American Journal of Analytical Chemistry
Vol. 3 No. 5 (2012) , Article ID: 19425 , 8 pages DOI:10.4236/ajac.2012.35046
Micelle Mediated Extraction of Iron and Its Determination in Geological, Geochemical, Hydrogeochemical, Biogeochemical and Process Solutions
Regional centre for Exploration and Research, AMDER/Department of Atomic Energy, Jamshedpur, India
Email: *ptarafder@sify.com
Received February 21, 2012; revised March 22, 2012; accepted March 27, 2012
Keywords: 2,3-Dihydroxynaphthalene; Cetyltrimethylammonium Ion; Spectrophotometric; Solvent Extraction; Determination
ABSTRACT
Iron irrespective of its valency states forms bluish 1:1 cationic, bluish-violet 1:3 neutral and purple 1:3 anionic complexes with 2,3-dihydroxynaphthalene, at pH 2 - 3, 4 - 5 and 8 - 10, respectively. The cationic bluish complex is highly insensitive and not extractable in organic solvents. Similarly, the bluish violet 1:3 neutral complex is of moderate sensitivity, and it is extractable in organic solvents with reduced sensitivity. The purple 1:3 anionic complex is as such not extractable in any organic solvents. However, in the surfactant medium of the cationic surfactant, cetyltrimethylammonium bromide it is extractable in organic solvents with enhanced sensitivity. The 1:3 anionic complex which is extractable in organic solvents as an ion associate in the presence of cetyltrimethyl ammonium bromide is chosen for application to Silicate rocks, Minerals, Soils, Stream sediments, Concentrates, Tobacco leaves, Cigarettes and Waters, for accurate and sensitive determination of iron owing to facile extraction of iron, enhanced sensitivity high selectivity and better reproducibility of results. The present paper describes a systematic study on the development of an extraction spectrophotometric method for the determination of iron in samples of diverse matrices.
1. Introduction
Iron, being a major element in the earth crust, is invariably present in all the natural products particularly geological (rocks, ores and minerals), geochemicals (soils and stream sediments), hydrogeochemicals (well, stream, river and ocean waters) and biogeochemical samples (plant samples) in both ferrous and ferric forms. Because of the ubiquitous presence of iron, either its removal/ separation as impurity while preparing high purity other metals/elements during hydrometallurgical/metallurgical processes or its accurate determination for ascertaining its actual content in the samples is important.
Most importantly, in almost all wet chemico-analytical methods for the accurate estimation of different elements, iron invariably interferes, for example presence of milligram level of iron poses serious problem in the fluorimetric determination of uranium in geological and geochemical samples, and similarly presence of microgram level of the same interferes seriously in the trace determination of uranium in water samples by laser induced fluorimetry.
Iron being a chromogenic element forms colour complexes with many reagents used in the colori-metric determination of different elements, thereby causing interference in their determinations. Therefore, separation of iron prior to their colorimetric determination is mandatory. Most colorimetric/spectrophotometric methods either determine Fe2+ or Fe3+.
For example, 1, 10 phenanthroline is a useful chelating reagent for the selective determination of Fe2+ ion in the presence of Fe3+ in solution. Similarly, there are reagents which can selectively determine Fe3+ ion in the presence of Fe2+ ion. However, while determining total iron, Fe3+ needs to be reduced to Fe2+ for reagents capable of determining only Fe2+ and, Fe2+ present in the sample/solution needs to be oxidized to Fe3+ for reagents capable of determining only Fe3+. No reagent is capable of determining both Fe2+ and Fe3+ together (total iron) as such. If some how oxidation of Fe2+ or reduction of Fe3+ is not complete, the results for total iron determination are never accurate. Often, Fe2+ undergoes auto oxidation with atmospheric oxygen, thereby affecting the spectrophotometric determination of ferrous ion adversely.
While investigating the complexing ability of the reagent 2,3-dihydroxynaphthalene [1], it was found that this reagent forms a series of different complexes with iron under different reaction conditions, irrespective of its oxidation states. It is assumed that the reagent 2,3- dihydroxynaphthalene itself acts as a reducing agent for Fe3+. It readily forms complex with Fe2+ ion, and when allowed to react with Fe3+, it instantaneously reduces Fe3+ into Fe2+ and then reacts with it to form Fe2+-H2ND complex.
Such type of reaction is not unprecedented [2]. As has already been reported, 2,3-H2ND forms a bluish-violet complex with manganese, irrespective of its valency states.
This type of system has got advantage over oxidation states specific reaction system because prior reduction/oxidation of iron species are not needed, and error due to incomplete oxida-tion/reduction is avoided. This prompted us to systematically investigate in detail the reaction of this reagent with iron.
Compounds having O-O’ type of functional groups on benzene or naphthalene ring like 1,2-dihydroxybenzene- 3,5-disulphonic acid, protocatechuic acid, chromotropic acid, 5-sulphosalicylic acid form a range of Fe complexes of different stoichiometries at different pH.
