American Journal of Analytical Chemistry
Vol.3 No.10(2012), Article ID:23620,8 pages DOI:10.4236/ajac.2012.310089
Utilization of Dispersive Liquid-Liquid Microextraction Coupled with HPLC-UV as a Sensitive and Efficient Method for the Extraction and Determination of Oleanolic Acid and Ursolic Acid in Chinese Medicinal Herbs
School of Pharmacy, Shanxi Medical University, Taiyuan, China
Email: *bxh246@163.com, *hurrah100@sohu.com
Received August 18, 2012; revised September 19, 2012; accepted September 26, 2012
Keywords: Dispersive Liquid-Liquid Microextraction; High Performance Liquid Chromatography; Enrichment Factor; Triterpenic Acids; Chinese Medicinal Herbs
ABSTRACT
Isomeric triterpenic acids of oleanolic acid (OA) and ursolic acid (UA) both have very low ultraviolet absorption and always exist in the same plant, so the separation and simultaneous determination of them have been a difficult task. In this study, a sensitive method combining dispersive liquid-liquid microextraction (DLLME) with HPLC-UV was developed for the extraction and determination of OA and UA in traditional Chinese medicinal herbs (CMHs). Variables influencing DLLME such as type and volume of extraction solvent, volume of dispersive solvent, ionic strength, aqueous phase pH, extraction time, centrifugation speed and time, and sample volume were investigated and optimized. Under the optimum conditions, both OA and UA attained favorable extraction efficiencies with enrichment factors 1378 and 933, respectively. The linear dynamic ranges of 0.07 - 30.4 μg·mL–1 for OA and 0.08 - 33.6 μg·mL–1 for UA were obtained with square correlation coefficients of 0.9963. The detection limits of OA and UA were both 0.02 μg·mL–1. The method recoveries ranged between 88.2% - 116.2% for OA and 85.7% - 108.2% for UA with the RSDs (n = 5) lower than 8.6%. The proposed method was successfully applied to concentrate and simultaneously determine these two triterpenic acids in Hedyotis diffusa and Eriobotrya japonica samples.
1. Introduction
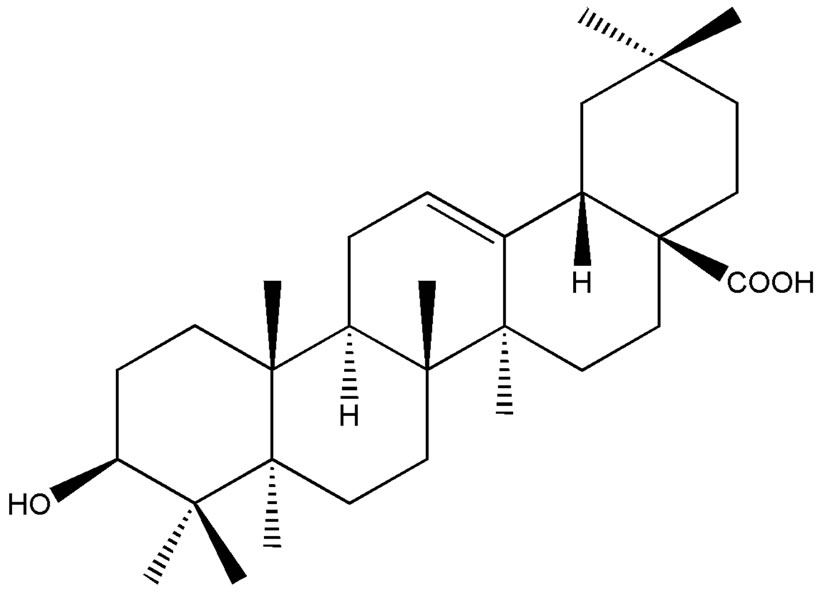
Oleanolic acid (OA) and ursolic acid (UA) are isomeric triterpenic acids with only one methyl’s position difference between them (Figure 1). They exist abundantly in the plant kingdom and always simultaneously exist in the same plant. Based on the closeness of their chemical structures, both components usually exert similar therapeutics and biological effects. They are well known for their hepatoprotective effects against both acute chemically induced liver injury and chronic liver fibrosis and cirrhosis [1]. Moreover, a variety of novel pharmacological effects produced by these triterpenoids have been reported, such as antioxidant [2,3], anti-inflammatory [4,5], antitumour [6,7], anti-HIV [8], antimicrobial [9,10], gastroprotective [11] and the antiobese potential [12,13]. Given their occurrence at low levels in many herbal medicines and very low UV absorptions, the determination of OA and UA has attracted increasing attentions in research of traditional Chinese medicinal herbs (CMHs).
At present, the separation and determination of the two components in plants are usually performed by using chromatographic method, such as Micellar Electrokinetic Capillary Chromatography (MECC) [14], Capillary Zone Electophoresis (CZE) [15], direct HPLC-UV detection [16] and HPLC-DAD [17], pre-column derivatization HPLC (pre-CD-HPLC) [18] and Iodine Derivatization then High Performance Thin-Layer Chromatography (IDHPTLC) [19]. However, these determination approaches suffer from some demerits either low sensitivity, poor precision or harsh derived reaction conditions. Thus, it is required that a simple and effective sample-preparation protocol should be designed to extract and preconcentrate then directly determine these two triterpenic acids from CMHs.
Dispersive liquid-liquid microextraction (DLLME), as a procedure of samples preparation inside “Green Chemistry” because of the small volumes of dissolvent employed [20], has attracted much attention in the recent years. DLLME is based on a ternary component solvent
 (a)
(a) (b)
(b)
Figure 1. Chemical structures of oleanolic acid (a) and ursolic acid (b).
system including disperser solvent, extraction solvent and aqueous sample containing interest analytes [21]. This new sample-preparation technique, that set extraction, purification and concentration of the target analytes in one step, has the following characteristics: simplicity of operation; high enrichment factor; low organic solvent consumption (generally a few to dozens of microliters); low labor cost and cheap equipment. Besides, it can achieve selective extraction and reduce matrix interfereence through the adjustment of solvent extraction of polarity or acidity and alkalinity [21]. Owing to above unique advantages, DLLME has been applied to the analyses of polycyclic aromatic hydrocarbons [22], organophosphorus pesticides [23], herbicides [24], cadmium [25] and cobalt [20,26] in water samples, cholesterol [27], amitraz [28] and cobalt [20] in food samples, psychotropic drugs [29], emodin and its metabolites [30] in urine samples. But so far, the method has been rarely applied in the extraction and concentration of active components in complex CMH samples [31], and there is no report that DLLME was used in determination of OA and UA.
