Computational Molecular Bioscience
Vol.1 No.1(2011), Article ID:16495,6 pages DOI:10.4236/cmb.2011.11001
Structural Characterization of the D-Tyr-tRNATyr Deacylase from Bacillus lichenformis, an Organism of Great Industrial Importance
Department of Biochemistry and Biophysics, University of Kalyani, Kalyani, India
E-mail: angshuman_bagchi@yahoo.com
Received November 5, 2011; revised December 2, 2011; accepted December 12, 2011
Keywords: Molecular Modeling, Molecular Docking, Molecular Dynamics, Industrially Important Microorganism, Ester hydrolysis, D-Tyr-tRNA Deacylase
Abstract
A new class of enzyme was established that hydrolyze the ester bond between D-Tyr bound onto its cognate t-RNA. The enzyme is called D-Tyr-tRNA deacylase. The three dimensional structure of the D-Tyr-tRNA deacylase from industrially important microorganism Bacillus lichenformis DSM13 was predicted by comparative modeling approach. Since the protein acts as a dimer a dimeric model of the enzyme was constructed. The interactions responsible for dimerization were also predicted. With the help of docking and molecular dynamics simulations the favourable binding mode of the enzyme was predicted. The probable biochemical mechanism of the hydrolysis process was elucidated. This study provides a rational framework to interpret the molecular mechanistic details of the removal of toxic D-Tyr-tRNA from the cells of industrially important microorganism Bacillus lichenformis DSM13 using the enzyme D-Tyr-tRNA deacylase.
1. Introduction
It has been observed previously that several organisms (for example E. coli and Bacillus lichenformis DSM13) possess an enzyme called D-Tyr-tRNA deacylase which is capable of accelerating the hydrolysis of the ester linkage between D-Tyr and its cognate t-RNA molecule [1,2]. However, the linkage between L-Tyr and its cognate t-RNA remain unaffected by the activity of the enzyme [2,3]. Involvement of the enzyme D-Tyr-tRNA deacylase in hydrolyzing the above mentioned ester linkage helps protecting the microbial organisms from the toxic effects of the D-Tyr [2,3]. However to date the detailed structural information about the mechanism of this hydrolysis reaction by the enzyme is not available.
In the present study an attempt has been made to predict the three dimensional structure of D-Tyr-tRNA deacylase from Bacillus lichenformis (Blich). Blich is an organism with great industrial potential [4]. The organism is a non-pathogenic organism and is used extensively for large scale industrial production of exoenzymes. Subtilisins that have their primary application as additives to household detergents are also manufactured from Blich. The complete genome sequence of Blich has recently been annotated [4].
The three dimensional structure of the protein has been predicted by comparative modeling approach. The active form of the protein is a dimer [2]. Therefore a dimeric model has been generated and the favourable binding interactions between the individual monomers have been predicted to establish the stability of the protein in the dimeric form. The binding site of the protein has been established and compared to other well defined proteins of the same family. The three dimensional coordinates of the predicted dimeric model of the protein have been used to dock the substrate D-Tyr-tRNA onto the protein. The docked structure so formed has been subjected to molecular dynamics calculations in order to establish the interactions between the substrate and the enzyme. From this analysis the molecular mechanistic details of the hydrolyzing activity of the protein D-Tyr-tRNA deacylase have been predicted. Since this is first report regarding the structural interactions of the D-Tyr-tRNA deacylase (from Blich) with its substrate D-Tyr-tRNA this study may shed light in elucidating the involvements of the various amino acid residues that are involved in the process. Since Blich is an industrially important microorganism, it is necessary to eliminate the toxic effects of the D-Tyr molecules for the proper growth and development of the organism. From that point of view this study would be important.
2. Materials and Methods
2.1. Sequence Analysis and Homology Modeling of Monomeric D-Tyr-tRNA Deacylase
The amino acid sequence of D-Tyr-tRNA deacylase of Blich was obtained from Entrez database (Accession No. NC_006322). The amino acid sequence was used to search Brookhaven Protein Data Bank (PDB) [5] using the software BLAST [6] for finding suitable template for homology modeling. The BLAST search result revealed that the protein had 52% sequence identity with X-ray crystal structure of D-Tyr-tRNA deacylase from E. coli (pdb code: 1JKE).