Similarly, in aqueous solution formation of 1:1, 1:2 and 1:3 complexes of 2,3-dihydroxy-naphthalene with Fe is indicated [3]. However, availability of data on the complex formation of Fe with this ligand is very scanty in the literature. The complex forming ability and the analytical potentiality of this ligand has been amply demonstrated [1]. Formation of Fe-2,3-dihydroxynaphthalene complex at pH-range, 4.5 - 4.7 and its estimation after extracting the anionic complex into CHCl3 under the influence of Bu3N as a counter cation had been reported in the past [4].
On raising the pH in the range, 8 to 10, a reddish purple 1:3 anionic complex was formed in aqu-eous solution having λmax at 450 - 455 nm. As such, it could not be extracted into organic solvents unlike the neutral complex formed at pH 4 - 5 [5]. On the contrary, in the micellar medium of the cationic surfactant, cetyltrimethylammonium bromide, it could be easily extracted into ethyl acetate without any shift in the λmax. The molar absorptivity of the system was 1.2 × 104 L∙mol–1·cm–1.
Surprisingly, the reddish purple 1:3 anionic complex which possesses all the characteristic features deemed necessary to qualify itself as an absorbing species suitable for the spectrophotometric determination of iron in real samples of diverse matrices and having sensitivity comparable with that of well known 1,10-phenanthroline method and more than that of thiocyanate and many other spectrophotometric methods for iron determination, as evident from the results obtained from the preliminary studies carried out in our lab, was not studied earlier for its possible analytical applications, to the best of our knowledge. Therefore, in what follows, an account of a detailed and systematic study on the complex formation of Fe with 2,3-H2ND at pH 8 - 10 in aqueous solution and its facile extraction into a suitable solvent in the micellar medium of the cationic surfactant, CTA with a view to develop a robust extraction spectrophotometric method for iron determination in diverse samples such as, silicate rock, mineral, soil, stream sediment, water, and biological samples is given.
2. Experimental
2.1. Apparatus
Elico model LI-120 pH meter with combined electrode was used for pH measurements and Chemito 2500-UVVIS-spectrophotometer with 1 cm quartz cuvettes was used for recording spectra and absorbance measurements.
2.2. Reagents
The stock solution of iron(III) (1 mg·ml–1) was prepared by dissolving 8.634 g of NH4Fe(SO4)2·12H2O in 1 L of distilled water containing concentrated sulfuric acid and it was standardized volumetrically [6]. The required concentration of iron(III) solution was prepared by appropriate dilution with double distilled water.
The 2,3-dihydroxynaphthalene, (3.12 × 10–2 mol·L–1) solution in ethyl acetate was prepared by dissolving 1.0 g of the reagent in 200 ml of ethyl acetate.
Cetyl-trimethylammonium bromide (CTA) (2.74 × 10–2 mol·L–1) aqueous solution was prepared by dissolving 1.0 g of the reagent in 100 ml of distilled water.
Sodium acetate trihydrate, solid as well as 0.7348 mol·L–1 solution.
2.3. Procedures
2.3.1. Dissolution of Sample
In a platinum dish weighed 0.5 g of 100 mesh rock sample and to it added 10 ml of hydrofluoric acid (40%) followed by 6 ml of 18 mol·L–1 sulphuric acid. Covered the dish and heated on a steam bath for 6 - 8 h. Removed the cover and heated for another 1 to 2 h. Again added 10 ml of 40% hydrofluoric acid and heated till the volume was reduced to near 3 ml. Transferred the dish on to a sand bath and heated until dense fume of sulphuric acid aerosol starts evolving. Cooled, and then added 1 ml of 16.0 mol·L–1 nitric acid and heated on a sand bath in order to remove by oxidation any organic matter present. Nitric acid treatment was repeated two times in order to completely remove the organic matter present. Added about 30 ml water and digested on a steam bath for about 20 min. Transferred the digested mass into a 250 ml beaker and added about 200 ml water. Boiled the solution and then cooled it. Filtered through a 541 whatman filter paper and transferred the filtrate into a 250 ml volumetric flask. Made up to the mark with water.
2.3.2. Spectrophotometric Determination in Aqueous Solution
An aliquot of sample solution containing iron in the range 2 to 250 μg was taken in a 125 ml separating funnel, the volume of aqueous layer was adjusted to 10 ml with distilled water. To it was added 10 ml of 3.12 × 10–2 mol·L–1 solution of 2,3-H2ND in ethyl acetate and mixed well. A 0.5 g solid sodium acetate was added and shaken for a while. To it was added 1 ml of aqueous ammonia (sp. gravity 0.91) and shaken for 2 minute. The final pH of the solution was in the range, 8.0 to 10.0. Allowed for phase separation. Drained out the aqueous layer and stored. Scrubbed the organic layer two times with water (5 ml each) containing 2 to 3 drops of ammonia and a pinch of sodium acetate. The scrubbed solutions were mixed up with the aqueous solution separated after solvent extraction. The organic layer was discarded. The aqueous layer was transferred into a 50 ml volumetric flask and made up the volume. Absorbance of the red anionic chelate of iron(III) with 2,3-H2ND was measured at 455 nm against a reagent blank.