In the present work, a DLLME prior to direct HPLCUV method was proposed for simultaneous extraction and determination of trace amounts of OA and UA in Hedyotis diffusa and Eriobotrya japonica samples. All variables affecting the enrichment were investigated and optimized. Under the optimum conditions, the advantages and disadvantages of the developed method and those of other methods for the extraction and determination of OA and UA were compared. It is found that DLLME coupled with HPLC is a simple, fast, sensitive, efficient and green method for the simultaneous analysis of OA and UA in CMH samples.
2. Experimental
2.1. Reagents
Standards of OA and UA were purchased from Chengdu must bio-technology Co. Ltd. (Purity ≥ 98%, Chengdu, China). Chloroform (CHCl3), carbon tetrachloride (CCl4), dichloromethane (CH2Cl2), 1, 2-dichloroethane (ClCH2 CH2Cl), chlorobenzene (C6H5Cl) were acquired from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Methanol of HPLC grade, ethanol, hydrochloric acid and phosphoric acid were purchased from Tianjin Chemical Reagent Factory (Tianjin, China). Other chemicals used were at least of analytical-reagent grade. Doubly distilled water was used throughout.
2.2. Preparation of Standard Solution and Real Samples
The mixture of stock solution of OA (0.20 mg·mL–1) and UA (0.26 mg·mL–1) was prepared in methanol and stored at 4˚C. 2 mL of stock solution was diluted to 5 mL with methanol as working solution before used.
CMHs of Hedyotis diffusa and Eriobotrya japonica were purchased from a local pharmacy (Taiyuan, China) and pulverized. 0.5 g of dry powder were accurately weighed and dissolved with 20 mL methanol, respectively. The obtained solutions were put in the ultrasonic bath for 40 min, and then filtered off the precipitate. The filtrates were diluted with methanol up to a volume of 50 mL. The solutions were stored around 4˚C for use.
2.3. Instrumentation
Chromatographic analysis was carried out on Agilent 1200 HPLC system equipped with two G1181A pumps, a G1186A thermostat and a VWD UV-detector (Agilent Technologies, California, USA). The chromatographic separation of the analytes was carried out on a Zorbax Eclipse XDB-C18 column (250 mm × 4.6 mm, 5 μm particle size). The mobile phase consisted of 13% water containing 0.2% phosphoric acid for adjusting to a suitable pH and 87% methanol. The flow-rate of 0.8 mL·min–1 was used in isocratic elution mode. The column was kept at 25˚C with detection wavelength at 210 nm. The total chromatographic run time was 32 min.
A centrifuge model 0406-1 (Shanghai, China) was used to accelerate the separation of sediment phase.
2.4. Dispersive Liquid-Liquid Microextraction Procedure
Into the 5.0 mL aqueous solution (acidified with 0.1 M HCl) containing the target analytes (100 μL working solution) and 10% (w/v) sodium chloride (NaCl), 300 μL of methanol (dispersive solvent) and 70 μL of chloroform (extraction solvent) were rapidly injected. Immediately, a cloudy solution was formed in the conical tube and then the mixture was gently hand-shaken for 30 s. The cloudy solution was kept at rest for 2 min until the distribution of analytes in both aqueous solution and extraction solvent reached equilibrium. In order to separate the extractant phase from the aqueous phase better, the above mixture was centrifuged for 4 min at 1500 rpm. After this process, the dispersed fine droplets of CHCl3 were deposited as sediment on the bottom of the test tube. The 30 μL of aggregation phase was withdrawn and transferred into a 2 mL small beaker, then evaporated to dryness in water bath. The residue was dissolved in 50 μL of HPLC-grade methanol and 20 μL was injected into HPLC system. All the experiments were performed in triplicate.
3. Results and Discussion
3.1. Optimization of the DLLME Procedure
3.1.1. Type and Volume of the Extraction Solvent
The selection of a suitable extraction solvent is critical for the DLLME process. In the DLLME, the extraction solvent should have following characteristics: 1) higher density than that of water, 2) low solubility in water, 3) the ability to extract interest analytes, and 4) good chromatographic behavior. In other words, the solvent peak should have no interference with the analyte peaks under the selected HPLC conditions. Based on these requirements, five organic solvent candidates, including C6H5Cl, CCl4, CH2Cl2, ClCH2CH2Cl and CHCl3 were investigated. The average peak areas (in triplicate) and the standard deviation for the different extraction solvents were tested. The results revealed that the peak areas obtained for the analytes using CHCl3 were higher than those with other solvents. In the case of CH2Cl2 as extraction solvent, the sedimented phase was hardly formed. Therefore, CHCl3 was selected as the extraction solvent for the study.
