Open Journal of Respiratory Diseases
Vol.2 No.3(2012), Article ID:21606,10 pages DOI:10.4236/ojrd.2012.23010
Systemic Inflammation in Patients with Chronic Obstructive Pulmonary Disease: Results from the Cosmic Study
1Department of Respiratory Medicine, Maastricht University Medical Center (MUMC), Maastricht, The Netherlands
2Program Development Center, Center of Expertise for Chronic Organ Failure, Horn, The Netherlands
3Department of Respiratory & Critical Care Medicine & Ludwig Boltzmann Institute for COPD and Pulmonary Epidemiology, Otto Wagner Hospital, Vienna, Austria
4Department of Biostatistics, Erasmus Medical Center, Rotterdam, The Netherlands
5Department of Pulmonology and Tuberculosis, GRIAC Research Institute, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands
Email: *marie.breyer@gmx.at, ericarutten@ciro-horn.nl, martijnspruit@ciro-horn.nl, w.hop@erasmusmc.nl, d.s.postma@umcg.nl, e.wouters@mumc.nl
Received April 20, 2012; revised May 28, 2012; accepted June 14, 2012
Keywords: Adipokines; Body Composition; CRP, Fibrinogen; Obesity; TNFα
ABSTRACT
Objective: The study aims to elucidate the association of host-related factors on systemic inflammation in COPD patients. Methods: In 295 clinically stable and optimally treated COPD patients from 39 outpatient centers, age, gender, and body composition (body mass index, BMI; fat-free mass index, FFMI; fat mass index, FMI) were related to inflammatory biomarkers: CRP, fibrinogen, TNFα, and its soluble receptors (s)TNFαR1 and sTNFαR2. Furthermore, forced expiratory volume in the first second (FEV1), BMI, FFMI, and FMI were stratified by quartiles to elucidate the influence on inflammatory biomarkers. Monovariate and multivariate regression analyses were performed for associations between inflammatory biomarkers. Results: Positive correlations were found for FFMI with sTNFαR1, FMI with CRP and age with TNFα, sTNFαR1 and sTNFαR2 (p < 0.01). FEV1 was not correlated with body composition and inflammatory markers. Monoand multivariate analysis showed weak correlations between the acute phase markers and the TNFα system after correcting for multiple co-variants. Conclusions: This study highlights the modest role of age and body composition on levels of systemic inflammatory biomarkers in COPD. Results show the degree of airflow limitation does not affect systemic inflammation. Last, a weak relationship between acute phase markers and markers of the TNFα system is present in COPD.
1. Introduction
Chronic obstructive pulmonary disease (COPD), until recently exclusively characterized by poorly reversible airflow limitation, is now approached as a heterogeneous, multi-component disease condition [1]. Supporting this notion, patients with COPD generally have low grade systemic inflammation compared with healthy elderly subjects [2-4].
With respect to the high heterogeneity of systemic inflammation, serum C-reactive protein (CRP), fibrinogen, and tumour necrosis factor alpha (TNFα) and its soluble receptors (s)TNFαR1 and 2 have been typically studied as systemic inflammatory biomarkers, yet in rather small-sized studies [3,5]. CRP, a systemic biomarker of the acute-phase response, is increased in COPD compared to healthy subjects [6] and is related to an increased risk for cardiac injury [7], hospitalizations [8], and worse survival [9]. Fibrinogen is associated with worse lung function and poor prognosis in COPD [5,10]. The TNFα system is often investigated in relation to the genesis of cachexia in COPD [11]. Together, available data show that increased systemic inflammation results in clinically relevant events in COPD.
Chronic low-grade systemic inflammation is present in all subjects with stable COPD and may reflect spill-over of the airways and parenchyma, promoting a generalised inflammatory reaction [2]. However, this concept has not been proven so far [3]. Indeed, others could only find little or no statistically significant association between disease severity based on airflow limitation and CRP and TNFα in COPD [12]. Besides lung inflammation, hostrelated factors, such as higher age, female sex, higher body mass index, and smoking contribute to increased systemic inflammation in COPD [13-15].
The aim of the present study was to evaluate the influence of host-related determinants, including age, gender, body composition, and smoking status on systemic inflammatory biomarkers in a large sample of clinically stable, optimally treated outpatients with COPD. Furthermore, the interrelationship between these circulating inflammatory biomarkers is studied.
2. Methods
Data were collected from 295 COPD patients participating in the COSMIC (COPD and Seretide: A multi-center intervention and characterization) study (post-bronchodilator FEV1 between 30% and 80% predicted; FEV1/FVC ratio < 70% predicted) at 39 outpatient centers in the Netherlands [16]. The central ethical committee in the Netherlands and the local medical ethics committees of the participating centers approved the study. All subjects gave written informed consent. Patients between 40 and 75 years of age, current or ex-smokers and with established COPD were included. Performance of lung function and measurement of body composition parameters have been reported elsewhere [17]. Briefly, body-, fat free-, and fat mass index (BMI, FFMI, FMI) were calculated. All patients were inhaling a combination therapy including a beta2agonist as well as a corticosteroid twice daily for at least 12 weeks prior to study entry (25 µg Salmeterol and 50 (COPD II), 125 (COPD III), and 250 µg (COPD IV) Fluticasone propionate = Seretide®). In this period, patients had no exacerbations to obtain maximum stability in the clinical condition of the COPD patients included. The clinical records were searched for the number of pack years (PY; the number of packs of cigarettes smoked per day multiplied by the number of years) and patients smoking status (current or former smoker).
Plasma high-sensitivity CRP was assessed by a particle-enhanced immune-nephelometry (BN Prospec, Dade Behring). The lower detection level for hs-CRP was 0.175 mg/L with a range of 0.175 - 1100 mg/L, depending on the dilution. Fibrinogen was assessed on Sysmex (Dade-Behring) CA-7000 coagulation analyzer according to the Clauss method and calculated from EDTA to citrate plasma values. The measuring range was 0.3 - 10.0 g/L. TNFα was determined with the Quantikine® high sensitivity ELISA from R&D systems (Minneapolis, USA). sTNFαR1 and R2 were measured by enzymelinked immunosorbent assay (ELISA) as described previously [18]. Lower detection level for TNFα was 0.5 pg/mL. Lower detection levels for the assays were 40 ng/mL for sTNFαR1 and 70 ng/mL for sTNFαR2.