2.3.3. Spectrophotometric Determination in Organic Solvent after Extraction
An aliquot of sample solution containing iron in the range 1.0 to 125 μg was taken in a 125 ml separating funnel and the volume of the aqueous layer was adjusted to 10 ml with distilled water. To it was added 10 ml of 3.12 × 10–2 mol·L–1 solution of 2,3-H2ND in ethyl acetate and mixed well. To it was added 1 ml of aqueous ammonia (sp. gr. 0.91) followed by 5 ml of 2.74 × 10–2 mol·L–1 CTA (the pH of the solution should be 8.0 to 10.0) and shaken for 2 minutes. Allowed for phase separation and then the aqueous layer was drained out and discarded. The organic layer was transferred into a 25 ml volumetric flask and made up the volume with ethyl acetate. The absorbance of the red chelate in ethyl acetate was measured at 455 nm against a reagent blank.
3. Results and Discussion
3.1. Reaction of Iron with 2,3-Dihydroxynaphthalene
We have investigated in detail the variables affecting the complex formation of Fe with this readily available reagent. We found that Fe irrespective of its oxidation states forms a 1:3 bluish-violet neutral complex with 2,3- dihydroxynaphthalene (2,3-H2ND) at pH 4 - 5 in aqueous solution having λmax at 470 nm and ε = ~ 1.0 × 104 L·mol–1·cm–1. However, this complex could be extracted into ethyl acetate without requiring any counter cation as opposed to the findings of V. Patrovsky.
These contradictory findings prompted us to undertake detailed investigations on the complex formation of Fe with 2,3-dihydroxynaphthalene (2,3-H2ND). Interestingly, we found that at pH 2 - 3, a 1:1 unstable bluish cationic complex which could not be extracted into organic solvent was formed in aqueous solution having λmax at 580 nm and ε = ~ 2.0 × 102 L·mol–1·cm–1. Similarly, as stated above, at pH 4 - 5 a 1:3 bluish-violet neutral complex was formed in aqueous solution having λmax 460 - 470 nm and the sensitivity (ε) was around 1.0 × 104 L·mol–1·cm–1. This complex could be extracted into ethyl acetate without the use of any counter cation. However, the λmax shifted to higher wavelength (~500 nm) and the sensitivity decreased to ~ 4.0 × 103 L·mol–1·cm–1. Extraction in the presence of a cationic surfactant, cetyltrimethylammonium bromide (CTA) recouped the sensitivity to ~ 1.0 × 104 L·mol–1·cm–1 without any shift in the λmax (~500 nm). On raising the pH to 8 - 10, a 1:3 reddish-orange anionic complex, extractable in ethyl acetate under the influence of cetyltrimethylammonium cation (CTA+) was formed.
Of the above three types of complexes of this reagent (2,3-H2ND) studied on this occasion, as stated above, the 1:1 complex is highly unstable as well as less sensitive, and is, therefore, not suitable for spectrophotometric measurements for iron determination. The 1:3 neutral complex has already been made use of by us [6] for iron determination.
In what follows are given details of systematic studies carried out on the hitherto unreported 1:3 reddish-orange anionic complex of iron with 2,3-H2ND with a view to develop a robust extraction spectrophotometric method for the determination of iron in diverse samples.
3.2. Solvent Selection
Various solvents were studied for the extraction of the ion-association system. Mention may be made of a few such as chloroform, methyl ethyl ketone, benzene, 1- butanol, n-butylacetate and ethyl acetate. Ethyl acetate gave the most satisfactory result. Therefore it was used in our extraction studies.
3.3. Optimum Conditions
3.3.1. Effect of pH
The strongly coloured 2,3-dihydroxynaphthalene anionic complex of Fe developed at a pH range of 8 - 10 in aqueous solution is suitable for spectrophotometric measurements. However, for better selectivity the complex was readily extracted into ethyl acetate with cetyltrimethylammonium ion, CTA+. The ion pair is stable for more than 24 h and only gentle shaking for 2 min was found to be sufficient for the quantitative extraction of iron.
The ion-pair was also found to be extracted in this pH range. The effect of pH on the percentage extraction of the complex into ethyl acetate is shown in Figure 1. Addition of a few drops of ammonia (sp. gravity 0.91) is sufficient to raise the pH to this level.
