International Journal of Clinical Medicine
Vol.05 No.20(2014), Article ID:51181,8 pages
10.4236/ijcm.2014.520158
Enzymatic Activity of Renal H-K-ATPase in the Outer Medullary Collecting Duct of Transgenic Mice
Tyler Downing, Amy Mangla, Michael Banta, Suguru Nakamura
Department of Biological Sciences, Jesse D. Jones College of Science, Engineering and Technology, Murray State University, Murray, USA
Email: snakamura@murraystate.edu
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 August 2014; revised 16 September 2014; accepted 15 October 2014
ABSTRACT
The H-K-ATPase (HKA), a potassium-dependent proton transporter in the outer medullary col- lecting duct (OMCD) plays an important role in acid-base homeostasis. The OMCD contains two HKA isoforms; gastric (HKAα1), dominant under normal dietary conditions (ND), and colonic (HKAα2), induced under a K-free diet (KD). The enzymatic activity (EA) of HKA in the OMCD is in- completely understood. The focus of the present study is elucidating the EA of the HKA in HKAα1 and HKAα2 knockout (KO) mice under ND and KD. KO mice were subjected to ND or KD for 10 days. Ten OMCD tubules were extracted, half placed in potassium-free media (Solution 2), half in potassium-containing media (Solution 3). Fluorescence measurements are based on the hydroly- sis of ATP to ADP, coupled with the oxidation of NADH. ADP is determined by a decrease in NADH fluorescence. In K presence, NADH fluorescence of HKAα1 KO mice read 13.5 ± 0.7 ppm for ND and 10.3 ± 0.2 ppm for KD, indicating stimulation of the colonic isoform. HKAα2 KO mice averaged 6.8 ± 0.3 ppm for ND and 5.4 ± 0.3 ppm for KD in solution 2 (p < 0.002). Solution 3 readings were 6.0 ± 0.3 ppm for ND and 4.6 ± 0.2 ppm for KD (p < 0.0005). K addition produced significant changes in NADH fluorescence of ND and KD KO mice. The results demonstrated potassium depletion’s asso- ciation with increased EA of H-K-ATPase in OMCD, consistent with the activation of HKAα2 isoform. A significant difference in ATP production in HKAα2 KO mice is likely due to enhanced EA of H- ATPase under potassium depletion.
Keywords:
H-K-ATPase (HKA), Outer Medullary Collecting Duct (OMCD), HKA Isoforms, Gastric (HKAα1) and Colonic (HKAα2), K-Free Diet (KD), Enzymatic Activity (EA), Normal Dietary Conditions [ND], Knockout (KO)

1. Introduction
The outer medullary collecting duct (OMCD) plays an important role in acid-base homeostasis by mediating transepithelial bicarbonate transport and urinary acidification. Bicarbonate absorption by type A intercalated cells (ICs) in the OMCD segment is thought to be mediated by an apical H-K-ATPase coupled to a basolateral Chloride-Bicarbonate exchanger (AE1) [1] . The evidence for a predominant role of H-K-ATPase(s) in luminal acidification is provided by studies that examine the effect of pharmacological agents and luminal K removal on the rate of net bicarbonate absorption (JtCO2) in the OMCD. Specifically, our previous studies indicate the pres- ence of at least two H-K-ATPase (HK) isoforms, HKAα1 (gastric) and HKAα2 (colonic), in OMCD cells. HKAα1 is sensitive to Sch-28080 and insensitive to ouabain, whereas HKAα2 is sensitive to ouabain and insensitive to Sch-28080. Under potassium-depleted conditions, HKAα2 increased in its mRNA or protein abundance and me- diated bicarbonate absorptionin hypokalemia associated with metabolic alkalosis. We also demonstrated active proton secretion (JtCO2), one-third of which was blocked by bafilomycin, a specific inhibitor of the H-ATPase. Molecular studies have demonstrated the third H-K-ATPase isoform (HKβ) in the kidney. It has been reported that HKβ subunit is required for acid-secretory activity of parietal cells in vivo [2] [3] .
The H-K-ATPase ion pump responds to changes in dietary potassium balance. Functional studies support a significant role for the gHKA isoform under normal diet conditions, but do not support the same conclusion for the cHKA isoform. Although much is known about the physiological function of the H-K-ATPase isoforms, very little has been determined about the enzymatic activity of the transporters. The transporters work via active transport of ATP, thus, the goal of the experiment was to determine the contribution of enzymatic activity to ion transport. We had expected to see the enzymatic function to be consistent with the physiological function. In or- der to determine the enzymatic activity, we used an ATPase assay that was successfully used in the past, in which the hydrolysis of ATP to ADP is coupled via the oxidation of NADH [4] .