3. Statistical Analyses
Values are given by median and 25th - 75th percentiles. The Kruskal-Wallis nonparametric test was performed to evaluate differences in median levels of systemic inflammatory biomarkers and body composition markers by quartiles. Afterwards, the post-hoc nonparametric Mann Whitney U Test was performed. Linear dependency between the systemic inflammatory biomarkers and the host-related determinants were tested using the Pearson correlation coefficient and non-parametric dependency within the systemic inflammatory biomarkers was calculated using the Spearman correlation coefficient. The stepwise multivariate linear regression model was performed with a single systemic inflammatory biomarker as dependent variables and the host-relating factors and the remaining inflammatory biomarkers as independent variables. In these analyses all systemic inflammatory biomarkers, except fibrinogen, were transformed logarithmically to approximate normal distributions; the body composition markers BMI, FFMI and FMI showed no co-linearity. Analyses were performed using Statistical Package for the Social Sciences (SPSS) version 15.01 for Windows. A priori, p < 0.05 (two-sided) was considered the level of significance.
4. Results
4.1. Characteristics
Patients were generally having mild to moderate COPD, a high BMI and a normal FFMI (Table 1). Median levels of systemic inflammatory biomarkers were 3.88 mg/L for CRP, 3.45 g/L for fibrinogen, 1.63 pg/mL for TNFα, 0.83 ng/mL for sTNFαR1, and 1.81 ng/mL for sTNFαR2. Female COPD patients were younger, more frequently current smokers, had a higher FMI and lower median levels of sTNFαR1 than males.
In order to evaluate an effect of disease severity on systemic inflammatory biomarkers and body composition, patients were stratified into quartiles based on FEV1% predicted. No differences were found for CRP, fibrinogen, TNFα and its soluble receptors or any of the body composition markers between FEV1 quartiles (Table 5 in the online supplement).
4.2. Influence of Host-Related Factors on Systemic Inflammatory Biomarkers (Table 2)