3.3.2. Effect of 2,3-Dihydroxynaphthalene Concentrations
The effect of 2,3-dihydroxynaphthalene concentration on the intensity of the colour formed and the subsequent extraction of the complex into ethyl acetate was studied by varying the concentration of the reagent over a wide range. A 10 ml of 6.24 × 10–3 mol·L–1 solution of the reagent was found to be optimum. However, in order to allow for reagent impurity a 10 ml of 3.12 × 10–2 mol·L–1 reagent was used.
3.3.3. Effect of Cetyltrimethylammonium Bromide Concentrations
Similarly, the effect of cetyltrimethylammonium bromide (CTA) on the extraction of the coloured anionic complex into ethyl acetate was studied by varying the CTA concentration over a wide range. A 5 ml of 5.48 × 10–3 mol·L–1 aqueous solution was found to be optimum for best results. However, in order to allow for reagent impurity, a 5 ml of 2.74 × 10–2 mol·L–1 aqueous solution was recommended.
3.3.4. Effect of Electrolyte, Time, Temperature, Volume Ratio and Standing Time
The equilibrium period necessary for complete extraction of Fe3+-2,3-H2ND-CTA complex into ethyl acetate was examined and found to be less than 1 min. The colour intensity of the complex is not affected by the addition of electrolytes such as NaCl, KCl, LiCl, NH4Cl or (NH4)2SO4 upto 2.0 mol·L–1.
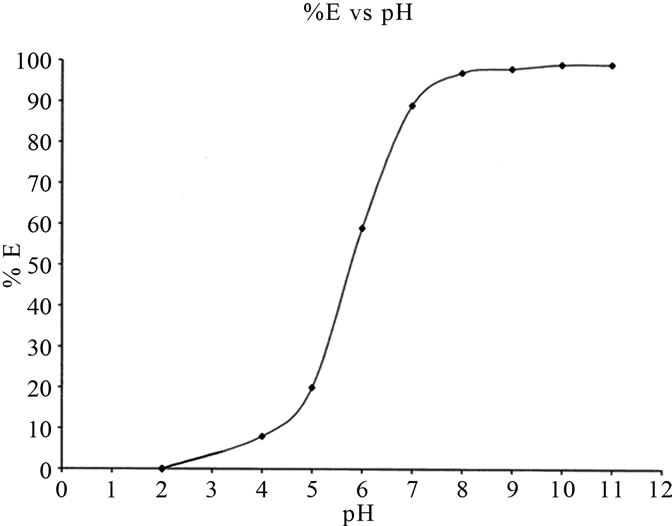
Figure 1. Effect of pH on the extraction of iron (plot of percentage extraction vs. pH). Conditions: [Fe] = 2.0 mg·L–1; [2,3-H2ND] = 0.0313 mol·L–1; [CTA] = 0.0086 mol·L–1.
The nature of the complex is not affected by variation in temperature from 20˚C to 40˚C and volume of aqueous phase from 10 to 50 ml. The complex is stable at least for 48 h at room temperature.
3.4. Absorption Spectra
The absorption spectra of the anionic complex in aqueous solution, the ion-association complex and its blank are shown in Figure 2. This shows the maximum absorbance of both the systems to be at 455 - 460 nm. The absorption due to the reagent blank at this wave length is negligible i.e., from 0.010 to 0.020.
3.5. Optimum Concentration Range, Molar Absorptivity, Sensitivity and Precision of the Method
Adherence to Beer’s law was tested and confirmed for both the systems i.e., aqueous as well as extractive by least square method. The Beer’s law was obeyed in the range 0.04 to 5.0 mg·L–1 of iron with 2,3-H2ND.
The optimum concentration range for the effective Spectrophotometric determination of iron, evaluated by Ringbom’s plot [6] was 0.7 to 4 mg·L–1 iron.
The molar absorptivity and Sandell’s sensitivity of the complex calculated at the region of least photometric error were 1.2 × 104 L·mol–1·cm–1 and 0.001 mg·cm–2, respectively. The precision of the method was tested by measuring the absorbance of 10 replicate sample solutions, each containing 1.0 mg·L–1. The mean absorbance of this concentration was found to be 0.21 with RSD of ±0.72%.

Figure 2. Absorption spectra against reagent blank of (a) ion associate complex, Q[Fe(OH)(HND)3] extracted into ethyl acetate under the influence of CTA (b) the anionic complex, [Fe(OH)(HND)3]–1 in aqueous solution and (c) reagent blank against ethyl acetate; Conditions: pH 10.0; concentra-tion of [Fe] =1.0 mg·L–1; [2,3-H2ND] = 0.0313 mol·L–1; [CH3COONa] = 0.367 mol·L–1; [CTA] = 0.0086 mol·L–1.