The three dimensional structure of the protein was built using the corresponding crystal structure as template. Homology modeling was performed using the program Modeller [7] that is incorporated in the Homology module of Insight II [Accelrys, San Diego, CA, USA] on a Silicon Graphics Indigo II workstation.
The modeled structure of D-Tyr-tRNA deacylase was then superimposed on the crystal template without altering the coordinate system of atomic positions in the template. The r.m.s deviation for the superimposition was found to be 0.5 Å. The mode of superimposition is presented in Figure 1. The model of the protein so generated was energy minimized fixing the backbones to ensure proper interactions. Conjugate gradient (CG) method was employed for minimization with the consistent valence force field (CVFF) [8] using the program DISCOVER until the structure reached the final derivative of 0.001 kcal/mol.
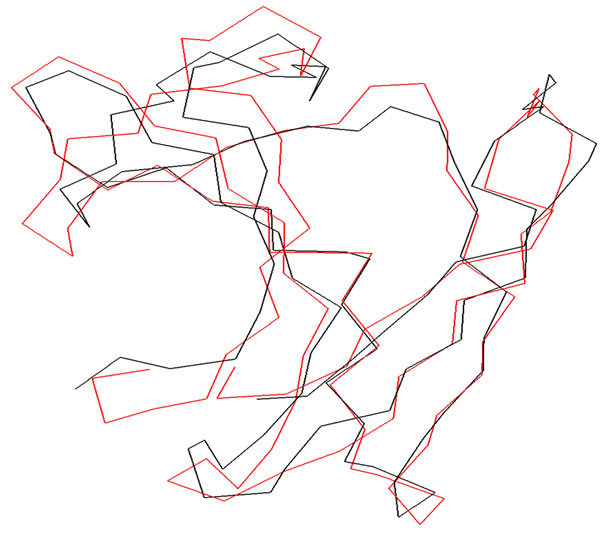
Figure 1. Superimposition of the α-carbon backbones of DTyr-tRNA deacylase (Black) on 1JKE (Red).
2.2. Validation of the Model
Regarding the main chain properties of the modeled protein, no considerable bad contacts nor Cα tetrahedron distortion nor hydrogen bond energy problems were found. Moreover, the average G factor, the measure of the normality degree of the protein properties, was of −1.04, which is inside the permitted values for homology models. Furthermore, there were no distortions of the side chain torsion angles found. The Z-scores calculated using the software PROSA 2003 [9] showed that the predicted model of the protein was well inside the range of a typical native structure [10]. The residue profiles of the three dimensional model was further checked by VERIFY3D [11]. PROCHECK [12] analysis was performed in order to assess the stereo-chemical qualities of the three dimensional model and Ramachandran plots [13] were drawn. No residues were found to be present in the disallowed regions of the Ramachandran plots.
2.3. Formation of the Dimeric Model of the Protein
It was known that the active form of D-Tyr-tRNA deacylase is a dimer [2]. In order to generate the dimeric model of the protein two monomeric models of D-Tyr-tRNA deacylase were superimposed separately on the corresponding monomeric units of the crystal structure and subsequently merged the two monomers together to form the dimeric model of D-Tyr-tRNA deacylase. The Biopolymer module of InsightII was used for the purpose. The r.m.s deviation for superimposition for each monomer was 0.5Å. The dimeric model of D-Tyr-tRNA deacylase thus built was subjected for energy minimization by CG using the program DISCOVER (MSI/Accelrys) with CVFF until the structure reached the final derivative of 0.001 kcal/mol. Procheck analysis was performed in order to identify the stereo chemical qualities of the final model and Ramachandran plots were drawn. Residue profiles were checked with Verify 3D.