The volume of extraction solvent usually has great influence on the extraction efficiency in DLLME. In this test, different volumes of CHCl3 (50, 60, 70, 80 and 90 μL) were evaluated. Figure 2 clearly showed that the peak areas of the analytes rose obviously with increasing of CHCl3 volume until 70 μL. When the volume of
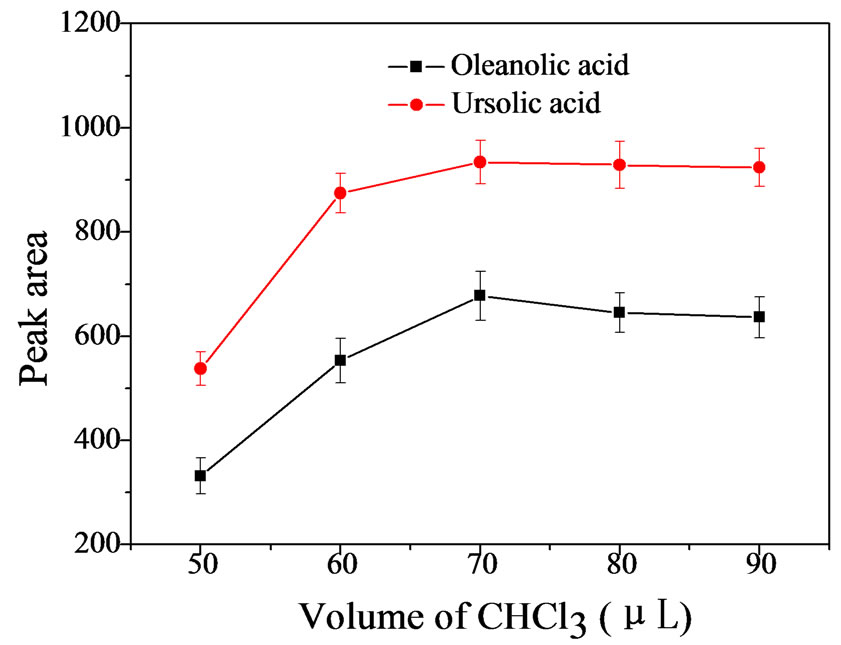
Figure 2. Effect of volume of CHCl3 (n = 3). Extraction conditions: [OA] = 1.60 µg·mL−1, [UA] = 2.08 µg·mL−1; sample volume, 5 mL; spiked working solution, 100 μL; 0.1 M HCl, 0.5 mL; NaCl concentration, 10%; methanol volume, 300 μL; shaking time, 50 s; stalling time, 2 min; cntrifugation speed, 1500 rpm; centrifugation time, 4 min.
CHCl3 continued to increase, the peak areas of both analytes decreased slightly. So, 70 μL was chosen as optimum extraction solvent volume for further experiments.
3.1.2. Effect of Disperser Solvent Volume
The disperser solvent must be miscible with both the extraction solvent and aqueous sample in order to form a distinct cloudy solution. Methanol is one of commonly used dispersants and was used for the solvent of stock solutions in the study, so we chose methanol as disperser solvent. The different volumes of methanol (0, 100, 200, 300, 400 and 500 μL) was tested. The results demonstrated that peak areas of OA and UA improved with the increasing of methanol volume up to 300 μL, and then the peak areas no longer had changes almost. Therefore, 300 μL of methanol was selected as the optimal disperser solvent volume for the subsequent experiments.
3.1.3. Effect of Sample pH
The pH value of sample solution determines the existing forms of the analytes (as ions or neutral molecules) because of the dissociation equilibrium, which could affect extraction efficiency. In this step, the effect of sample pH was evaluated within the pH range of 1 - 7 by adding equal volume (0.5 mL) of various concentrations of hydrochloric acid (1, 10–1, 10–2, 10–3, 10–4, 10–5 and 10–6 M) to aqueous samples. It can be observed from Figure 3 that the peak areas of OA and UA were significantly enhanced when pH was changed from 1 to 2. However, the peak areas had significant drop on increasing pH value in the range of pH 2 - 7. This may be due to the fact that the analytes existed most in ionic forms in aqueous solution with higher pH values. Hence, pH 2 was selected as the optimum sample pH.
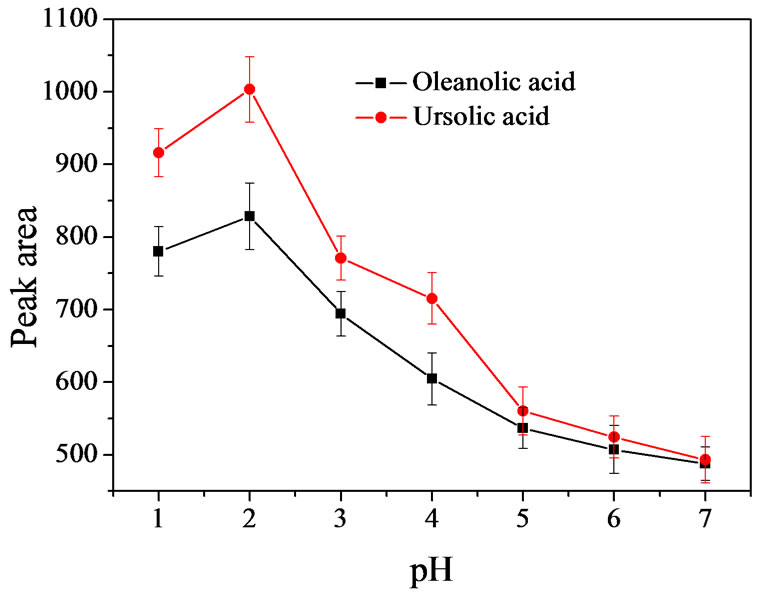
Figure 3. Effect of sample pH (n = 3). Extraction conditions: [OA] =1.44 μg·mL–1, [UA] =1.84 μg·mL–1. Sample volume, 5 mL; spiked working solution, 100 μL; NaCl concentration, 10 %; chloroform volume, 70 μL; methanol volume, 300 μL; shaking time, 50 s; stalling time, 2 min; centrifugation speed, 1500 rpm; centrifugation time, 4 min.
3.1.4. Effect of Ionic Strength
The influence of the ionic strength of the solution was evaluated by adding NaCl amounts from 0 to 20% (w/v) into the sample solution. The results (see Figure 4) showed that the ionic strength had an obvious effect on extraction efficiency. When salt concentration was below 10%, peak areas increased with the increasing of the salt concentration. However, higher than 10% of salt amount, peak areas greatly reduced. Maximum peak areas were obtained when 10% of salt was added. So, 10% of NaCl was used to the following experiments.
3.1.5. Effect of Extraction Time
In the DLLME, the extraction time is referred to as the time interval between injection the mixture of disperser solvent and extraction solvent and the start of centrifugation. Therefore, we examined the effect of hand-shaking time (5, 15, 25, 35 and 45 s) and resting time from 0 min to 4 min with fixed other experimental conditions. The results showed that the analytes had the best enrichments when the hand-shaking was performed for 50 times (about 25 s) before resting 2 min.
3.1.6. Effect of Centrifugation Speed and Time
Centrifugation is essential to separate extraction solvent from aqueous solution in DLLME, because centrifugation speed and time may affect the volume of settled phase. The effect of the centrifugation speed and time on the extraction efficiencies of these two triterpenic acids was examined from 500 to 3500 rpm and from 2 to 6 min. The experimental results showed that the best performance was obtained at 1500 rpm for 4 min.