The detection and determination limits for Fe were 0.08 and 0.25 µg·mL–1, respectively. The limit of detection defined as CL = 3.3 SB/m, where SB and m denote standard deviation of the blank signal and the slop of the calibration plot, respectively. Similarly the limit of determination was defined as CL = YB + 10 × SB, where, YB denotes the sinal of the blank sample.
The minimum detectable amount for the rock and soil samples was 100 ppm (100 µg Fe per gram of sample) and 0.2 mg·L–1 for water samples.
3.6. Extraction Equilibria and Probable Composition of the Complex
The mechanisim by which Fe3+ ion was extracted from an aqueous phase into ethyl acetate by 2,3-dihydroxynaphthalene (2,3-H2ND) and CTA follows the reaction as given below:
 (1)
(1)
Where H2ND stands for 2,3-dihydroxynaphthalene and CTA+ stands for cetyl-trimethylammonium ion. The equilibrium constant, K of the reaction can be written as,
 (2)
(2)
or
 (3)
(3)
Where,
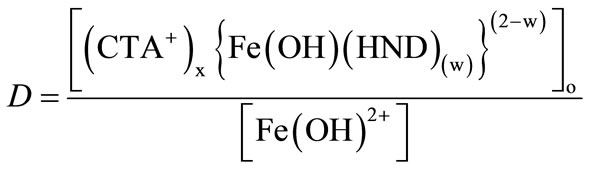
Equation (3) can be re-written as
 (4)
(4)
Analysing the experimental values of distribution ratio (D) as a function of equilibrium pH and extractant concentrations at constant value of other parameters allows estimation of the number of extractant molecules associated with the extracted metal. The “slope analysis” was made use of in the study of distribution behaviour of Fe3+ by plotting the logarithmic value of distribution ratio of Fe3+ vs. experimental variables (curve fitting method) [7]. The plot of log D value vs. logarithmic concentration of 2,3-H2ND in the organic phase at a constant  concentration are shown in Figure 3. The slope of 3 was obtained. It showed that three 2,3-H2ND molecules were used in the extraction process. This result indicates, w = 3.
concentration are shown in Figure 3. The slope of 3 was obtained. It showed that three 2,3-H2ND molecules were used in the extraction process. This result indicates, w = 3.
Then the number of CTA cation used to extract the chelate into organic phase was determined. A slope of 1 was obtained for plot of log D vs. logarithmic concentration of  in the organic phase at a constant H2ND(o) (Figure 4). This result indicates that one CTA+ ion takes
in the organic phase at a constant H2ND(o) (Figure 4). This result indicates that one CTA+ ion takes
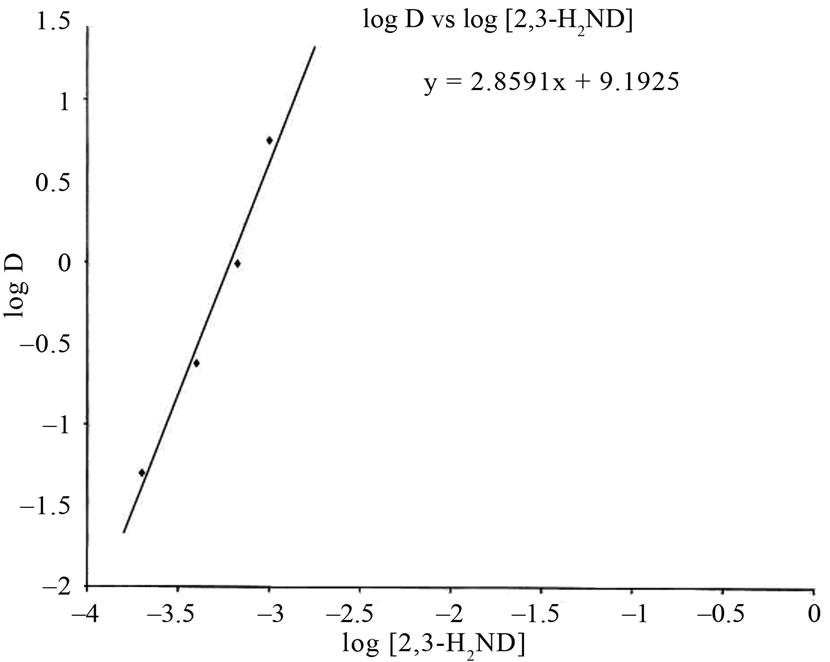
Figure 3. Application of Curve-fitting method for the determination of the stoichiometry of the complex: dependency of distribution ratio of iron on the concentrations of 2,3-dihydroxynaphthalene (H2ND), (Plot of log D vs. Logarithmic concentration of 2,3-dihydroxynaphthalene). Conditions: [Fe] = 2.0 mg·L–1; [CH3COONa] = 0.367 mol·L–1; [CTA] = 0.0086 mol·L–1.
