strand Breaks. Oncogene, 26, 7749-7758.
http://dx.doi.org/10.1038/sj.onc.1210880 - 33. Di Domenico, E.G., Romano E., Del Porto, P. and Ascenzioni, F. (2014) Multifunctional Role of ATM/Tel1 Kinase in Genome Stability: From the DNA Damage Response to Telomere Maintenance. BioMed Research International, 2014, Article ID: 787404.
http://dx.doi.org/10.1155/2014/787404 - 34. Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W. and Lipman, D.J. (1997) Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs. Nucleic Acids Research, 25, 3389-3402.
http://dx.doi.org/10.1093/nar/25.17.3389 - 35. Marchler-Bauer, A., Lu, S., Anderson, J.B., Chitsaz, F., Derbyshire, M.K., DeWeese-Scott, C., et al. (2011) CDD: A Conserved Domain Database for the Functional Annotation of Proteins. Nucleic Acids Research, 39, D225-D229.
http://dx.doi.org/10.1093/nar/gkq1189 - 36. Thompson, J.D., Higgins, D.G. and Gibson, T.J. (1994) CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Research, 22, 4673-4680.
http://dx.doi.org/10.1093/nar/22.22.4673 - 37. Jaroszewski, L., Rychlewski, L., Li, Z., Li, W. and Godzik, A. (2005) FFAS03: A Server for Profile-Profile Sequence Alignments. Nucleic Acids Research, 33, W284-W288.
http://dx.doi.org/10.1093/nar/gki418 - 38. Jaroszewski, L., Li, Z., Cai, X.H., Weber, C. and Godzik, A. (2011) FFAS Server: Novel Features and Applications. Nucleic Acids Research, 39, W38-W44.
http://dx.doi.org/10.1093/nar/gkr441 - 39. Rice, P., Longden, I. and Bleasby, A. (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics, 16, 276-277.
http://dx.doi.org/10.1016/S0168-9525(00)02024-2 - 40. Diella, F., Cameron, S., Gemünd, C., Linding, R., Via, A., Kuster, B., Sicheritz-Pontén, T., Blom, N. and Gibson, T.J. (2004) Phospho.ELM: A Database of Experimentally Verified Phosphorylation Sites in Eukaryotic Proteins. BMC Bioinformatics, 5, 79.
http://dx.doi.org/10.1186/1471-2105-5-79 - 41. Dinkel, H., Chica, C., Via, A., Gould, C.M., Jensen, L.J., Gibson, T.J. and Diella, F. (2011) Phospho.ELM: A Database of Phosphorylation Sites-Update 2011. Nucleic Acids Research, 39, D261-D267.
http://dx.doi.org/10.1093/nar/gkq1104 - 42. Gnad, F., Gunawardena, J. and Mann, M. (2011) PHOSIDA 2011: The Posttranslational Modification Database. Nucleic Acids Research, 39, D253-D260.
http://dx.doi.org/10.1093/nar/gkq1159 - 43. Zvárová, J. (2001) Biomedical Statistics. I. The Fundamentals of Statistics for Biomedical Fields. Karolinum, Prague.
- 44. Mefford, J. (2012) Bayesian Statistics and Bias Analysis.
http://slideplayer.com/slide/8525698/ - 45. Lowry, R. (2001) 2x2 Contingency Table.
http://vassarstats.net/tab2x2.html - 46. Dembo, A., Karlin, S. and Zeitouni, O. (1994) Limit Distribution of Maximal Non-Aligned Two-Sequence Segmental Score. The Annals of Probability, 22, 2022-2039.
http://dx.doi.org/10.1214/aop/1176988493 - 47. Karlin, S. and Altschul, S.F. (1990) Methods for Assessing the Statistical Significance of Molecular Sequence Features by Using General Scoring Schemes. Proceedings of National Academy of Sciences of the United States of America, 87, 2264-2268.
http://dx.doi.org/10.1073/pnas.87.6.2264 - 48. Potter, M., Padlan, E. and Rudikoff, S. (1976) Localized Deletion-Insertion Mutations: A Major Factor in the Evolution of Immunoglobulin Structural Variability. The Journal of Immunology, 117, 626-629.
- 49. Kabat, E.A., Wu, T.T., Perry, H.M., Gottesman, K.S. and Foeller, C. (1991) Sequences of Proteins of Immunological Interest. NIH publication No. 91-3242, Bethesda.