3.1.7. Effect of Volume of Sample Phase
The volume of sample phase has a great influence on the extraction efficiency of the analytes. In this study, different
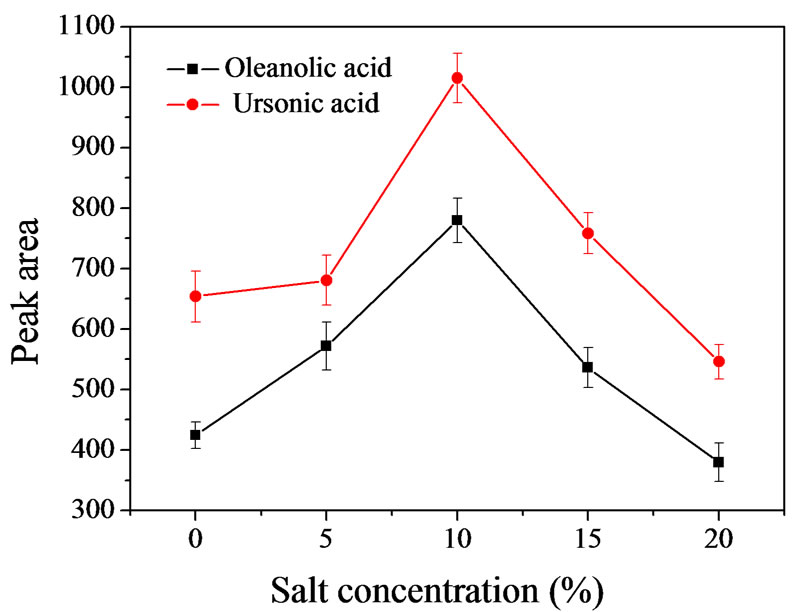
Figure 4. Effect of concentration of NaCl (n=3). Extraction conditions: [OA] = 1.44 μg·mL–1, [UA]=1.84 μg·mL–1. Sample volume, 5 mL; spiked working solution, 100 μL; 0.1 M HCl, 0.5 mL; chloroform volume, 70 μL; methanol volume, 300 μL; shaking time, 50 s; stalling time, 2 min; centrifugation speed, 1500 rpm; centrifugation time, 4 min.
volumes of sample phase (2, 4, 5, 6 and 8 mL) were investigated. Results indicated that the peak areas of OA and UA increased with increasing of sample phase volume up to 5 mL, then decreased remarkably with the volume of sample phase from 5 to 8 mL. Finally, 5 mL of the sample phase was chosen for the study.
3.2. Analytical Performance
3.2.1. Linearity and Limits of Detection
The method of DLLME coupled with HPLC for the determination of OA and UA was evaluated under the optimized conditions. The 7-point calibration curves were observed linear over the range of 0.07 - 30.4 μg·mL–1 for OA and 0.08 - 33.6 μg·mL–1 for UA, respectively. The regression equations between the peak area (y) and concentration (c) and square correlation coefficients (r2) were got as follows:
OA: 
UA: 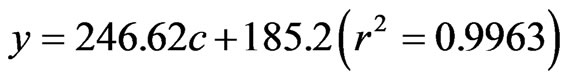
The limits of detection (LODs, at the signal to noise ratio of 3) were found to be both 0.02 μg·mL–1. The limits of quantitation (LOQs, at the signal to noise ratio of 10) were 0.07 μg·mL–1 for OA and 0.08 μg·mL–1 for UA, respectively.
3.2.2. Precision
Precision of the method was examined based on nine replicates of three levels. The RSDs for the intra-day and inter-day precision were displayed in Table 1.
3.2.3. Stability
At the optimum conditions, the stability of two CMH samples was performed by preparing a group of real
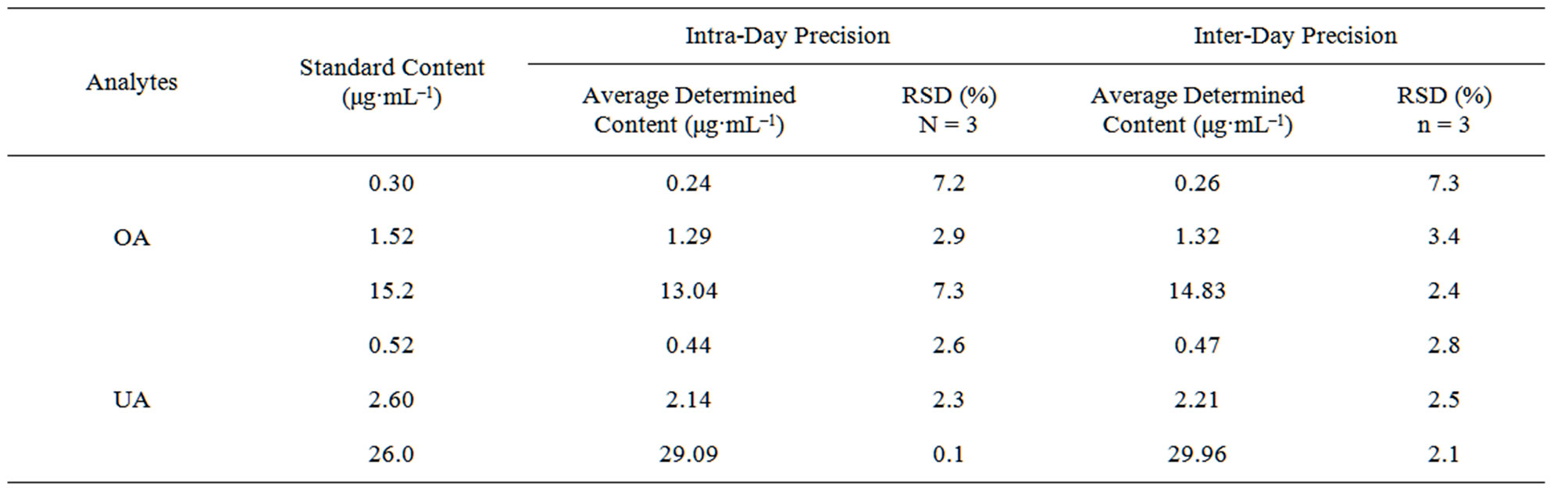
Table 1. Results of intra-day and inter-day precision test of analytes (n = 3).
samples according to DLLME procedures in Section 2.4. In the study, the samples were analyzed at 0, 3, 6, 9, 12, 18 and 24 h, respectively. It was found that the peak areas of the two target analytes remained unchanged (RSD < 8.5%). It means that the samples were in stability within 24 h.