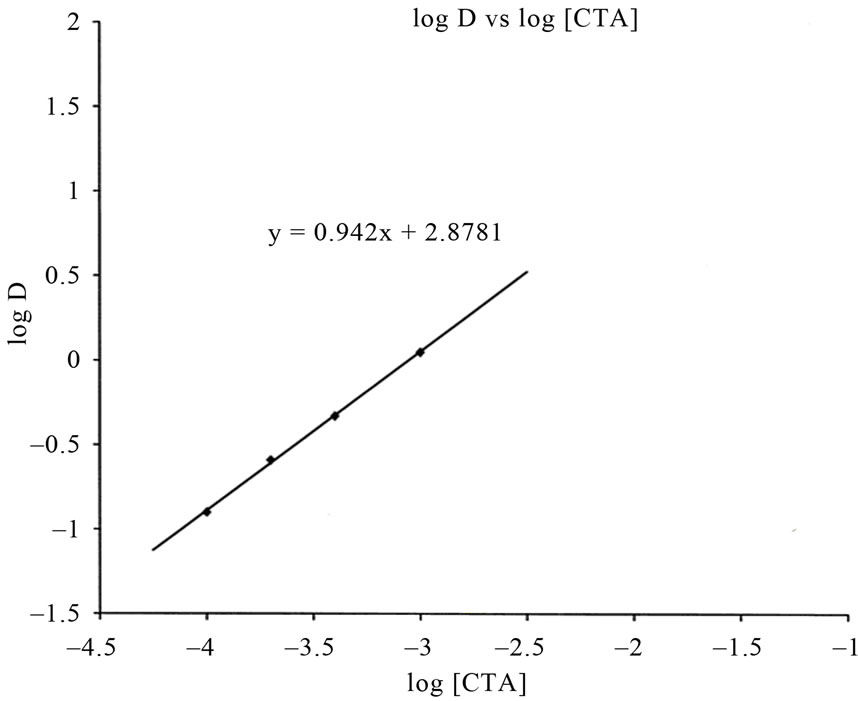
Figure 4. Application of Curve-fitting method for the determination of the stoichiometry of the complex: dependency of distribution ratio of iron on the concentrations of Cetyltrimethylammonium bromide (CTA) (Plot of log D vs. Logarithmic concentration of Cetyltrimethylammonium bromide). Conditions: [2,3-H2ND] = 3.12 × 10–2 mol·L–1, [Fe] = 2.0 mg·L–1; [CH3COONa] = 0.367 mol·L–1.
part in the extraction system and x is 1 in Equation (1). Thus the stoichiometry of the complex is 1:3:1 (Fe3+: 2,3-H2ND:CTA+). Figure 1 shows that 3 hydrogen atoms (slope of the plot was 3) were released during complexation. Thus the extracted complex under optimized reaction conditions i.e., pH 8 - 10 is an ion associate Q[Fe(OH)(HND)3] where Q = CTA+. Similar species is however, not unprecedented.
3.7. Tentative Reaction Scheme
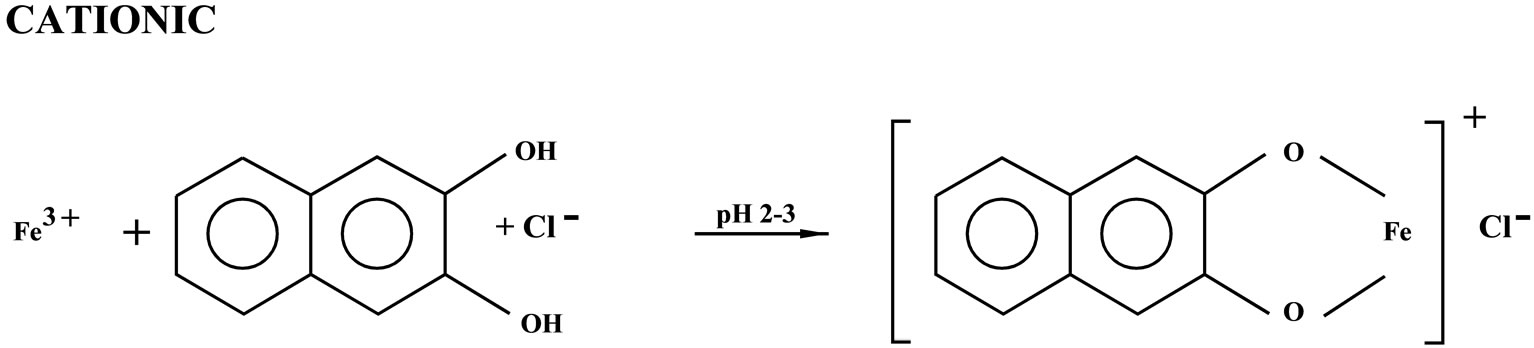

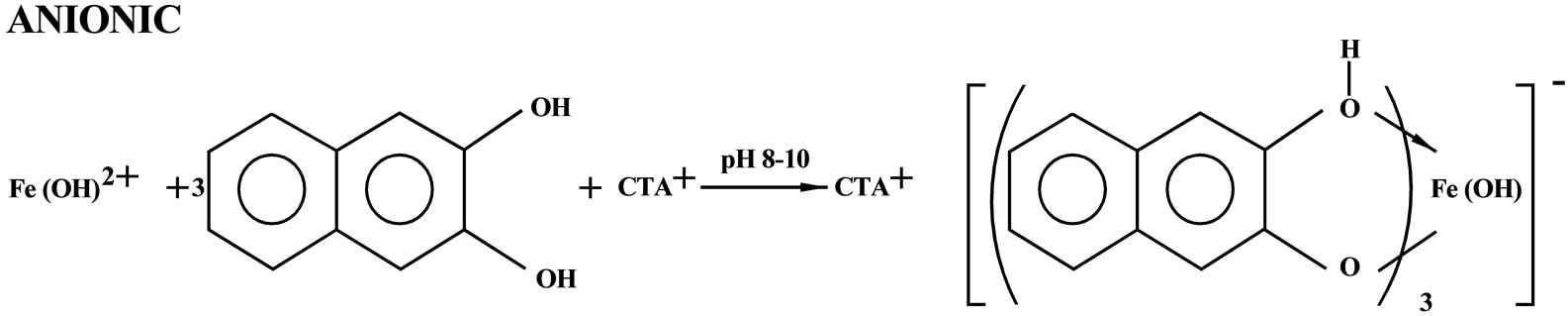
3.8. Effect of Diverse Ions
An aliquot portion of solution containing 100 μg of iron (Fe3+) was transferred into a 125 ml separating funnel, various elements were added individually, and iron was extracted as described in the procedure. At least 10,000 mg·L–1 of ,
,  ,
,  ,
, 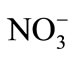 , Li, Na, K and 5000 mg·L–1 of Ca2+, Mg2+, Be2+, Ba2+, Rb+, Sr+, do not interfere in the present method. Milligram level of Cu, Ni, Co, Ag, Hg, Zn, As, Sb, Bi, Al, Th, La, are