- 50. Schmid, F.X. (1993) Prolyl Isomerase: Enzymatic Catalysis of Slow Protein-Folding Reactions. Annual Review of Biophysics and Biomolecular Structure, 22, 123-142.
http://dx.doi.org/10.1146/annurev.bb.22.060193.001011 - 51. Snorek, M. (2002) Neural Networks and Neurocomputers. CVUT Publishing, Prague.
- 52. Waegeman, W., De Baets, B. and Boullart, L. (2008) Learning Layered Ranking Functions with Structured Support Vector Machines. Neural Networks, 21, 1511-1523.
http://dx.doi.org/10.1016/j.neunet.2008.07.008 - 53. Gao, S., Zhang, N., Duan, G.Y., Yang, Z., Ruan, J.S. and Zhang, T. (2009) Prediction of Function Changes Associated with Single-Point Protein Mutations Using Support Vector Machines (SVMs). Human Mutation, 30, 1161-1166.
http://dx.doi.org/10.1002/humu.21039 - 54. Rangwala, H., Kauffman, C. and Karypis, G. (2009) SvmPRAT: SVM-Based Protein Residue Annotation Toolkit. BMC Bioinformatics, 10, 439.
http://dx.doi.org/10.1186/1471-2105-10-439 - 55. Ansari, H.R. and Raghava, G.P. (2010) Identification of Conformational B-Cell Epitopes in an Antigen from Its Primary Sequence. Immunome Research, 6, 6.
http://dx.doi.org/10.1186/1745-7580-6-6 - 56. Oprea, M., Cowell, L.G. and Kepler, T.B. (2001) The Targeting of Somatic Hypermutation Closely Resembles That of Meiotic Mutation. The Journal of Immunology, 166, 892-899.
http://dx.doi.org/10.4049/jimmunol.166.2.892 - 57. Yoder, J.A., Turner, P.M., Wright, P.D., Wittamer, V., Bertrand, J.Y., Traver, D. and Litman, G.W. (2010) Developmental and Tissue-Specific Expression of NITRs. Immuno-genetics, 62, 117-122.
http://dx.doi.org/10.1007/s00251-009-0416-5 - 58. Wilson, P., Liu, Y.J., Banchereau, J., Capra, J.D. and Pascual, V. (1998) Amino Acid Insertions and Deletions Contribute to Diversify the Human Ig Repertoire. Immunological Reviews, 162, 143-151.
http://dx.doi.org/10.1111/j.1600-065X.1998.tb01437.x - 59. Papavasiliou, F.N. and Schatz, D.G. (2000) Cell-Cycle-Regulated DNA Double-Stranded Breaks in Somatic Hypermutation of Immunoglobulin Genes. Nature, 408, 216-221.
http://dx.doi.org/10.1038/35041599 - 60. Shen, H.M. (2007) Activation-Induced Cytidine Deaminase Acts on Double-Strand Breaks in Vitro. Molecular Immunology, 44, 974-983.
http://dx.doi.org/10.1016/j.molimm.2006.03.015 - 61. Holliday, R. (1995) Understanding Ageing. Cambridge University Press, New York.
http://dx.doi.org/10.1017/CBO9780511623233 - 62. Crawford, T.O., Skolasky, R.L., Fernandez, R., Rosquist, K.J. and Lederman, H.M. (2006) Survival Probability in Ataxia Telangiectasia. Archives of Disease in Childhood, 91, 610-611.
http://dx.doi.org/10.1136/adc.2006.094268 - 63. Boohaker, R.J. and Xu, B. (2014) The Versatile Functions of ATM Kinase. Biomedical Journal, 37, 3-9.
http://dx.doi.org/10.4103/2319-4170.125655 - 64. Ohlin, M. and Borrebaeck, C.A.K. (1998) Insertions and Deletions in Hypervariable Loops of Antibody Heavy Chains Contribute to Molecular Diversity. Molecular Immunology, 35, 233-238.
http://dx.doi.org/10.1016/S0161-5890(98)00030-3 - 65. Jeng, Y.M., Wu, M.Z., Mao, T.L., Chang, M.H. and Hsu, H.C. (2000) Somatic Mutations of Beta-Catenin Play a Crucial Role in the Tumorigenesis of Sporadic Hepatoblastoma. Cancer Letters, 152, 45-51.
http://dx.doi.org/10.1016/S0304-3835(99)00433-4 - 66. Radivojac, P., Baenziger, P.H., Kann, M.G., Mort, M.E., Hahn, M.W. and Mooney, S.D. (2008) Gain and Loss of Phosphorylation Sites in Human Cancer. Bioinformatics, 24, i241-i247.