3.2.4. Recoveries
Accuracy was assessed by investigating the recoveries of the target analytes in Hedyotis diffusa and Eriobotrya japonica samples spiked with OA and UA (five replicates for each sample), respectively. The resulted recoveries were shown in Table 2. The average recoveries of the both target analytes indicated that the method had a good accuracy.
3.3. Calibration of Enrichment Factor
In order to evaluate the performance of the proposed method, Enrichment Factor (EF) was applied and calculated by following equation.
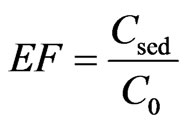
where Csed and C0 were the concentration of analyte in sedimented phase and the initial concentration of analyte in sample solution, respectively. Based on the equation, the average EFs of OA and UA were 1378 and 933, respectively.
3.4. Application
To investigate the applicability of the developed method to real samples, OA and UA in Hedyotis diffusa and Eriobotrya japonica were concentrated and assayed by DLLME–HPLC, and the analytical results were listed in Table 3. The chromatograms in Figure 5 showed that the target analytes had excellent enrichment after the process of DLLME.
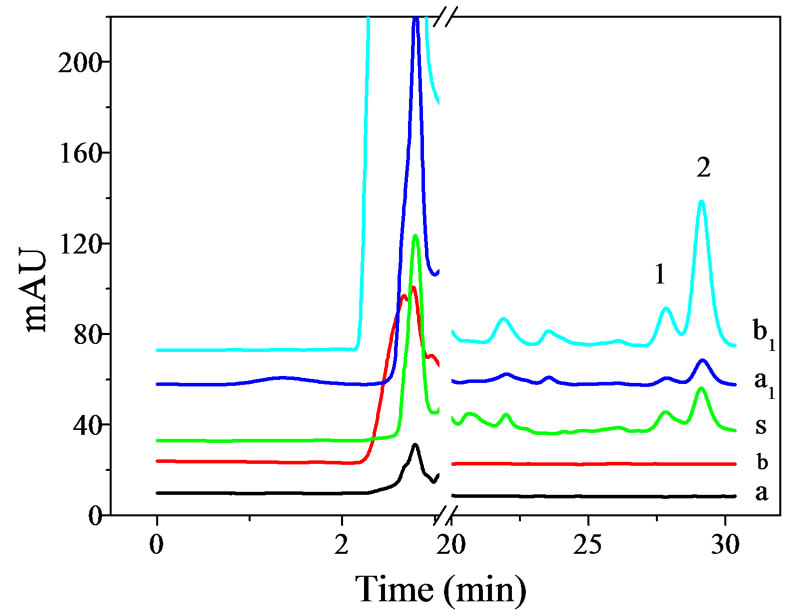
Figure 5. Typical chromatograms for analytes in Hedyotis diffusa and Eriobotrya japonica. 1: Oleanolic acid; 2: Ursolic acid. a: before DLLME of Hedyotis diffusa; a1: after DLLME of Hedyotis diffusa; b: before DLLME of Eriobotrya japonica; b1: after DLLME of Eriobotrya japonica; s: standard analytes.
3.5. Comparison of DLLME-HPLC with Other Analysis Method
As listed in Table 4, comparing to the other analysis methods encompassing chromatographic separation techniques [14-19] for the determination of OA and UA, the proposed method attained a higher sensitivities (see slopes of regression equation in Table 4) and better linear dynamic ranges, for which major reason is that the DLLME-based work involves high enrichment factors for the OA and UA. HPLC-DAD [17] obtained higher sensitivities but with poor LODs and linear ranges. Though the lower LODs were obtained by derivatizationHPLC [18] and ID-HPTLC [19], the derived reaction conditions were relatively strict and not easy to control.
4. Conclusion
In the present study, an original and simple method for the separation and determination of closely related isomeric triterpenic acid was presented. Under the optimum
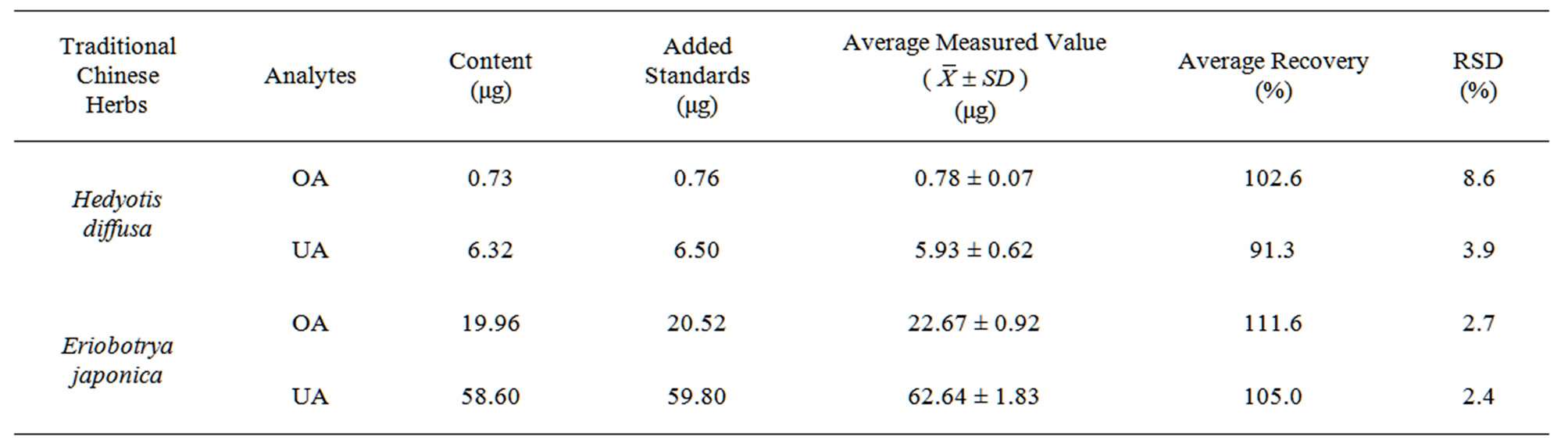
Table 2. Recovery experiments of two analytes in Hedyotis diffusa and Eriobotrya japonica samples by the DLLME-HPLC method (n = 5).