2. Methods
2.1. Micro Dissection of Tubule Segments
After the kidneys were removed from the animals, they were placed on plastic dissecting dishes and cut into thin horizontal slices. The slices were placed immediately into a dissecting solution containing NaCl 119 mM, KCL 3 mM, CaCl2 1 mM, MgSO4 1.2 mM, K2HPO4 1 mM, KH2PO4 1 mM, Glucose 5 mM, L-alanine 6 mM, and NaHCO3 25 mM (Solution 1). The dissecting solution was maintained at 14˚C. Ten individual tubules from the OMCD were dissected from the darkened renal medulla [5] .
2.2. Preparation of Nephron Segments for Enzyme Assay
Scientists have observed that to get the maximal ATPase activity in the micro-dissected renal segments, the tis- sue must be given an osmotic and temperature shock [4] [6] - [9] . The increase in ATPase activity was due to an increase in the permeability of cells to ATP. Therefore, the micro-dissected OMCD segments were transferred to a micro-centrifuge containing 100 μl of a hypotonic solution (1.0 mM Imidazole, 1.0 mM MgCl2, and 0.50 mM of EDTA) and placed in a −24˚C freezer for 40 min. After 40 min, the sample was thawed. Half of the sample was then placed in a K+ free imidazole buffer (25.0 mM Imidazole, 30.0 mM MgCl2, 7.50 mM Sodium Azide, Solution 2) while the remaining sample was put into a K+ imidazole buffer (other chemicals were same as above, Solution 3). Without K+ in the imidazole buffer, the K-ATPase function would be inhibited. Additionally, So- dium Azide was added to the buffer mixture to decrease the extraneous ATPases found within the tubules [4] [6] - [9] .
2.3. Incubation of Micro-Dissected Nephron Segments for ATP Hydrolysis
The ATP hydrolysis reaction was initiated by the addition of a starting buffer to each of the separated samples (50 mM Imidazole, 1.8 mM Phosphoenolpyruvate, 3.3 mM Na2ATP, 0.05 mM NADH, 2.5 μl Lactate dehydro- ganse, and 17.3 μl pyruvate kinase). Each sample was then placed in a 37˚C water bath for 30 min. After 30 min, one of each sample was put into a micro-cuvette and tested in a TD-700 Turner Microflurometer in which the NADH fluorescence was measure [4] [6] - [9] .
2.4. ATPase Assay
The measurement of net ATP production is based on the hydrolysis of ATP to ADP with the subsequent oxida- tion of NADH as shown in the equation below:
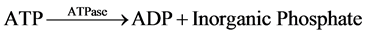
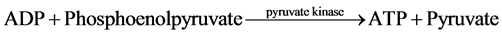
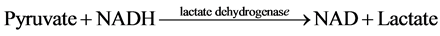
The NADH fluorescence of the starting buffer was measured without the presence of tubule samples in order to calibrate the machine and standardized the enzymatic relationship of NADH to ATP production. Previous stu- dies demonstrated that the NADH fluorescence is inversely proportional to ADP generation [4] [6] - [9] . Thus, ADP generated in the assay was determined by a decrease in NADH fluorescence.
3. Statistics
The data is expressed as means +/− standard error where appropriate. Prior to the experiment it was determined that a one-tailed, two sample t-test would be employed to analyze the data. The statistical analysis was per- formed using “R” software. p values < 0.05 were considered statistically significant.
4. Materials
The test species included genetically altered transgenic mice. The Brown Swiss HKAα1 KO mice did not con- tain the gastric isoform while the HKAα2 KO mice did not contain the colonic isoform [10] [11] . Animals were housed in the College of Science Animal Facility and were given a normal mouse diet ad lib. All animals were cared for in accordance with United States Department of Agriculture animal guidelines. The mice were eutha- nized by intraperitoneal injection of sodium pentobarbital (50 mg/ml) and for EA measurements; the kidneys were used on the same day the animals were sacrificed.