2.4. Docking and Molecular Dynamics Calculations
To study the interaction between D-Tyr-tRNA deacylase and its substrate D-Tyr-tRNA, a search in the whole PDB was done to obtain the crystal structure of D-Tyr-tRNA. But none was found to be available. The crystal structure of L-Tyr-tRNA (pdb code: 1J1U) was found. The L-Tyr molecule of the crystal was converted to D-form using the Biopolymer module of Insight II. The model of DTyr-tRNA so formed was subjected to energy refinement using the program DISCOVER (MSI/Accelrys) with CVFF until the structure reached the final derivative of 0.001 kcal/mol. The model of the D-Tyr-tRNA deacylase was then docked to the D-Tyr-tRNA using the Patchdock server [14]. The docked structures, that yielded the best score were selected and analyzed visually using the software Insight II. Molecular dynamics (MD) simulations were performed on the docked structures to predict the favourable binding interactions. The docked complexes of D-Tyr-tRNA deacylase and its substrate D-Tyr-tRNA, were solvated by a 10 Å water layer by the program SOAK of Insight II. The resulting systems were subsequently energy minimized in two steps. In the first the step, the whole protein along with its substrate was kept fixed and in the next round of minimization backbones of the complex was fixed to perform rigorous minimization. In the next step of energy minimization, the whole system (i.e., both the protein with its substrate and water) was allowed to move. The minimization process was done initially by Steepest Descent (SD) and then by CG using CVFF with the help of the program DISCOVER3 of Insight II.
PROCHECK analysis of the complex showed that no residues were in the disallowed regions of the Ramachandran plot. The minimized systems were equilibrated for period of 210 ps with constant volume and temperature (NVT ensemble) through the velocity verlet integrator [15]. The temperature was kept at 298K, as Blich is a mesophilic bacteria [4]. The time step for integration was 1fs using the RATTLE algorithm, a velocity version of SHAKE [16]. The last 120 ps trajectories were analyzed saving the coordinates at every 0.2 ps interval. All the simulations were performed with CVFF force fields; for non-bonded calculations the cell multipole method along with a dielectric constant of 1 was used. The stereo-chemical qualities of the final structures were analyzed with PROCHECK.
2.5. Calculations of the Interactions
To find out the interactions responsible for the dimerization of the protein as well as between the protein and its substrate (i.e., D-Tyr-tRNA deacylase and its substrate D-Tyr-tRNA), What If software package [17] as well as the Biopolymer module of Insight II were used.
3. Results and Discussion
3.1. Description of the Structure of Monomeric D-Tyr-tRNA Deacylase
D-Tyr-tRNA deacylase protein is a 147 amino acid residues long protein. The modeled structure is identical with the X-ray crystal structure of D-Tyr-tRNA decaylase from E. coli (pdb code: 1JKE). The predicted structure consists of a mixture of alpha helices and beta strands culminating in two beta sheets approximately 45˚ to each other (Figure 2). The N-terminal part of the protein starts with three beta sheets (amino acid residues 2 to 15, 18 to
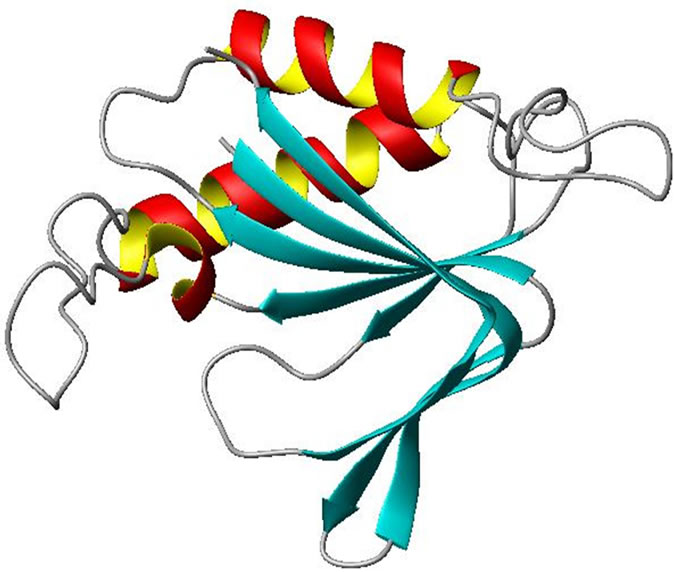
Figure 2. Ribbon representation of monomeric D-Tyr-tRNA deacylase. α-helices and β-sheets are shown as helices (Red and Yellow) and ribbons (Cyan) respectively. The rest are shown as loops (White). The figure was prepared by MOL SCRIPT [19].