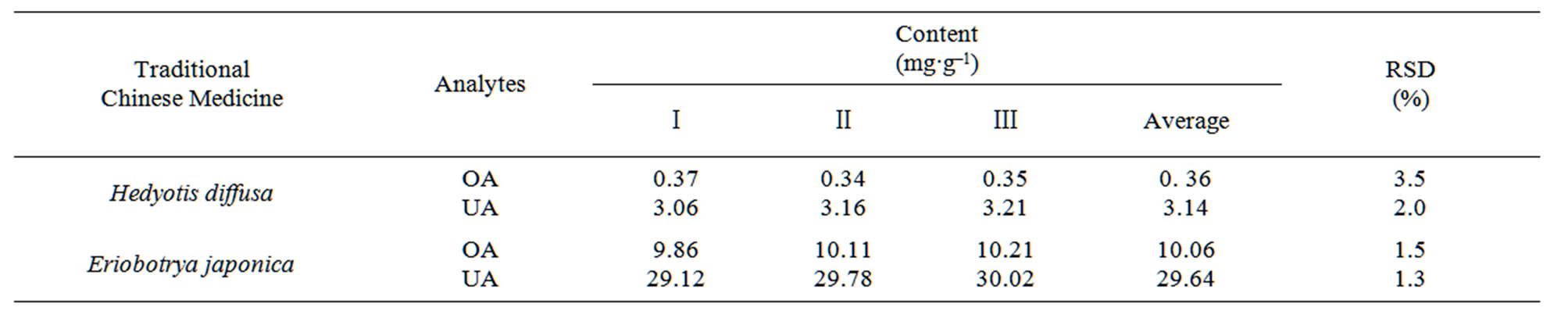
Table 3. Analytical results of OA and UA in CMHs real samples by DLLME-HPLC (n = 3).
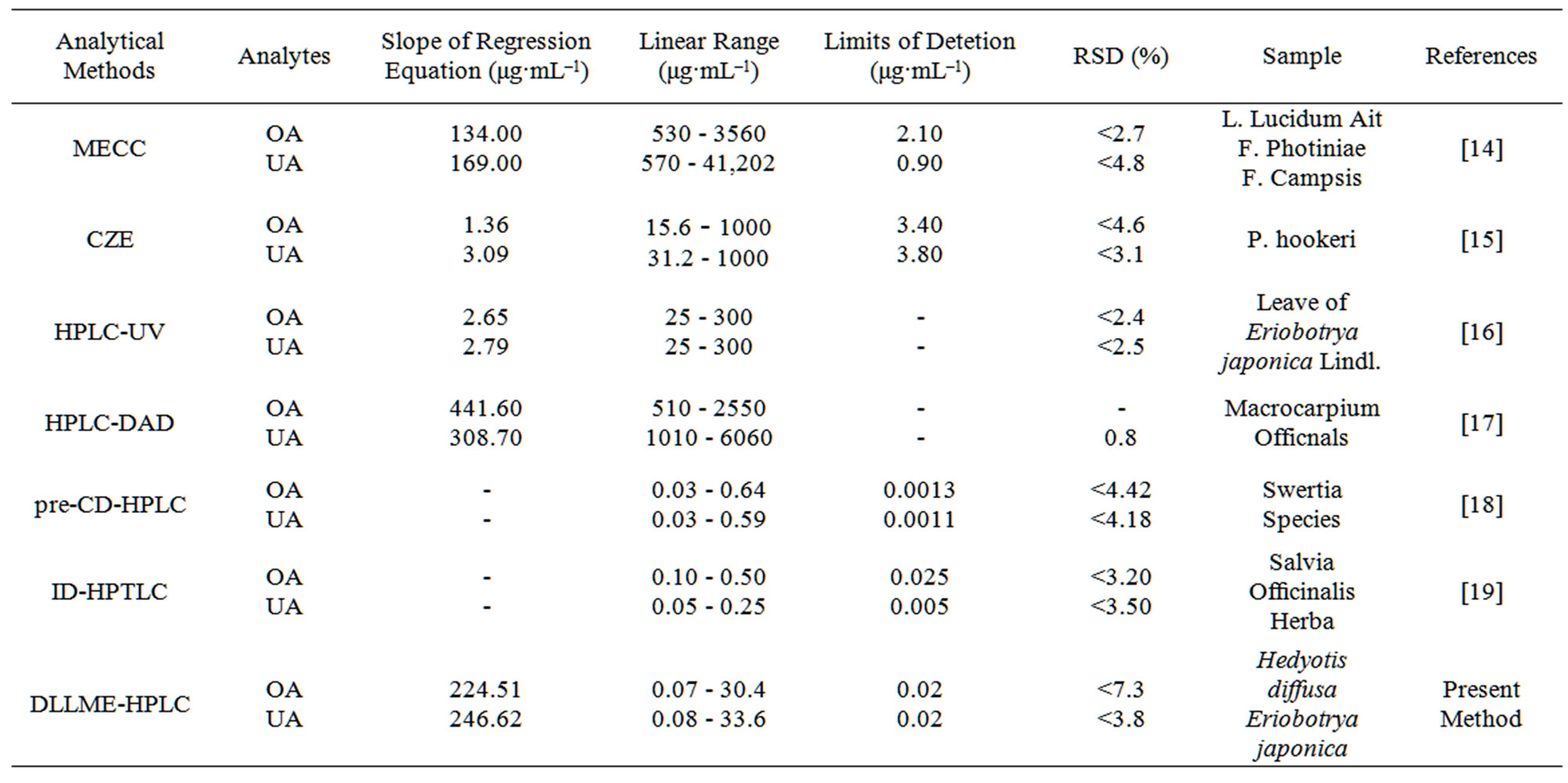
Table 4. Comparison of performance of the proposed method with that of other analytical methods.
conditions, both OA and UA attained favorable extraction efficiencies, high enrichment factor, wide linear range, and low detection limit. Furthermore, the method successfully applied to simultaneous quantification of both triterpenes acids in Hedyotis diffusa and Eriobotrya japonica. Comparing to the other analysis methods, DLLME-HPLC is a feasible technique used for the separation, enrichment and determination of trace analytes in complex CMH samples.