tolerated. Titanium, niobium, molybdenum, zirconium and chromium when present up to ten fold in excess to iron do not interfere. However, above 1:5 level vanadium, cerium and manganese interfere. In silicate rocks these elements are present at much below level to iron; hence their interference is not encountered.
, Li, Na, K and 5000 mg·L–1 of Ca2+, Mg2+, Be2+, Ba2+, Rb+, Sr+, do not interfere in the present method. Milligram level of Cu, Ni, Co, Ag, Hg, Zn, As, Sb, Bi, Al, Th, La, are tolerated. Titanium, niobium, molybdenum, zirconium and chromium when present up to ten fold in excess to iron do not interfere. However, above 1:5 level vanadium, cerium and manganese interfere. In silicate rocks these elements are present at much below level to iron; hence their interference is not encountered.
3.9. Analytical Applications
The method thus developed was thoroughly applied to a range of silicate rock, mineral, soil, stream sediment, water, drug formulations, and plant ash samples, and results obtained as tabulated in Tables 1 to 3 have been found to be favourably comparable with those obtained from 1,10-phenanthroline and AAS methods.
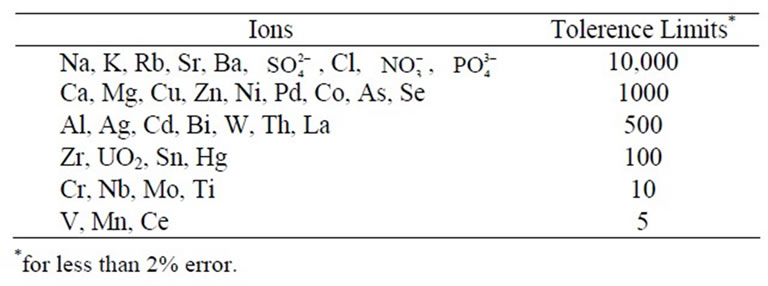
Table 1. Effect of diverse ion in the determination of 1 mg·L–1 iron.
However, in order to make the method completely free from interference, the following procedure for the determination of iron in any type of samples is recommended.