23 and 26 to 32 respectively) connected to an alpha helix (amino acid residue 39 to 51) by short turn regions. The C-terminal part of the protein is made up of one alpha helix (amino acid residues 99 to 114) and two beta strands (amino acid residues 120 to 124 and 130 to 144 respectively) along with several turns and bends. A short beta sheet (amino acid residues 72 to 77) and 310 helix (amino acid residues 79 to 83) along with several turns connect the C-terminal part of the protein with the N-terminal region.
3.2. Dimerization of Monomeric D-Tyr-tRNA Deacylase
It has been a well established fact that the active form of the protein is a dimer. The two monomers show extensive intermolecular contact in the dimeric form. The contact surface area is of the order of 1100 Å. The interactions between the monomers are basically hydrogen bonding (H-bonding). In the dimeric form of the molecule there is formation of a six stranded antiparallel beta sheet which covers the N-terminal part of the protein (Figure 3). The amino acid residues which are involved in the H-bonding interactions in the dimerization of the protein are presented in Table 1. There are also hydrophobic packing interactions between the non-polar amino acid residues in the dimeric form of the protein. It is interesting to note that the dimerization of the protein creates a positively charged cluster of amino acid residues on the molecular surface of the protein which may be an essential criterion for the activity of the molecule as a decylase (Figure 4).
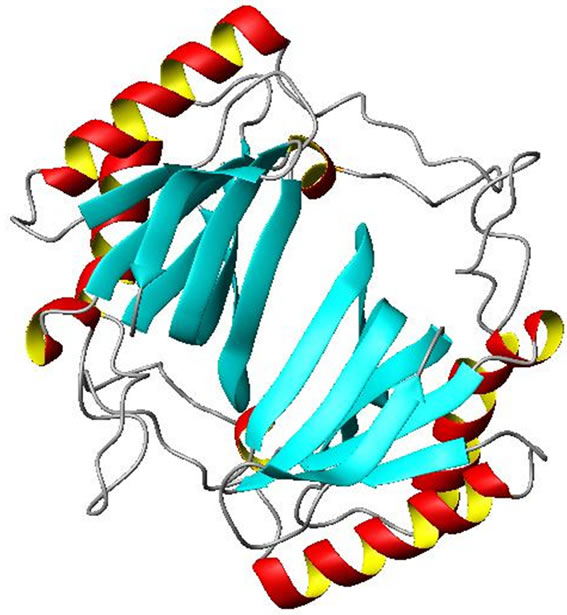
Figure 3. Ribbon representation of dimeric D-Tyr-tRNA deacylase. α-helices and β-sheets are shown as helices (Red and Yellow) and ribbons (Cyan) respectively. The rest are shown as loops (White). The figure was prepared by MOL SCRIPT [19].
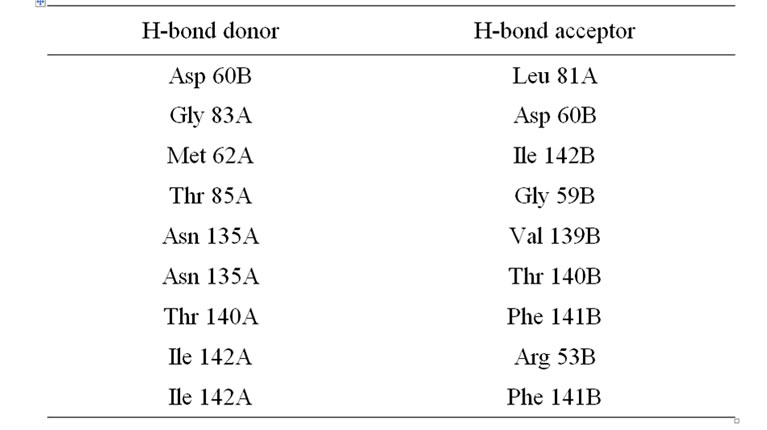
Table 1. Inter-subunit hydrogen bonding interactions in DTyr-tRNA deacylase*.