5. Acknowledgements
This research was supported by the National Nature Science Foundation of China (No.81041084), the Nature Science Foundation of Shanxi Province, China (No. 2011011035-2), the Innovation Foundation of University Student of Shanxi Province for the financial support and the Foundation of Shanxi Medical University, China.
REFERENCES
- J. Liu, “Pharmacology of Oleanolic Acid and Ursolic Acid,” Journal of Ethnopharmacology, Vol. 49, No. 2, 1995, pp. 57-68. doi:10.1016/0378-8741(95)90032-2
- V. Mandal and S. C. Mandal, “Design and Performance Evaluation of a Microwave Based Low Carbon Yielding Extraction Technique for Naturally Occurring Bioactive Triterpenoid: Oleanolic Acid,” Biochemical Engineering Journal, Vol. 50, No. 1-2, 2010, pp. 63-70. doi:10.1016/j.bej.2010.03.005
- A. T. Dinkova-Kostova, K. T. Liby, K. K. Stephenson, W. D. Holtzclaw, X. Q. Gao, N. Suh, C. Williarrli, R. Risingsong, T. Honda, G. W. Gribble, M. B. Sporn and P. Talalay, “Extremely Potent Triterpenoid Inducers of the Phase 2 Response: Correlations of Protection against Oxidant and Inflammatory Stress,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 102, No. 12, 2005, pp. 4584-4589. doi:10.1073/pnas.0500815102
- S. Schwaiger, I. Zeller, P. Pölzelbauer, S. Frotschnig, G. Laufer, B. Messner, V. Pieri, H. Stuppner and D. Bernhard, “Identification and Pharmacological Characterization of the Anti-Inflammatory Principal of the Leaves of Dwarf Elder (Sambucus ebulus L.),” Journal of Ethnopharmacology, Vol. 133, No. 2, 2011, pp. 704-709. doi:10.1016/j.jep.2010.10.049
- J. P. Benincá, J. B. Dalmarco, M. G. Pizzolatti and T. S. Fröde, “Analysis of the Anti-Inflammatory Properties of Rosmarinus Officinalis L. in Mice,” Food Chemistry, Vol. 124, No. 2, 2011, pp. 468-475.
- K. Mazumder, E. Siwu, S. Nozaki, Y. Watanabe and K. Tanaka, “Ursolic Acid Derivatives from Bangladeshi Medicinal Plant, Saurauja Roxburghii: Isolation and Cytotoxic Activity against A431 and C6 Glioma Cell Lines,” Phytochemistry Letters, Vol. 4, No. 3, 2011, pp. 287-291. doi:10.1016/j.phytol.2011.04.019
- S. L. Yan, C. Y. Huang, S. T. Wu and M. C. Yin, “Oleanolic acid and Ursolic Acid Induce Apoptosis in Four Human Liver Cancer Cell Lines,” Toxicology in Vitro, Vol. 24, No. 3, 2010, pp. 842-848. doi:1016/j.tiv.2009.12.008
- Y. Kashiwada, H. K. Wang, T. Nagao, S. Kitanaka, I. Yasuda, T. Fujioka, T. Yamagishi, L. M. Cosentino, M. Kozuka, H. Okabe, Y. Ikeshiro, C. Q. Hu, E. Yeh and K. H. Lee, “Anti-AIDS Agents. 30 Anti-HIV Activity of Oleanolic Acid, Pomolic Acid, and Structurally Related Triterpenoids,” The Journal of Natural Products, Vol. 61, No. 9, 1998, pp. 1090-1095. doi:10.1021/np9800710
- J. F. Rivero-Cruz, M. Zhu, A. D. Kinghorn and C. D. Wu, “Antimicrobial Constituents of Thompson Seedless Raisins (Vitis vinifera) against Selected Oral Pathogens,” Phytochemistry Letters, Vol. 1, No. 3, 2008, pp. 151-154. doi:10.1016/j.phytol.2008.07.007
- K. Horiuchi, S. Shiota, T. Hatano, T. Toshita, T. Kuroda and T. Tsuchiya, “Antimicrobial Activity of Oleanolic acid from Salvia Officinalis and Related Compounds on Vancomycin-Resistant Enterococci (VRE),” Biological and Pharmaceutical Bulletin, Vol. 30, No. 6, 2007, pp. 1147-1149. doi:10.1248/bpb.30.1147
- J. A. Rodriguez, L. Astudillo and G. Schmeda-Hirschmann, “Oleanolic Acid Promotes Healing of Acetic AcidInduced Chronic Gastric Lesions in Rats,” Pharmacological Research, Vol. 48, No. 3, 2003, pp. 291-294. doi:10.1016/S1043-6618(03)00155-5
- C. L. Meloa, M. Queiroza, S. Fonsecaa, A. Bizerrab, T. Lemosb, T. Meloc, F. Santosc and V. Raoc, “Oleanolic Acid, a Natural Triterpenoid Improves Blood Glucose Tolerance in Norma Mice and Ameliorates Visceral Obesity in Mice Fed a High-Fat Diet,” Chemico-Biological Interactions, Vol. 185, No. 1, 2010, pp. 59-65. doi:10.1016/j.cbi.2010.02.028
- I. Kazmi, M. Rahman, M. Afzal, A. G. Gupta, S. Saleem, O. Afzal, M. A. Shaharyar, U. Nautiyal, S. Ahmed and F. Anwar, “Anti-Diabetic Potential of Ursolic Acid Stearoyl Glucoside: A New Triterpenic Gycosidic Ester from Lantana Camara,” Fitoterapia, Vol. 83, No. 1, 2012, pp. 142- 146.
- H. X. Liu, Y. H. Shi, D. X. Wang, G. L. Yang, A. M. Yu and H. Q. Zhang, “MECC Determination of Oleanolic Acid and Ursolic Acid Isomers in Ligustrum lucidum Ait,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 32, No. 3, 2003, pp. 479-485.