Procedure: An aliquot of sample solution containing not more than 125 μg of iron is taken into a 125 ml separating funnel. To it was added10 ml of 3.12 × 10–2 mol·L–1 2,3-H2ND in ethyl acetate followed by 10 ml of 0.7348 mol·L–1 sodium acetate trihydrate. The separating funnel was stoppered and shaken for 2 - 3 minutes. After complete phase separation the aqueous layer was drained out and discarded. To the separating funnel containing ethyl acetate loaded with neutral complexes of iron, titanium, vanadium, aluminum, thorium etc added 10 ml of distilled water containing a few drops (~1 ml) of aqueous ammonia and a pinch of solid sodium acetate (sodium acetate here acts as an electrolyte to maintain ionic strength) and then the separating funnel was shaken for few seconds. The reddish-orange (1:3) anionic complex of iron with 2,3-H2ND immediately passes into the aqueous layer. The ethyl acetate layer containing neutral complexes of Ti, V, Th and Al, and entrapped anionic complex of Fe(III) was scrubbed two times with 5 ml portion of water containing a few drops of ammonia. The scrubbed solutions were mixed with the aqueous layer. Then the organic layer was discarded. To the aqueous solution containing the reddish-orange anionic chelate of
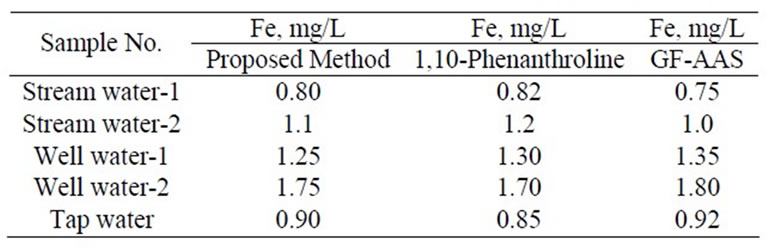
Table 3. Determination of iron in water samples n = 5.

Table 2. determination of iron as Fe2O3 in silicate rocks, minerals, soils, stream sediments, uranium concentrates, tobacco leaves and cigarettes, n = 5.
iron(III) was added 10 ml of 3.12 × 10–2 mol·L–1 2,3- H2ND in ethyl acetate followed by 5 ml of 2.74 × 10–2 mol·L–1 CTA. This was shaken in a separating funnel for 2 min. Allowed for phase separation and the aqueous layer was discarded. The absorbance of the bright reddish-orange ion associate of CTA+ with the anionic (Fe- 2,3-H2ND) complex in ethyl acetate was measured at 450 nm against a reagent blank. This way interference of manganese, vanadium and cerium is completely removed.
4. Conclusions
The proposed method is simple, rapid and selective over other existing methods as follows:
1) Irrespective of its oxidation states, Fe forms complexes with 2,3-H2ND.
2) Concentration of 2,3-H2ND (10 ml of 3.12 × 10–2 mol·L–1) required is much less as compared with reagents like 8-hydroxyquinoline, acetylacetone and thenoyl trifluoroacetone.
3) Various metals showed different extent of extraction with 2,3-H2ND in ethyl acetate at different pH. These factors permitted the separation of iron(III) from commonly associated elements like, Ti, V, Cr, Nb, Mo, etc.
4) Equilibrium time required is much less i.e., 1 min.
5) The chelate formed is highly stable.
6) The Beer’s law range is very wide.
7) A single extraction removes move than 99% iron from the aqueous solution.
5. Acknowledgements
The authors (PKT & RKM) are thankful to Shri V. J. Katti, Regional Director, AMD, Eastern Region and Dr. K. Satyarayana, Head, Chemistry Group, AMD Hyderabad for providing facility to carry out the work. The same authors also express their gratitude to Dr. Anjan Chaki, Director, AMD for giving permission to publish the work.
REFERENCES
- P. K. Tarafder and R. K. Mondal, “A Review on the Complex Forming Ability of O-O’ Type Ligands with Transition Metals: Introducing 2,3-Dihydroxynaphthalene as a Potential Analytical Reagent,” Reviews in Analytical Chemistry, Vol. 30, No. 2, 2011, pp. 73-81. doi:10.1515/revac.2011.016
- P. K. Tarafder and R. K. Mondal, “Liquid-Liquid Extraction and Spectrophotometric Determination of Mn(II) in Geochemical Samples,” Chemia Analityczna (Warsaw), Vol. 49, 2004, pp. 251-260.
- L. Sommer, “Analytical Absorption Spectrophotometry in the Visible and Ultraviolet,” Elsevier, Budapest, 1989.
- V. Patrovsky, “2,3-Dihydroxynaphthalene as a Photometric Reagent for the Determination of Certain Metals,” Collection of Czechoslovak Chemical Communications, Vol. 35, 1970, pp. 1599-1604.
- P. K. Tarafder and A. K. Sardana, “A Rapid Method for the Spectrophotometric Determination of Iron in Silicate Rocks,” Chemia Analityczna (Warsaw), Vol. 45, 2000, pp. 145-151.
- A. I. Vogel, “A Text Book of Quantitative Chemical Analysis,” 15th edition, Longman, Essex, 1989.
- L. G. Sillen, “Some Graphical Methods for Determining Euilibrium Constants II. On ‘Curve-Fitting’ Methods for Two-Variable Data,” Acta Chemica Scandinavica, Vol. 10, 1956, pp. 185-202.
NOTES
*Corresponding author.