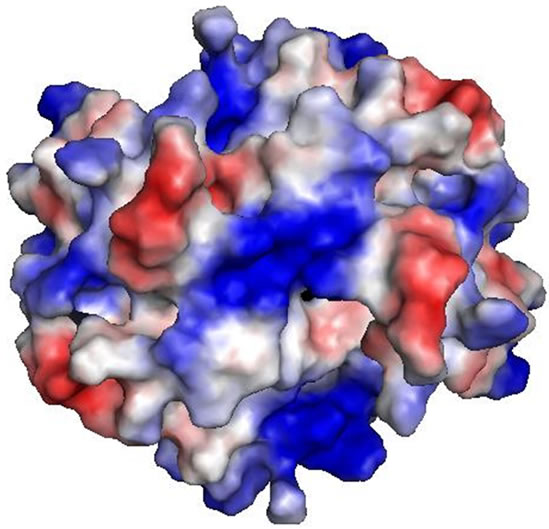
Figure 4. Electrostatic surface of D-Tyr-tRNA deacylase showing the distribution of charges on the surface. At the center of the surface there is a positively charged cluster which is essential for the activity of the protein.
3.3. Comparison with Other D-Tyr-tRNA Deacylase Protein
The D-Tyr-tRNA deacylase has strong structural similarities with other known proteins of the same family as, viz., Aqufex (2DBO), Hemophilus influnzae (1J7G). The r.m.s. deviations of the superimpositions were fond to be 0.5Å , 0.7 Å respectively. The mode of superimposition is similar to that with the template 1JKE (Figure 1).
3.4. Docking of D-Tyr-tRNA Deacylase with Its Substrate D-Tyr-tRNA
In order to predict the favourable binding interactions of the D-Tyr-tRNA deacylase with its substrate D-Tyr-tRNA, the three dimensional coordinates of the two structures were subjected to molecular docking analyses. It was observed that the D-Tyr-tRNA binds near the positively charged cluster on the surface of the molecule. The negative charges on the surface of the tRNA molecule, specifically the sugar-phosphate backbone of the CCA triplet on the tRNA molecule, help in the binding by chargecharge interaction with the protein. The side chains of Lys61, Arg 90, Gln 101 and Arg 124 from the protein are specifically involved in the binding of the substrate to it. The covalent bond between the carboxyl group of D-Tyr and hydroxyl group of the tRNA molecule is then cleaved by the positive atmosphere created by the aforementioned amino acid residues as the polar covalent bond between the O-atom of the carboxyl group of D-Tyr and the Catom of the t-RNA molecule is not strong enough to withstand the strong positive influence created by the side chains of the basic amino acid residues of the protein. Interestingly when the crystal structure of the L-Tyr-tRNA was used for the docking purpose the binding was not found to be as in the previous case. In this case the tRNA portion of the substrate L-Tyr-tRNA gets bound to the protein keeping the L-Tyr molecule away from the positively charged cluster of the enzyme thus keeping the L-Tyr molecule unaffected by the enzyme. The binding interactions between D-Tyr-tRNA and L-Tyr-tRNA with the enzyme were calculated separately using the Structural Thermodynamics Calculator (STC) program [18]. The results are presented in Table 2. The calculated free energy change on binding of the D-isomer is more than that of the L-isomer with the enzyme. This result also points to the fact that the enzyme has more affinity towards the D-isomer than towards the L-isomer.