http://dx.doi.org/10.1093/bioinformatics/btn267 - 67. Hu, Z., Wan, X., Hao, R., Zhang, H., Li, L., Li, L., et al. (2015) Phosphorylation of Mutationally Introduced Tyrosine in the Activation Loop of HER2 Confers Gain-of-Function Activity. PLoS ONE, 10, e0123623.
http://dx.doi.org/10.1371/journal.pone.0123623 - 68. Ndegwa, N., Côté, R.G., Ovelleiro, D., D’Eustachio, P., Hermjakob, H., Vizcaíno, J.A. and Croft, D. (2011) Critical Amino Acid Residues in Proteins: A BioMart Integration of Reactome Protein Annotations with PRIDE Mass Spectrometry Data and COSMIC Somatic Mutations. Database, 2011, bar047.
http://dx.doi.org/10.1093/database/bar047 - 69. Reimand, J., Wagih, O. and Bader, G.D. (2013) The Mutational Landscape of Phosphorylation Signaling in Cancer. Scientific Reports, 3, 2651.
http://dx.doi.org/10.1038/srep02651 - 70. Soucek, P., Gut, I., Trneny, M., Skovlund, E., Kristensen, T., Børresen-Dale, A.-L. and Kristensen, V.N. (2003) Multiplex Single-Tube Screening for Mutations in the Nijmegen Breakage Syndrome (NBS1) Gene in Hodgkin’s and Non-Hodgkin’s Lymphoma Patients of Slavic Origin. European Journal of Human Genetics, 11, 416-419.
http://dx.doi.org/10.1038/sj.ejhg.5200972 - 71. Soucek, P., Borovanova, T., Pohlreich, P., Kleibl, Z. and Novotny, J. (2007) Role of Single Nucleotide Polymorphisms and Haplotypes in BRCA1 in Breast Cancer: Czech Case-Control Study. Breast Cancer Research and Treatment, 103, 219-224.
http://dx.doi.org/10.1007/s10549-006-9367-9 - 72. Mohelnikova-Duchonova, B., Havranek, O., Hlavata, I., Foretova, L., Kleibl, Z., Pohlreich, P. and Soucek, P. (2010) CHEK2 Gene Alterations in the Forkhead-Associated Domain, 1100delC and del5395 Do Not Modify the Risk of Sporadic Pancreatic Cancer. Cancer Epidemiology, 34, 656-658.
http://dx.doi.org/10.1016/j.canep.2010.06.008 - 73. Smith, G.P. (1985) Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science, 228, 1315-1317.
http://dx.doi.org/10.1126/science.4001944 - 74. Schmitz, R., Baumann, G. and Gram, H. (1996) Catalytic Specificity of Phosphotyrosine Kinases Blk, Lyn, c-Src and Syk as Assessed by Phage Display. Journal of Molecular Biology, 260, 664-677.
http://dx.doi.org/10.1006/jmbi.1996.0429 - 75. Fack, F., Hugle-Dorr, B., Song, D., Queitsch, I., Petersen, G. and Bautz, E.K.F. (1997) Epitope Mapping by Phage Display: Random Versus Gene-Fragment Libraries. Journal of Immunological Methods, 206, 43-52.
http://dx.doi.org/10.1016/S0022-1759(97)00083-5 - 76. Reche, P.A. and Reinherz, E.L. (2005) PEPVAC: A Web Server for Multi-Epitope Vaccine Development Based on the Prediction of Supertypic MHC ligands. Nucleic Acids Research, 33, W138-W141.
http://dx.doi.org/10.1093/nar/gki357 - 77. Shehzadi, A., Rehman, S. and Indrees, M. (2011) Promiscuous Prediction and Conservancy Analysis of CTL Binding Epitopes of HCV 3a Viral Proteome from Punjab Pakistan: An in Silico Approach. Virology Journal, 8, 55.
http://dx.doi.org/10.1186/1743-422X-8-55 - 78. Kubrycht, J., Sigler, K. and Soucek, P. (2012) Virtual Interactomics of Proteins from Biochemical Standpoint. Molecular Biology International, 2012, Article ID: 976385.
http://dx.doi.org/10.1155/2012/976385 - 79. Jing, H., Takagi, J., Liu, J.H., Lindgren, S., Zhang, R.G., Joachimiak, A., Wang, J.H. and Springer, T.A. (2002) Archaeal Surface Layer Proteins Contain Beta Propeller, PKD, and Beta Helix Domains and Are Related to Metazoan Cell Surface Proteins. Structure, 10, 1453-1464.
http://dx.doi.org/10.1016/S0969-2126(02)00840-7
NOTES

*Corresponding author.