While none of the systemic inflammatory biomarkers correlated with gender, and smoking status, fibrinogen, TNFα and its soluble receptors correlated positively with age. Additionally, sTNFαR1 levels correlated positively with gender. Both BMI and FMI were positively correlated with plasma levels of CRP, sTNFαR1 and sTNFαR2, while FFMI was positively correlated with sTNFαR1.
Stratifying BMI, FFMI and FMI into quartiles, median levels of CRP were lower in the first quartile of FMI compared to the third and fourth quartile (Figure 1(a)). Median levels of sTNFαR1 were significantly lower in the first and second quartiles of BMI compared to the fourth quartile (Figure 1(b)), and were lower in the first quartile of FFMI compared to the third and fourth quartile (Figure 1(c)). No differences in fibrinogen, TNFα, and sTNFαR2 levels were observed with quartiles in body composition markers (Table 6 for BMI, Table 7 for FFMI, and Table 8 for FMI in the online supplement).
4.3. Association between the Different Systemic Inflammatory Biomarkers (Table 3)
A moderate correlation between CRP and fibrinogen was found, while weaker correlations were found between CRP and the TNFα system. TNFα correlated weakly with its soluble receptors. Moreover, the TNFα system correlated weakly with fibrinogen. All correlations showed a positive direction.
4.4. Stepwise Multivariate Regression Analysis of Individual Systemic Inflammatory Biomarkers
Fibrinogen and BMI explained 33% of the variance in CRP (Table 4), while CRP and age explained 32% of the variance in fibrinogen. When analysing TNFα as dependent variable, only sTNFαR2 remained in the model; using sTNFαR1 as dependent variable, TNFαR2, age, CRP, BMI and fibrinogen remained in the final model, while sTNFαR1 and TNFα were independent determinants of sTNFαR2.
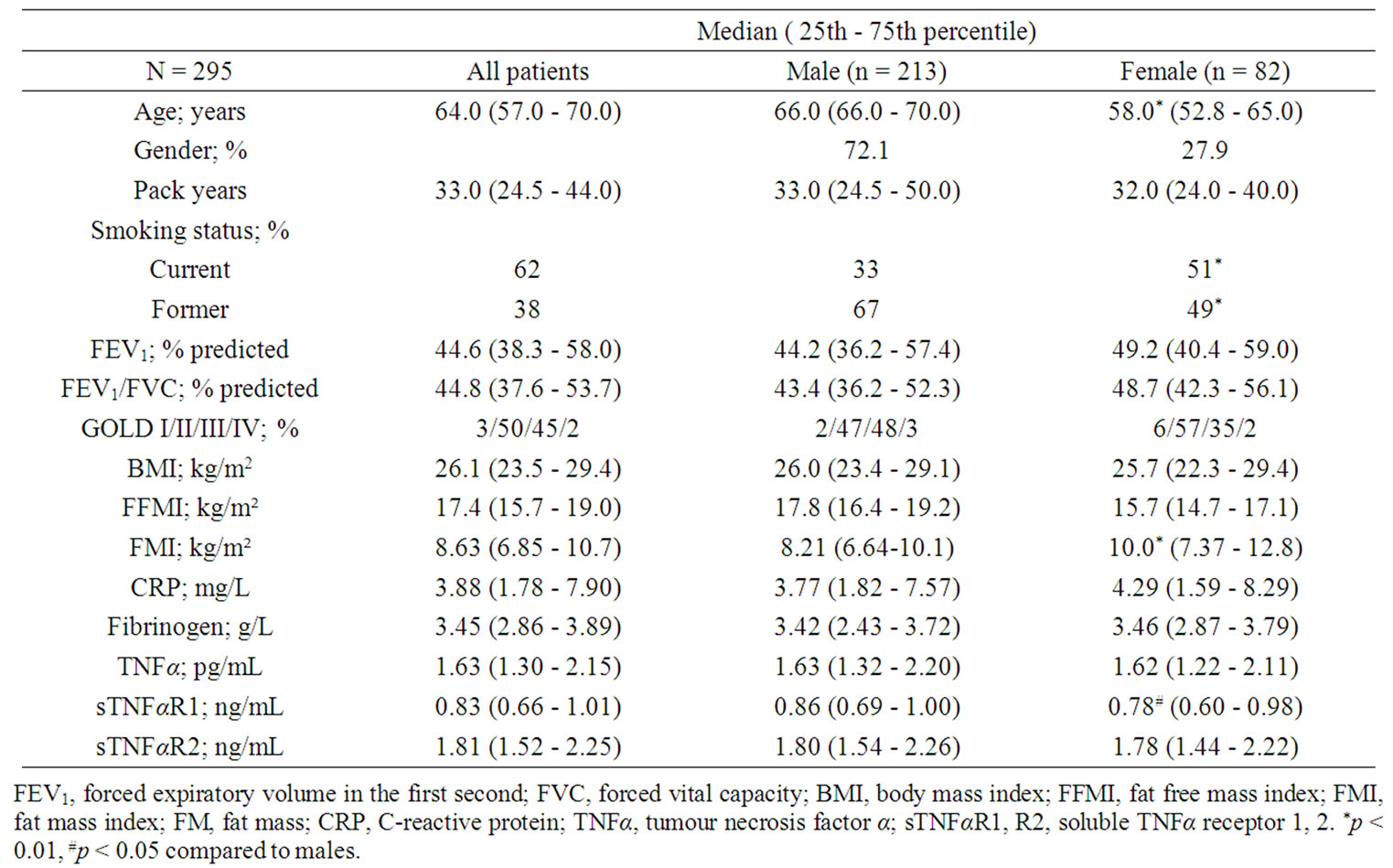
Table 1. Baseline characteristics.
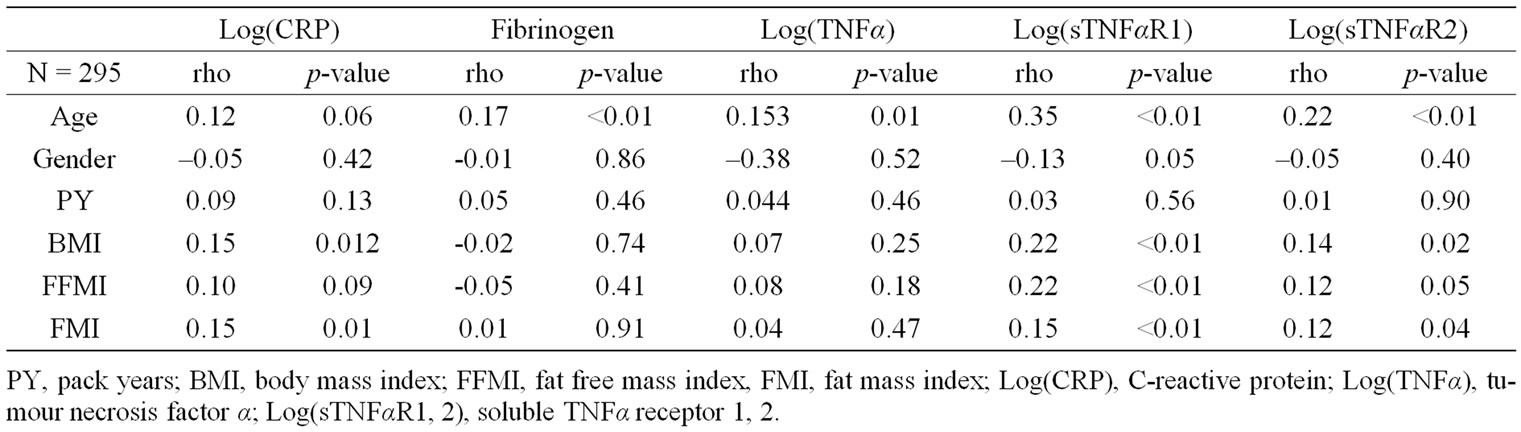
Table 2. Pearson monovariate correlation coefficients of the studied systemic inflammatory biomarkers with host-related determinants.
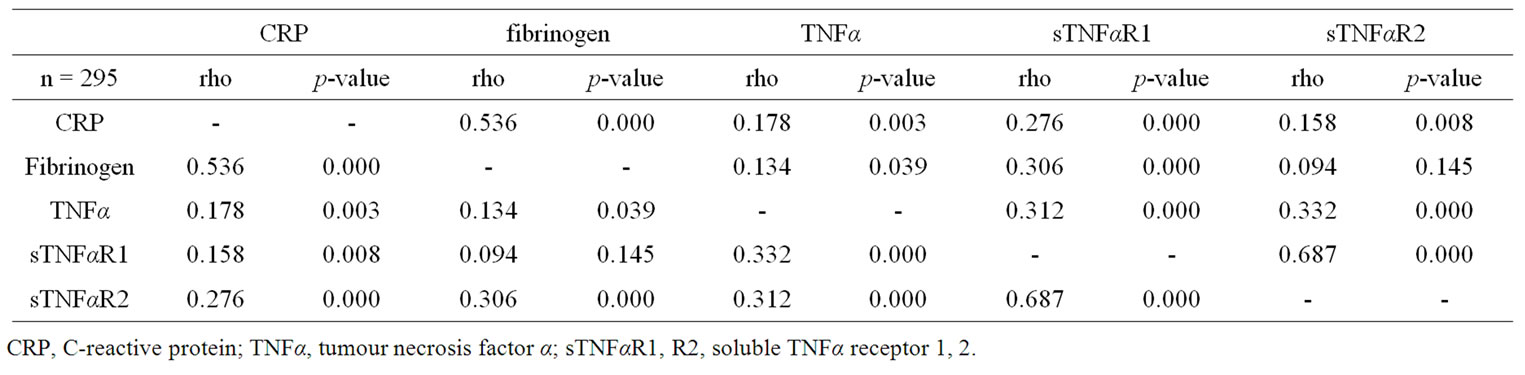
Table 3. Spearman monovariate correlation coefficients of the studied systemic inflammatory biomarkers.