4. Conclusions
In this study, an attempt has been made to elucidate the structural basis of the involvements of D-Tyr-tRNA deacylase to bind with its substrate D-Tyr-tRNA, and thereby
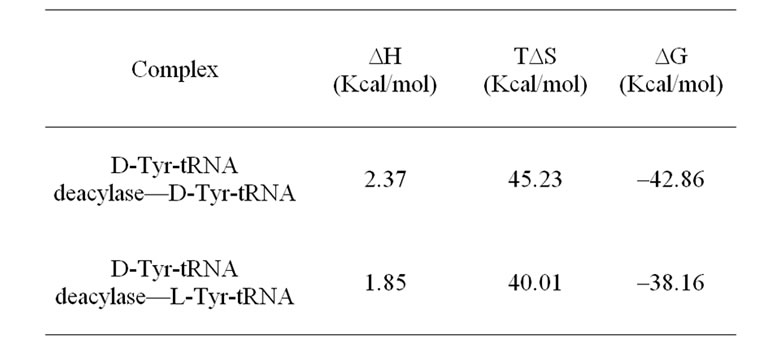
Table 2. Comparative binding interactions of the D-TyrtRNA and L-Tyr-tRNA molecules with the D-Tyr-tRNA deacylase at T = 300 K.
cleaving the ester bond to release the free D-Tyr molecule. The predicted three-dimensional structures of D-Tyr-t RNA deacylase has been described. The putative active site geometry of the protein has been established. The protein acts in the dimeric form. Therefore a dimeric model of the protein has been generated and the binding interactions for the dimerization process have been calculated. The docking of D-Tyr-tRNA with D-Tyr-tRNA decaylase has been performed in order to predict the molecular mechanism of the hydrolysis process by the enzyme. The structural basis of the formation of D-Tyr-tRNA decaylase—D-Tyr-tRNA and D-Tyr-tRNA deacylase—L-TyrtRNA complexes has also been demonstrated to analyze the binding modes of the protein and to predict the plausible biochemical hydrolysis reaction pathway. Results from this study will be important for the understanding of the structure of D-Tyr-tRNA deacylase. This model provides a rational framework for designing experiments aimed at determining the contribution of various amino acid residues in the complex formation between the D-TyrtRNA deacylase and D-Tyr-tRNA as well as dimerization of the protein in this industrially important microorganism Blich. The results from this study may be useful for proper maintenance of Blich.
5. Acknowledgements
The author is thankful to Dr. T. C. Ghosh of Bioinformatics Center, Bose Institute, Kankurgachi, Kolkata—54, India, for rendering help to prepare the manuscript.
6. References
[1] R. Calender and P. Berg, “Valyl t-RNA Synthetase from Staphylococcus aureaus,” Biochemistry, Vol. 5, No. 5, 1966, pp. 1681-1690.
[2] M. L. F-Fioni, E. Schmitt, J. Soutourina, P. Plateau, Y. Mechulam and S. Blanquet, “Structure of Crystalline DTyr-t-RNATyr Deacylase,” Journal of Biological Chemistry, Vol. 14, 2001, pp. 47285-47290.