5. Results
The experiment groups of mice we selected were HKAα1 KO and HKAα2 KO. These two types of transgenic mice were under ND or KD diet. After 10 days of ND or KD diet, mice were sacrificed and tubules were dissect- ed. Follow the procedure of EA measurements (mentioned in the method section). The ADP generated in the as- say system is determined by a decrease in NADH fluorescence. There is a 1:1 relationship between ADP utiliza- tion and NADH oxidation [4] [6] .
As shown in Figure 1, in the presence of K+ (Solution 3) the NADH fluorescence of HKAα1 KO mice read 13.5 ± 0.7 ppm for ND and 10.3 ± 0.2 ppm for KD, indicating stimulation of the colonic isoform. In the absence of K+ (Solution 2), the NADH fluorescence of HKAα1 KO mice read 14.81 ± 1.26 ppm for ND and 10.20 ± 0.3 ppm KD, which indicated an increased H-K-ATPase activity under low K+ diet (See Table 1).
As shown below in Figure 2, HKAα2 KO mice averaged 6.8 ± 0.3 ppm for ND and 5.4 ± 0.3 ppm for KD in K+ free solution (Solution 2) (p < 0.002). K+ containing solution (Solution 3) readings were 6.0 ± 0.3 ppm for ND and 4.6 ± 0.2 ppm for KD (p < 0.0005). In HKAα2 KO mice in which the HKAα2 gene was disrupted, the results show under K+ containing solution (Solution 3) it is presumably mediated by the gastric H-K-ATPase. Compared to K+ free solution (Solution 2), the activity we observed presumably is due to H-ATPase (See Table 1).
Figure 3 shows a comparison of HKAα1 KO mice and HKAα2 KO mice under ND. Through this comparison, we understand the difference and the regulation between gHKA and cHKA under ND. The results indicate in HKAα2 KO mice, the activity of gHKA (observed in K+ containing solution) is closer to H-ATPase activity (observed in K+ free solution). As well as in HKAα1 KO mice, the activity of cHKA (observed in K+ containing solution) is closer to H-ATPase activity (observed in K+ free solution). However, we see a significant increase in activities of H-ATPase and cHKA in HKAα1 KO mice. In other words, without gHKAα1 gene there is up regu- lation of H-ATPase and cHKA.
The results in Figure 4 show a comparison between HKAα1 KO mice and HKAα2 KO mice under KD. The

Figure 1. A comparison of NADH fluorescence of HKAα1 KO under a normal or K+ deprivation diet. The hydrolysis of ATP to ADP with the subsequent oxidation of NADH. No K+ and K+ refer to the solution type in which the tubules were incubated. (p < 0.03 for comparison of HKAα1 KO ND vs. KD with no K+; p < 0.02 for comparison of HKAα1 KO ND vs KD with K+).

Figure 2. A comparison of NADH fluorescence of HKAα2 KO under normal or K+ deprivation diet. The hydrolysis of ATP to ADP with the subsequent oxidation of NADH. No K+ and K+ refer to solution type in which the tubules were incubated. (p < 0.001 for comparison of HKAα2 KO ND vs. KD with no K+ as well as with K+).
data has shown a significant increase in cHKA and H-ATPase activities in the HKAα1 KO mice. It indicates that a lack of HKAα1 gene can be compensated by an increase in activities mediated by cHKA and H-ATPase under low K+ conditions.
6. Discussion
It has been previously observed that gHKA is responsible for H+ secretion and K+ reabsorption under normal die- tary conditions [3] [12] . cHKA does not play a role under normal conditions; however, it shows up regulation under K+ depletion.
Our previous data have shown that HCO3-reabsorption increases in the OMCD during chronic hypokalemia [2] . Two possible mechanisms through which enhanced
 reabsorption can occur in the OMCD are via the vacuolar-type proton pump (H-ATPase) and the H-K-ATPase, both of which are present in intercalated cells (ICs). Hypokalemia is very commonly associated with metabolic alkalosis.
reabsorption can occur in the OMCD are via the vacuolar-type proton pump (H-ATPase) and the H-K-ATPase, both of which are present in intercalated cells (ICs). Hypokalemia is very commonly associated with metabolic alkalosis.
Hypokalemia causes metabolic alkalosis by three mechanisms. The initial effect is by causing a transcellular shift in which K leaves and H+ enters the cells, thereby raising the extracellular pH. The second effect is by

Table 1. ND indicates normal diet, KD indicates potassium depletion diet, no K+ re- presents potassium free media/solution, and K+ represents potassium-containing me- dia/solution for the experiment. The data is expressed as means +/− standard error where appropriate. p values < 0.05 were considered statistically significant. Mean NADH values are inversely proportional to the ADP generated in the assay. N represents the number of measurements.