Table 4. Stepwise multivariate regression analysis with various systemic inflammatory biomarkers as dependent variable.
 (a)
(a)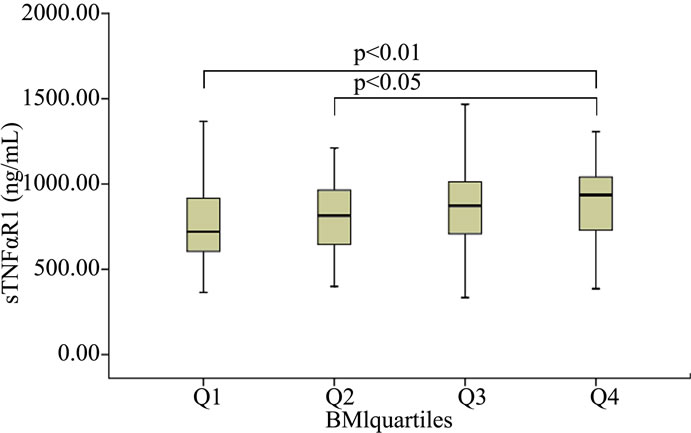 (b)
(b) (c)
(c)
Figure 1. Median levels of CRP after stratification of FMI by quartiles (a), median levels of sTNFαR1 after stratification of BMI by quartiles (b) as well as FFMI (c).
5. Discussion
The present study confirms moderate associations of host-related factors, i.e. BMI and age, with systemic inflammatory biomarkers, such as CRP, fibrinogen and sTNFαR1. Otherwise, the degree of airflow limitation, gender, fat free mass index, fat mass index and smoking status do not seem to significantly explain the variance observed in the studied systemic inflammatory biomarkers in patients with mild-to-moderate COPD. Last, a weak relationship between acute phase markers and markers of the TNFα system is present in COPD patients.
5.1. Disease-Related Factors
The current study confirms previous reports that inflammatory markers cannot discriminate on disease stages of COPD [12,19]. However, one study found decreasing levels of sTNFαR1 with lower FEV1 values in multiple regression analysis [20]. These data could not be confirmed in the present study. Others have reported that by applying protein microarray platform technology, systemic biomarkers are associated with different clinical variables including the degree of airflow limitation and carbon monoxide transfer factor [21]. Carbon monoxide transfer factor was not assessed in the present study. The last mentioned finding implies an association between systemic inflammation and the level of emphysema, which was not assessed in the present study. In addition, the study by Pinto-Plata and colleagues [21] included other biomarkers as in the present study. Based on the concept of spill-over [2] and the assumed progressive inflammation with more impaired lung function [22], a more pronounced systemic inflammatory response with progressive airflow limitation could be hypothesised. However, an important overlap in airway inflammation has been reported between different GOLD stages as well as within each GOLD stage category [23]. The same authors reported no relationship between local and systemic inflammation, confirming previous reported data that challenge the spill-over theory [3]. These data favour the concept that additional confounders contribute to the systemic inflammation in COPD. The absence of a relationship between progressive airflow limitation and systemic inflammation we and others have found cannot rule out a contributing role of emphysema in this respect. Further studies using state of the art diagnostic procedures of emphysema are needed to unravel the influence of emphysema on systemic inflammation.
5.2. Host-Related Factors
The results from the present study show increasing CRP levels with increasing BMI and FMI values. Furthermore, BMI remained in the final multivariate regression model on CRP which is in line with a positive relationship of increased levels of CRP in obese patients with COPD (BMI ≥ 30 kg/m2) compared to normal peers [13]. Levels of fibrinogen showed no relation with body composition markers. The soluble TNFα receptor levels correlated positively with BMI in monovariate analyses and even after correcting for confounders in the multiple regression analyses, BMI independently determined levels of sTNFαR1. In line, Takabatake and colleagues reported an association between both sTNFα receptors and the percentage of fat mass in COPD [24]. A recent manuscript concluded that both circulating CRP and sTNFαR1 were positively associated with fat mass rather than fat free mass [20], data which are confirmed in the present study.
Aging induces an inflammatory response and this has been proposed as a key for age-related diseases [25,26]. An age-dependent increase in the TNFα system in the elderly population has been reported [27]. Despite the small age range of the present study (25th - 75th percentile: 57 - 70 years), our analysis showed that age remained an independent determinant for TNFαR1 and fibrinogen in the multivariate regression models. Epidemiological data suggest an age-related increase of serum levels of fibrinogen [28], based on an age-related slower rate of disposal of fibrinogen [29].
We did not find a gender effect on the analysed markers of systemic inflammation, which was also found in other studies [20]. However, worth noting, differences in other mediators as the Adipokines seem gender related. Increased levels of leptin have been reported in obese COPD women compared to non-obese counterparts [30]. In addition, increased levels of circulating leptin might lead to an increased CRP production from hepatocytes due to direct signalling pathways [31]. This is in line with an observed increase in CRP levels with increasing body weight (reflecting an increased amount of adipose tissue) in female COPD patients [32], implying that, although no gender differences could be detected in the present study, the signalling pathways of systemic inflammation could be gender dependent. Indeed, it has recently been shown that other inflammatory biomarkers like Interleukin-6, Interleukin-16 and the vascular endothelial growth factor are gender dependent in patients with COPD [33].
The direct adverse effects of inhaled cigarette smoke on lung health [34] and on other organ systems as the result of an indirect exposure are well established [35]. Although cigarette smoke is the main risk factor for COPD, the relationship between smoke exposure and the degree of airflow limitation is only poor [36]. In the present study no effect of pack years or smoking status was found on FEV1. In the general population a dose-response as well as a dose-effect (number of pack years) relationship between cigarette smoking and increased levels of CRP and fibrinogen is reported [37-39]. Interestingly, until 19 years after smoking cessation CRP levels are still increased and do not revert to levels of never smokers [40]. Indeed, no differences in CRP levels were found between current and former smokers in the present study (data not shown). However, a sample-size effect cannot be ruled out. Limited data are available on the effects of cigarette smoke on levels of TNFα. While some report increased levels of TNFα in healthy smokers [41] others failed to confirm these results [42]. We also found no effect of smoking status or pack years on levels of the TNFα system.