- P. Yang, Y. Q. Li, X. Liu and S. X. Jiang, “Determination of Free Isomeric Oleanolic Acid and Ursolic Acid in Pterocephalus Hookeri by Capillary Zone Electrophoresis,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 43, No. 4, 2007, pp. 1331-1334.
- X. H. Xu, Q. Su and Z. H. Zang, “Simultaneous Determination of Oleanolic Acid and Ursolic Acid by RPHPLC in the Leave of Eriobotrya japonica Lindl,” Journal of Pharmaceutical Analysis, Vol. 2, 2012, pp. 238- 240. doi:10.1016/j.jpha.2012.01.006
- H. H. Wang, Z. Z. Wang and W. B. Guo, “Comparative Determination of Ursolic Acid and Oleanolic Acid of Macrocarpium officinalis (Sieb. et Zucc.) Nakai by RPHPLC,” Industrial Crops and Products, Vol. 28, No. 3, 2008, pp. 328-332. doi:10.1016/j.indcrop.2008.03.004
- G. L. Li, X. L. Zhang, J. M. You, C. H. Song, Z. W. Sun, L. Xia and Y. R. Suo, “Highly Sensitive and Selective Pre-Column Derivatization High Performance Liquid Chromatography Approach for Rapid Determination of Triterpenes Oleanolic and Ursolic Acids and Application to Swertia Species: Optimization of Triterpenic Acids Extraction and Pre-Column Derivatization Using Response Surface Methodology,” Analytica Chimica Acta, Vol. 688, No. 2, 2011, pp. 208-218. doi:10.1016/j.aca.2011.01.010
- M. Wójciak-Kosior, “Separation and Determination of Closely Related Triterpenic Acids by High Performance Thin-Layer Chromatography after Iodine Derivatization,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 45, No. 2, 2007, pp. 337-340.
- C. B. Ojeda, F. S. Rojas and J. M. C. Pavón, “Determination of Cobalt in Food, Environmental and Water Samples with Preconcentration by Dispersive Liquid-Liquid Micro Extraction,” American Journal of Analytical Chemistry, Vol. 3, 2012, pp. 125-130. doi:10.4236/ajac.2012.32018
- S. Berijani, Y. Assadi, M. Anbia, M. R. M. Hosseini and E. Aghaee, “Dispersive Liquid-Liquid Microextraction Combined with Gas Chromatography-Flame Photometric Detection: Very Simple, Rapid and Sensitive Method for the Determination of Organ Phosphorus Pesticides in Water,” Journal of Chromatography A, Vol. 1123, No. 1, 2006, pp. 1-9.
- M. I. Leong, C. C. Chang, M. R. Fuh and S. D. Huanga, “Low Toxic Dispersive Liquid-Liquid Microextraction Using Halosolvents for Extraction of Polycyclic Aromatic Hydrocarbons in Water Samples,” Journal of Chromatography A, Vol. 1217, No. 34, 2010, pp. 5455-5461. doi:10.1016/j.chroma.2010.06.056
- L. J. He, X. L. Luo, H. X. Xie, C. J. Wang, X. Jiang and K. Lu, “Ionic Liquid-Based Dispersive Liquid-Liquid Microextraction Followed High-Performance Liquid Chromatography for the Determination of Organophosphorus Pesticides in Water Sample,” Analytica Chimica Acta, Vol. 655, No. 1-2, 2009, pp. 52-59.
- S. Q. Gao, J. Y. You, X. Zheng, Y. Wang, R. B. Ren, R. Zhang, Y. P. Bai and H. Q. Zhang, “Determination of Phenylurea and Triazine Herbicides in Milk by Microwave Assisted Ionic Liquid Microextraction High-Performance Liquid Chromatography,” Talanta, Vol. 82, No. 4, 2010, pp. 1371-1377.
- E. Z. Jahromi, A. Bidari, Y. Assadi, M. R. M. Hosseini and M. R. Jamali, “Dispersive Liquid-Liquid Microextraction Combined with Graphite Furnace Atomic Absorption Spectrometry: Ultra Trace Determination of Cadmium in Water Samples,” Analytica Chimica Acta, Vol. 585, No. 2, 2007, pp. 305-311.
- H. F. Zhang and Y. P. Shi, “Temperature-Assisted Ionic Liquid Dispersive Liquid-Liquid Microextraction Combined with High Performance Liquid Chromatography for the Determination of Anthraquinones in Radix et Rhizoma Rhei Samples,” Talanta, Vol. 82, No. 3, 2010, pp. 1010-1016. doi:10.1016/j.talanta.2010.06.008
- A. Daneshfar, T. Khezeli and H. J. Lotfi, “Determination of Cholesterol in Food Samples Using Dispersive Liquid-Liquid Microextraction followed by HPLC-UV,” Journal of Chromatography B, Vol. 877, No. 4, 2009, pp. 456-460. doi:10.1016/j.jchromb.2008.12.050
- M. Bashiri-Juybari, A. Mehdinia, A. Jabbari and Y. Yamini, “Determination of Amitraz in the Honey Samples by Dispersive Liquid-Liquid Microextraction Followed by Gas Chromatography-Flame Ionization Detection C,” American Journal of Analytical Chemistry, Vol. 2, 2011, pp. 632-637.
- M. Xiong, J. L. Ruan, Y. L. Cai and Y. Tang, “Extraction and Determination of Some Psychotropic Drugs in Urine Samples using Dispersive Liquid-Liquid Microextraction Followed by High-Performance Liquid Chromatography,” Journal of Pharmaceutical and Biomedical Analysis, Vol. 49, No. 2, 2009, pp. 572-578.
- J. Tian, X. Chen and X. H. Bai, “Comparison of Dispersive Liquid-Liquid Microextraction Based on Organic Solvent and Ionic Liquid Combined with High-Performance Liquid Chromatography for Analysis of Emodin and Its Metabolites in Urine Samples,” Journal of Separation Science, Vol. 35, No. 1, 2012, pp. 145-152.
- L. H. Li, H. F. Zhang, S. Hu and X. H. Bai, “Dispersive Liquid-Liquid Microextraction Coupled with High-Performance Liquid Chromatography for Determination of Coumarin Compounds in Radix Angelicae Dahuricae,” Chromatographia, Vol. 75, No. 3, 2012, pp. 131-137. doi:10.1007/s10337-011-2177-1
NOTES
*Corresponding authors.