Figure 3. A comparison of NADH fluorescence of HKAα1 and 2 KO mice under a normal diet. No K+ and K+ refer to the solution type in which the tubules were incu- bated. (p < 0.001 for no K+ comparison of HKAα1 KO and HKAα2 KO as well as for K+ comparison of HKAα1 KO and HKAα2 KO).
causing a transcellular shift in the cells of the proximal tubules resulting in an intracellular acidosis, which pro- motes ammonium production and excretion. Thirdly, in the presence of hypokalemia, hydrogen secretion in the proximal and distal tubules increases. This leads to further reabsorption of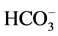 . The net effect is an increase in the net acid excretion. During potassium depletion, which occurs as a consequence of a low-K+ diet, JtCO2 increases in the OMCD. Much of the enhanced H+ secretion associated with the K+―restricted diet was inhi- bited by ouabain, a specific blocker of the cHKA.
. The net effect is an increase in the net acid excretion. During potassium depletion, which occurs as a consequence of a low-K+ diet, JtCO2 increases in the OMCD. Much of the enhanced H+ secretion associated with the K+―restricted diet was inhi- bited by ouabain, a specific blocker of the cHKA.

Figure 4. A comparison of NADH fluorescence of both HKA isoforms under K+ deprivation diet. No K+ and K+ refer to the solution type in which the tubules were incubated (p < 0.001 for no K+ comparison of HKAα1 KO and HKAα2 KO as well as for K+ comparison of HKAα1 KO and HKAα2 KO).
Our current results are consistent with our previous outcomes by micro-perfusion that cHKA is increased and mediated by K+ depletion. It is most likely that cHKA plays an important role in disease states such as hypoka- lemia [2] [12] - [14] . Based on our results that show gHKA had an increased ATP production under low K+ diet, it remains a possibility that there is another H-K-ATPase involved besides gHKA since the gHKA was presumed to primarily operate under normal dietary conditions. It has been reported by other studies that non-gastric H- K-ATPase presented under low K+ conditions, [15] - [22] compared to our observation that a significant change in ATP production under K+ depletion, is likely due to an enhanced EA, a non-gastric H-K-ATPase, and H-ATPase, a K+ independent ATPase located in the OMCD tubule. To address this issue, additional experiments will be needed to determine whether a non-gastric H-ATPase also plays a functional role along with gHKA during K+ depletion.
The design for our current experiment is by using K+ free solution (Solution 2) and K+ containing solution (Solution 3) to assess H-K-ATPase, a K+ dependent transporter and H-ATPase, a K+ independent transporter. The increase of EA with K+ free solution is the indication of K+ independent ATP activity. So it is suggested that H-ATPase is increased under a K+ depletion condition, which is also consistent with our previous micro-perfu- sion study that showed H-ATPase had a significant increase under a low K+ diet [1] [3] [5] . Intriguingly, we see a difference between HKAα2 KO mice and HKAα1 KO mice with K+ free solution.
The contribution of bafilomycin-sensitive H-ATPase to net acid secretion in tubules under low K+ circums- tances was unclear. Recently, it was shown that chronic K+ depletion induces an increase in plasma membrane H-ATPase activity in individual intercalated cells (ICs). Immunocytochemical localization of the proton translo- cating H-ATPase showed that K+-deprivation produced an increase of intercalated cells (ICs) with H-ATPase localized in the apical membrane [3] [12] .
Our results have shown H-ATPase has a higher EA in HKAα1 KO mice, this indicates that H-ATPase plays a significant role equal to the gHKA. When gHKA (HKAα1 gene) is disrupted, a compensatory mechanism through H-ATPase activity is recognizable. Where as in HKAα2 KO mice (HKAα2 gene disruption), both H-ATPase and gHKA contribute significantly whether under ND or KD conditions.
A further approach for H-ATPase is using a H-ATPase inhibitor to specifically evaluate activity of H-ATPase by EA measurements, so that we can determine gHKA versus H-ATPase under ND and cHKA versus H-ATPase under KD. A non-gastric H-K-ATPase has been reported in a variety of tissues; [23] - [25] additionally it was shown by other studies that non-gastric H-K-ATPase was up regulated under low K+ conditions [26] - [29] . The non-gastric H-K-ATPase is not sensitive to Sch-28020, a specific inhibitor to gHKA. Research also shows that non-gastric H-K-ATPase is associated with Na-K-ATPase that is sensitive to ouabain, an inhibitor of cHKA and Na-K-ATPase [23] [24] [30] .