Consensus is growing that the inflammatory processes that underlie COPD are probably mediated by a multitude of cytokines. In the present study we have focused on the acute phase markers CRP and fibrinogen and on markers of the TNFα system. These markers are most typically studied in COPD patients. Increased levels of CRP and fibrinogen have been reported in stable COPD patients compared to healthy controls [2], and both seem to rise even more during acute COPD exacerbations [4,43]. Furthermore, increased CRP and fibrinogen levels have both been shown to be associated with an increased risk for cardiovascular disease and mortality in COPD [7,9]. More conflicting is the role of the TNFα system in COPD: Levels of TNFα have previously shown to be elevated in patients with COPD compared to healthy controls [44,45], whereas more recent studies have not confirmed this [11,46-48]. Even more recently, reported absolute levels appeared even lower and no differences could be found based on severity of COPD [49]. These differences could be related to assay specificity in the past years [11]. Interestingly, a clinical trial of shortterm anti-TNFα treatment in patients with COPD found no effect on the TNFα system [50]. Furthermore, using a broad panel of inflammation associated analytes, antiTNFα treatment does not affect systemic inflammatory profile in COPD [51]. The present study is the first investigating the correlation between different markers for systemic inflammation, namely markers of the acute phase response and markers of the TNFα system. A previous research in a small cohort (n = 58) found no correlation between CRP and the TNFα system [52]. These data illustrate the complexity of the role of systemic inflammation in the pathogenesis of COPD and COPD related co-morbidities.
5.3. Limitations
The following limitations have to be recognised: Firstly, CRP, fibrinogen and TNFα measures are only parts of the pathological pathways proposed in the low-grade systemic inflammation in patients with COPD and other pathways might become identified to have a more prominent role in the low-grade systemic inflammation in patients with COPD. Secondly, the lack of a control group, however this is the first study taking blood samples from patients receiving standardized combination therapy as well as having a controlled exacerbation-free period. Thirdly, in the present study no information is included about direct or indirect assessment of activity patterns. Exercise is however suggested to have a benefit role on systemic inflammation in COPD, and as it is previously shown, physical activity is related to fibrinogen levels in COPD [53], and registration of physical activity levels would be interesting.
5.4. Conclusion
In conclusion, host-related factors like body composition and age have a modest role in the systemic inflammation in optimally treated and clinically stable mild-to-moderate COPD patients. In addition, our study underscores the high heterogeneity among various biomarkers of systemic inflammation.
6. Acknowledgements
The Ludwig Boltzmann Institute for COPD and pulmonary epidemiology, Vienna, Austria funded MKB.
REFERENCES
- K. F. Rabe, et al., “Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Pulmonary Disease: GOLD Executive Summary,” American Journal of Respiratory and Critical Care Medicine, Vol. 176, No. 6, 2007, pp. 532-555. doi:10.1164/rccm.200703-456SO
- W. Q. Gan, et al., “Association between Chronic Obstructive Pulmonary Disease and Systemic Inflammation: A Systematic Review and a Meta-Analysis,” Thorax, Vol. 59, No. 7, 2004, pp. 574-580. doi:10.1136/thx.2003.019588
- J. H. Vernooy, et al., “Local and Systemic Inflammation in Patients with Chronic Obstructive Pulmonary Disease: Soluble Tumor Necrosis Factor Receptors Are Increased in Sputum,” American Journal of Respiratory and Critical Care Medicine, Vol. 166, No. 9, 2002, pp. 1218-1224. doi:10.1164/rccm.2202023
- M. A. Spruit, et al., “Muscle Force during an Acute Exacerbation in Hospitalised Patients with COPD and Its Relationship with CXCL8 and IGF-I,” Thorax, Vol. 58, No. 9, 2003, pp. 752-756. doi:10.1136/thorax.58.9.752
- M. Dahl, et al., “Elevated Plasma Fibrinogen Associated with Reduced Pulmonary Function and Increased Risk of Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 164, No. 6, 2001, pp. 1008-1011.