[3] J. Soutourina, P. Plateau and S. Blanquet, “Metabolism of D-Aminoacyl-tRNAs in Escherichia coli and Saccharomyces cerevisiae Cells,” Journal of Biological Chemistry, Vol. 275, 2000, pp. 32535-32542. doi:10.1074/jbc.M005166200
[4] B. Veith, C. Herzberg, S. Steckel, J. Feesche, K. H. Maurer, P. Ehrenreich, S. Baumer, A. Henne, H. Liesegang, R. Merkl, A. Ehrenreich and G. Gottschalk, “The Complete Genome Sequence of Bacillus licheniformis DSM13, an Organism with Great Industrial Potential,” Journal of Molecular Microbiology and Biotechnology, Vol. 7, No. 4, 2004, pp. 204-211. doi:10.1159/000079829
[5] M. H. Berman, J. Westbrook, J. Feng, G. Gillilang, T. N. Bhat, H. Weissig and I. N. Sindyalov, “The Protein Data Bank,” Nucleic Acids Research, Vol. 28, No. 1, 2000, pp. 235- 242. doi:10.1093/nar/28.1.235
[6] S. F. Altschul, W. Gisli, W. Miller, E. W. Mayer and D. J. Lipman, “Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs,” Nucleic Acids Research, Vol. 25, No. 17, 1997, pp. 3389-3402. doi:10.1093/nar/25.17.3389
[7] A. Sali and T. L. Blundell, “Comparative Protein Modeling by Satisfaction of Spatial Restraints,” Journal of Molecular Biology, Vol. 234, No. 3, 1993, pp. 779-815. doi:10.1006/jmbi.1993.1626
[8] P. Dauber-Osguthorpe, V. A. Roberts, D. J. Osguthorpe, J. Wolff, M. Genest and A. T. Hagler, “Structure and Energetics of Ligand Binding to Proteins: Escherichia coli Dihydrofolate Reductase Trimethoprim, a Drug Receptor System,” Proteins, Vol. 4, No. 1, 1988, pp. 31-47. doi:10.1002/prot.340040106
[9] M. J. Sippl, “Recognition of Errors in Three-Dimensional Structures of Proteins,” Proteins, Vol. 17, No. 4, 1993, pp. 355-362. doi:10.1002/prot.340170404
[10] M. Wiederstein, P. Lackner and F. Kieberger, “Direct in Silico Mutagenesis,” in: S. Brankmann and A. Schwienhorst, Eds., Evolutionary Methods in Biotechnology, WileyVCH, New York, 2004
[11] D. Eisenberg, R. Luthy and J. U. Bowie, “VERIFY3D: Assessment of Protein Models with Three-Dimensional Profiles,” Methods in Enzymology, Vol. 277, 1997, pp. 396-404. doi:10.1016/S0076-6879(97)77022-8
[12] R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, “PROCHECK: A Program to Check the Stereochemistry of Protein Structures,” Journal of Applied Crystallography, Vol. 26, 1993, pp. 283-291. doi:10.1107/S0021889892009944
[13] G. N. Ramachandran and V. Sashisekharan, “Conformation of Polypeptides and Proteins,” Advances in Protein Chemistry, Vol. 23, 1968, pp. 283-438. doi:10.1016/S0065-3233(08)60402-7
[14] D. Schneidman-Duhovny, Y. Inbar, V. Polak, M. Shatsky, I. Halperin, H. Benyamini, A. Barzilai, O. Dror, N. Haspel, R. Nussinov and H. J. Wolfson, “Taking Geometry to Its Edge: Fast Unbound Rigid (and Hinge-Bent) Docking,” Proteins, Vol. 52, No. 1, 2003, pp. 107-112. doi:10.1002/prot.10397
[15] L. Verlet, “Computer Experiments on Classical Fluids. I. Thermodynamical Properties of Lenard-Jones Molecules,” Physical Review, Vol. 159, No. 1, 1967, pp. 98-103. doi:10.1103/PhysRev.159.98
[16] J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, “Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of nAlokanes,” Advances in Protein Chemistry, Vol. 23, No. 3, 1977, pp. 327-331. doi:10.1016/0021-9991(77)90098-5
[17] G. Vriend, “WHAT IF: A Molecular Modeling and Drug Design Program,” Journal of Molecular Graphics, Vol. 8, No. 1, 1990, pp. 52-58. doi:10.1016/0263-7855(90)80070-V
[18] P. Lavigne, J. R. Bagu, R. Boyko, L. Willard, C. F. Holmes and B. D. Sykes, “Structure-Based Thermodynamic Analysis of the Dissociation of Protein Phosphatase-1 Catalytic Subunit and Microcystin-LR Docked Complexes,” Protein Science, Vol. 9, No. 2, 2000, pp. 252-264. doi:10.1110/ps.9.2.252
[19] P. J. Kraulis, “Molscript: A Program to Produce Both Detailed and Schematic Plots of Protein Structures,” Journal of Applied Crystallography, Vol. 24, No. 1991, pp. 946-950.