Our previous study has confirmed that ouabain can have inhibitory effects on cHKA and Na-K-ATPase be- cause the structures of cHKA and Na-K-ATPase share substantial (63% - 65%) homology [1] - [3] [5] [31] [32] . Our future plan is to further investigate the activity of non-gastric H-K-ATPase by EA measurements. If we re- move Na+ from the media, we could possibly observe an activity due to a non-gastric H-K-ATPase compared to in the presence of ouabain. This will allow us to see if the non-gastric H-K-ATPase is sensitive to ouabain. If so, we are able to distinguish the activity whether it is from Na-K-ATPase or from the non-gastric H-K-ATPase. In summary, the EA of gHKA is up regulated under a normal diet in HKAα2 KO mice, and the EA of cHKA is up regulated under a low K+ diet in HKAα1 KO mice. A significant difference in ATP production in gHKA isoform is likely due to the enhanced EA of H-ATPase under K+ depletion.
Acknowledgements
This study was supported by grants from CISR (Committee on Institutional Studies and Research at Murray State University), URSA (Undergraduate Research and Scholarly Activities at Murray State University), and the American Heart Association. We would like to thank Dr. Gary E. Shull (University of Cincinnati, Cincinnati, OH) for providing the transgenic mice used in our research as well as Donna Crabtreeand Veronica Ingland (undergraduate and graduate students at Murray State University) for providing technical assistance.
References
- Nakamura, S., Amlal, H., Schultheis, P., Galla, J., Shull, G. and Soleimani, M. (1999) HCO3-Reabsorption in Renal Collecting Duct of NHE-3 Deficient Mouse: A Compensatory Response. American Journal of Physiology, 276, 914- 921.
- Nakamura, S., Wang, Z., Galla, J. and Soleimani, M. (1998) K+ Depletion Increases HCO3-Reabsorption in OMCD by Activation of Colonic H-K-ATPase. American Journal of Physiology, 274, 687-692.
- Nakamura, S., Amlal, H., Soleimani, M. and Galla, J. (2000) Pathways for HCO3-Reabsorption in Mouse Medullary Col- lecting Duct Segments. Journal of Laboratory and Clinical Medicine, 136, 218-223. http://dx.doi.org/10.1067/mlc.2000.108750
- Garg, L. and Narang, N. (1988) Oubain-Insensitive K-Adenosine Triphosphatase in Distal Nephron Segments of the Rabbit. The Journal of Clinical Investigation, 81, 1204-1208.
- Nakamura, S., Amlal, H., Soleimani, M. and Galla, J. (1999)
 Secretion in Inner Medullary Collecting Duct in Po- tassium Deprivation: Role of Colonic H+-K+-ATPase. Kidney International, 56, 2160-2167. http://dx.doi.org/10.1046/j.1523-1755.1999.00780.x
Secretion in Inner Medullary Collecting Duct in Po- tassium Deprivation: Role of Colonic H+-K+-ATPase. Kidney International, 56, 2160-2167. http://dx.doi.org/10.1046/j.1523-1755.1999.00780.x - Garg, L., Knepper, M. and Burg, M. (1981) Mineralocorticoid Effects of Na-K-ATPase in Individual Nephron Seg- ments. American Journal of Physiology, 240, 536-544.
- Zemkova, H., Teisinger, J. and Vyskocil, F. (1988) Inhinition of the Electrogenic Na,K Pump and Na,K-ATPase Ac- tivity by Tetraethlyammonium, Tetrabutylammonium, and Apamin. Journal of Neuroscience Research, 19, 497-503. http://dx.doi.org/10.1002/jnr.490190414
- Nunez, R., Calva, E. and Marsh, M. (1975) Nicotinamide Adenine Dinucleotide Degradation in Infarcted Cardiac Mu- scle. Recent Advances in Studies on Cardiac Structure and Metabolism, 10, 241-250.