- D. G. Yanbaeva, et al., “IL6 and CRP Haplotypes Are Associated with COPD Risk and Systemic Inflammation: A Case-Control Study,” BMC Medical Genetics, Vol. 10, 2009, p. 23. doi:10.1186/1471-2350-10-23
- D. D. Sin and S. F. Man, “Why Are Patients with Chronic Obstructive Pulmonary Disease at Increased Risk of Cardiovascular Diseases? The Potential Role of Systemic Inflammation in Chronic Obstructive Pulmonary Disease,” Circulation, Vol. 107, No. 11, 2003, pp. 1514-1519. doi:10.1161/01.CIR.0000056767.69054.B3
- M. Dahl, et al., “C-Reactive Protein as a Predictor of Prognosis in Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 175, No. 3, 2007, pp. 250-255. doi:10.1164/rccm.200605-713OC
- S. F. Man, et al., “C-Reactive Protein and Mortality in Mild to Moderate Chronic Obstructive Pulmonary Disease,” Thorax, Vol. 61, No. 10, 2006, pp. 849-853. doi:10.1136/thx.2006.059808
- K. H. Groenewegen, et al., “Increased Systemic Inflammation Is a Risk Factor for COPD Exacerbations,” Chest, Vol. 133, No. 2, 2008, pp. 350-357. doi:10.1378/chest.07-1342
- P. D. Wagner, “Possible Mechanisms Underlying the Development of Cachexia in COPD,” European Respiratory Journal, Vol. 31, No. 3, 2008, pp. 492-501. doi:10.1183/09031936.00074807
- L. G. Franciosi, et al., “Markers of Disease Severity in Chronic Obstructive Pulmonary Disease,” Pulmonary Pharmacology & Therapeutics, Vol. 19, No. 3, 2006, pp. 189- 199. doi:10.1016/j.pupt.2005.05.001
- M. K. Breyer, et al., “Highly Elevated C-Reactive Protein Levels in Obese Patients with COPD: A Fat Chance?” Clinical Nutrition, Vol. 28, No. 6, 2009, pp. 642-647. doi:10.1016/j.clnu.2009.05.005
- D. D. Sin and S. F. Man, “Impaired Lung Function and Serum Leptin in Men and Women with Normal Body Weight: A Population Based Study,” Thorax, Vol. 58, No. 8, 2003, pp. 695-698. doi:10.1136/thorax.58.8.695
- E. F. Wouters, E. C. Creutzberg and A. M. Schols, “Systemic Effects in COPD,” Chest, Vol. 121, No. 5, 2002, pp. 127S-130S. doi:10.1378/chest.121.5_suppl.127S
- E. F. Wouters, et al., “Withdrawal of Fluticasone Propionate from Combined Salmeterol/Fluticasone Treatment in Patients with COPD Causes Immediate and Sustained Disease Deterioration: A Randomised Controlled Trial,” Thorax, Vol. 60, No. 6, 2005, pp. 480-487. doi:10.1136/thx.2004.034280
- M. A. Vermeeren, et al., “Prevalence of Nutritional Depletion in a Large Out-Patient Population of Patients with COPD,” Respiratory Medicine, Vol. 100, No. 8, 2006, pp. 1349-1355. doi:10.1016/j.rmed.2005.11.023
- J. F. Leeuwenberg, M. A. Dentener and W. A. Buurman, “Lipopolysaccharide LPS-Mediated Soluble TNF Receptor Release and TNF Receptor Expression by Monocytes. Role of CD14, LPS Binding Protein, and Bactericidal/ Permeability-Increasing Protein,” Journal of Immunology, Vol. 152, No. 10, 1994, pp. 5070-5076.
- M. Cazzola, et al., “Outcomes for COPD Pharmacological Trials: From Lung Function to Biomarkers,” European Respiratory Journal, Vol. 31, No. 2, 2008, pp. 416- 469. doi:10.1183/09031936.00099306
- T. M. Eagan, et al., “Body Composition and Plasma Levels of Inflammatory Biomarkers in COPD,” European Respiratory Journal, Vol. 36, No. 5, 2010, pp. 1027-1033. doi:10.1183/09031936.00194209
- V. Pinto-Plata, et al., “Profiling Serum Biomarkers in Patients with COPD: Associations with Clinical Parameters,” Thorax, Vol. 62, No. 7, 2007, pp. 595-601. doi:10.1136/thx.2006.064428
- J. C. Hogg, et al., “The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease,” New England Journal of Medicine, Vol. 350, No. 26, 2004, pp. 2645-2653. doi:10.1056/NEJMoa032158
- D. Singh, et al., “Induced Sputum Genes Associated with Spirometric and Radiological Disease Severity in COPD Ex-Smokers,” Thorax, Vol. 66, No. 6, 2011, pp. 489-495. doi:10.1136/thx.2010.153767
- N. Takabatake, et al., “The Relationship between Chronic Hypoxemia and Activation of the Tumor Necrosis Factor-Alpha System in Patients with Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 161, No. 4, 2000, pp. 1179-1184.
- F. Licastro, et al., “Innate Immunity and Inflammation in Ageing: A Key for Understanding Age-Related Diseases,” Immunity and Ageing, Vol. 2, 2005, p. 8. doi:10.1186/1742-4933-2-8
- M. Provinciali, M. Cardelli and F. Marchegiani, “Inflammation, Chronic Obstructive Pulmonary Disease and Aging,” Current Opinion in Pulmonary Medicine, Vol. 17, No. 1, 2011, pp. S3-S10. doi:10.1097/01.mcp.0000410742.90463.1f
- H. Bruunsgaard and B. K. Pedersen, “Age-Related Inflammatory Cytokines and Disease,” Immunology and Allergy Clinics of North American, Vol. 23, No. 1, 2003, pp. 15-39. doi:10.1016/S0889-8561(02)00056-5
- A. R. Folsom, “Epidemiology of Fibrinogen,” European Heart Journal, Vol. 16, No. A, 1995, pp. 21-24.
- A. Fu and K. S. Nair, “Age Effect on Fibrinogen and Albumin Synthesis in Humans,” American Journal of Physiology, Vol. 275, No. 6, 1998, pp. E1023-E1030.
- M. K. Breyer, et al., “Gender Differences in the Adipose Secretome System in Chronic Obstructive Pulmonary Disease (COPD): A Pivotal Role of Leptin,” Respiratory Medicine, Vol. 105, No. 7, 2011, pp. 1046-1053. doi:10.1016/j.rmed.2011.02.001
- G. Fantuzzi, “Adipose Tissue, Adipokines, and Inflammation,” Journal of Allergy and Clinical Immunology, Vol. 115, No. 5, 2005, pp. 911-920. doi:10.1016/j.jaci.2005.02.023
- P. O. Bridevaux, et al., “Sex-Specific Effect of Body Weight Gain on Systemic Inflammation in Subjects with COPD: Results from the SAPALDIA Cohort Study 2,” European Respiratory Journal, Vol. 34, No. 2, 2009, pp. 332-339. doi:10.1183/09031936.00162608
- J. P. de Torres, et al., “Gender Differences in Plasma Biomarker Levels in a Cohort of COPD Patients: A Pilot Study,” PLoS One, Vol. 6, No. 1, 2011, Article ID: e16021. doi:10.1371/journal.pone.0016021
- J. Crofton and K. Bjartveit, “Smoking as a Risk Factor for Chronic Airways Disease,” Chest, Vol. 96, No. 3, 1989, pp. 307S-312S. doi:10.1378/chest.96.3.307S
- P. Boyle, “Cancer, Cigarette Smoking and Premature Death in Europe: A Review including the Recommendations of European Cancer Experts Consensus Meeting, Helsinki, October 1996,” Lung Cancer, Vol. 17, No. 1, 1997, pp. 1-60. doi:10.1016/S0169-5002(97)00648-X
- A. Agusti, et al., “Characterisation of COPD Heterogeneity in the ECLIPSE Cohort,” Respiratory Research, Vol. 11, 2010, p. 122.
- W. B. Kannel, R. B. D’Agostino and A. J. Belanger, “Fibrinogen, Cigarette Smoking, and Risk of Cardiovascular Disease: Insights from the Framingham Study,” American Heart Journal, Vol. 113, No. 4, 1987, pp. 1006-1010. doi:10.1016/0002-8703(87)90063-9
- L. A. Bazzano, et al., “Relationship between Cigarette Smoking and Novel Risk Factors for Cardiovascular Disease in the United States,” Annals of Internal Medicine, Vol. 138, No. 11, 2003, pp. 891-897.
- J. Heinrich, et al., “Fibrinogen and Factor VII in the Prediction of Coronary Risk. Results from the PROCAM Study in Healthy Men,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 14, No. 1, 1994, pp. 54-59. doi:10.1161/01.ATV.14.1.54
- S. G. Wannamethee, et al., “Associations between Cigarette Smoking, Pipe/Cigar Smoking, and Smoking Cessation, and Haemostatic and Inflammatory Markers for Cardiovascular Disease,” European Heart Journal, Vol. 26, No. 17, 2005, pp. 1765-1773. doi:10.1093/eurheartj/ehi183
- P. H. Wirtz, et al., “Enhanced Glucocorticoid Sensitivity of Cytokine Release from Circulating Leukocytes Stimulated with Lipopolysaccharide in Healthy Male Smokers,” Brain, Behavior, and Immunity, Vol. 18, No. 6, 2004, pp. 536-543. doi:10.1016/j.bbi.2004.01.002
- M. L. Gander, et al., “Effect of the G-308A Polymorphism of the Tumor Necrosis Factor (TNF)-Alpha Gene Promoter Site on Plasma Levels of TNF-Alpha and CReactive Protein in Smokers: A Cross-Sectional Study,” BMC Cardiovascular Disorders, Vol. 4, No. , 2004, p. 17. doi:10.1186/1471-2261-4-17
- D. Stolz, et al., “Copeptin, C-Reactive Protein, and Procalcitonin as Prognostic Biomarkers in Acute Exacerbation of COPD,” Chest, Vol. 131, No. 4, 2007, pp. 1058- 1067. doi:10.1378/chest.06-2336
- C. E. Bolton, et al., “Associated Loss of Fat-Free Mass and Bone Mineral Density in Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 170, No. 12, 2004, pp. 1286-1293. doi:10.1164/rccm.200406-754OC
- N. Takabatake, et al., “Circulating Leptin in Patients with Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 159, No. 4, 1999, pp. 1215-1219.
- S. Yende, et al., “Inflammatory Markers Are Associated with Ventilatory Limitation and Muscle Dysfunction in Obstructive Lung Disease in Well Functioning Elderly Subjects,” Thorax, Vol. 61, No. 1, 2006, pp. 10-16. doi:10.1136/thx.2004.034181
- K. Ito and P. J. Barnes, “COPD as a Disease of Accelerated Lung Aging,” Chest, Vol. 135, No. 1, 2009, pp. 173- 180. doi:10.1378/chest.08-1419
- A. A. Eid, et al., “Inflammatory Response and Body Composition in Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 164, No. 8, 2001, pp. 1414-1418.
- H. A. Van Helvoort, et al., “Exercise-Induced Systemic Effects in Muscle-Wasted Patients with COPD,” Medicine & Science in Sports & Exercise, Vol. 38, No. 9, 2006, pp. 1543-1552. doi:10.1249/01.mss.0000228331.13123.53
- H. van der Vaart, et al., “First Study of Infliximab Treatment in Patients with Chronic Obstructive Pulmonary Disease,” American Journal of Respiratory and Critical Care Medicine, Vol. 172, No. 4, 2005, pp. 465-469. doi:10.1164/rccm.200501-147OC
- M. J. Loza, et al., “Systemic Inflammatory Profile and Response to Anti-Tumor Necrosis Factor Therapy in Chronic Obstructive Pulmonary Disease,” Respiratory Research, Vol. 13, No. 1, 2012, p. 12. doi:10.1186/1465-9921-13-12
- U. Kolsum, et al., “The Repeatability of Interleukin-6, Tumor Necrosis Factor-Alpha, and C-Reactive Protein in COPD Patients over One Year,” International Journal of Chronic Obstructive Pulmonary Disease, Vol. 4, 2009, pp. 149-156. doi:10.2147/COPD.S5018
- B. K. Pedersen and H. Bruunsgaard, “Possible Beneficial Role of Exercise in Modulating Low-Grade Inflammation in the Elderly,” Scandinavian Journal of Medicine & Science in Sports, Vol. 13, No. 1, 2003, pp. 56-62. doi:10.1034/j.1600-0838.2003.20218.x
Competing Interest
MKB, EPAR and MAS have no competing interests.
WCJH is a regular biostatistical consultant to GSK but has no competing interest.
DP has received money to her institution for consulting with AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Nycomed, and Teva, and grants to her institution by AstraZeneca, Chiesi and GSK.
EFMW is receiving fees for membership on the board of Nycomed and Glaxo Smith Kline, speaking fees from AstraZeneca, GlaxoSmithKline, Boehringer Ingelheim, Danone and Novartis, and grants from AstraZeneca, GlaxoSmithKline and Nycomed.
Online Supplement
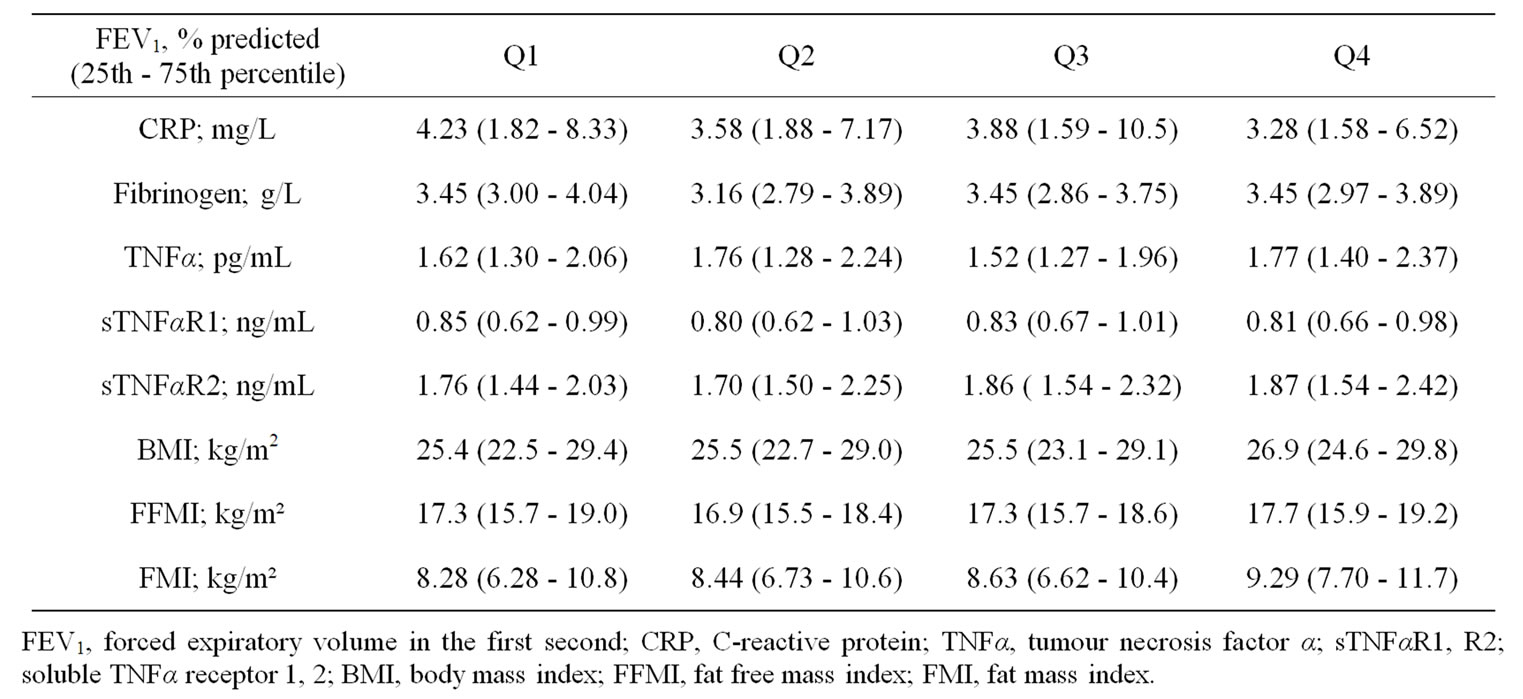
Table 5. Systemic inflammatory and body composition markers after stratification for disease severity (FEV1) by quartiles (Q1 - Q4).
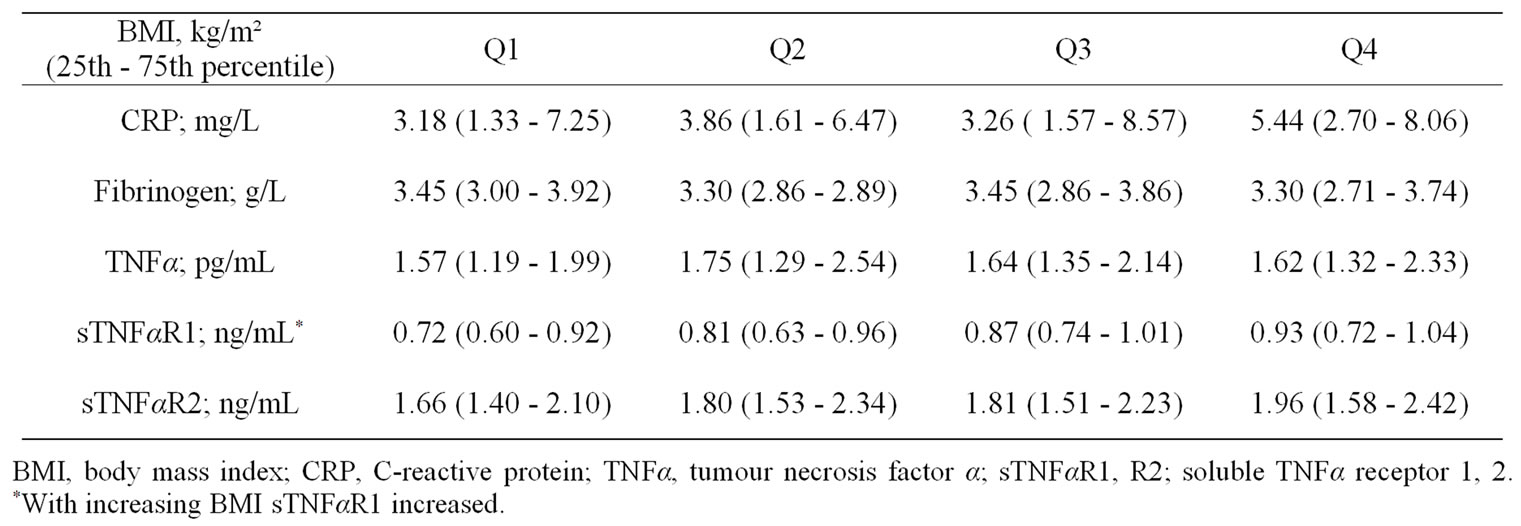
Table 6. Systemic inflammatory biomarkers after stratification for BMI by quartiles (Q1 - Q4).

Table 7. Systemic inflammatory biomarkers after stratification for FFMI by quartiles (Q1 - Q4).

Table 8. Systemic inflammatory biomarkers after stratification for FMI by quartiles (Q1 - Q4).
NOTES
*Corresponding author.