- Wilder, M., Do, T., Atmomarsono, M., Tran, T., Truong, Q. and Yang, W. (2000) Characterization of Na/K-ATPase in Macrobrachiumrosenbergii and the Effects of Changing Salinity on Enzymatic Activity. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 123, 377-388. http://dx.doi.org/10.1016/S1095-6433(00)00162-8
- Meneton, P., Schultheis, P. and Greeb, J. (1998) Increased Sensitivity to K+ Deprivation in Colonic H,K-ATPase De- ficient Mice. Journal of Clinical Investigation, 101, 536-542. http://dx.doi.org/10.1172/JCI1720
- Spicer, Z., Miller, M., Andringa, A., Riddle, T., Duffy, J., Doetschman, T. and Shull, G. (2000) Stomachs of Mice Lacking the Gastric H,K-ATPase Alpha-Subunit Have Achlorhydria, Abnormal Parietal Cells and Ciliated Metaphasia. The Journal of Biological Chemistry, 275, 21555-21565. http://dx.doi.org/10.1074/jbc.M001558200
- Nakamura, S. (2006) H+-ATPase Activity in Selective Disruption of H+-K+-ATPase Alpha 1 Gene of Mice under Nor- mal and K-Depleted Conditions. The Journal of Laboratory and Clinical Medicine, 147, 45-51. http://dx.doi.org/10.1016/j.lab.2005.08.013
- Guojun, W., Ravelletee, J. and Nakamura, S. (2013) Potassium Depletion Stimulates Beta Subunit of Colonic H+- K+-ATPase in Mice. International Journal of Clinical Medicine, 4, 244-250.
- Xiaming, Z., Nakamura, S., Xia, S. and Wingo, C. (2001) Increase CO2 Stimulates K/Rb Reabsorption Mediated by H-K-ATPase in CCD of Potassium-Restricted Rabbit. American Journal of Physiology, 281, 366-373.
- Swarts, H., Koenderink, J., Willems, P. and De Pont, J. (2007) The Human Non-Gastric H,K-ATPase Has a Different Cation Specificity than the Rat Enzyme. Biochimica et Biophysica Acta, 1768, 580-589. http://dx.doi.org/10.1016/j.bbamem.2006.10.010
- Swarts, H., Koenderink, J., Willems, P. and De Pont, J. (2005) The Non-Gastric H,K-ATPase Is Oligomycin-Sensitive and Can Function as an H+,
 -ATPase. Journal of Biological Chemistry, 280, 33115-33122.http://dx.doi.org/10.1074/jbc.M504535200
-ATPase. Journal of Biological Chemistry, 280, 33115-33122.http://dx.doi.org/10.1074/jbc.M504535200 - Streif, D., Iglseder, E., Hauser-Kronberger, C., Fink, K., Jakab, M. and Ritter, M. (2011) Expression of the Non-Gas- tric H+/K+ ATPase ATP12A in Normal and Pathological Human Prostate Tissue. Cellular Physiology and Biochemi- stry, 28, 1287-1294. http://dx.doi.org/10.1159/000335860
- Novak, I., Wang, J., Henriksen, K., Haanes, K., Krabbe, S., Nitschke, R. and Hede, S. (2011) Pancreatic Bicarbonate Secretion Involves Two Proton Pumps. The Journal of Biological Chemistry, 286, 288-289. http://dx.doi.org/10.1074/jbc.M110.136382
- Fischer, H. and Widdicombe, J. (2006) Mechanisms of Acid and Base Secretion by the Airway Epithelium. Journal of Membrane Biology, 211, 139-150. http://dx.doi.org/10.1007/s00232-006-0861-0
- Lerner, M., Lemke, D., Bertram, H., Schillers, H., Oberleithner, H., Caplan, M. and Reinhardt, J. (2006) An Extracellular Loop of the Human Non-Gastric H,K-ATPase Alpha-Subunit Is Involved in Apical Plasma Membrane Polarization. Cellular Physiology and Biochemistry, 18, 75-84. http://dx.doi.org/10.1159/000095169
- Qiu, L., Swarts, H., Tonk, E., Willems, P., Koenderink, J. and De Pont, J. (2006) Conversion of the Low Affinity Ouabain-Binding Site of Non-Gastric H,K-ATPase into a High Affinity Binding Site by Substitution of Only Five Amino Acids. The Journal of Biological Chemistry, 281, 13533-13539. http://dx.doi.org/10.1074/jbc.M600551200
- Crambert, G., Horisberger, J.D., Modyanov, N.N. and Geering, K. (2002) Human Nongastric H+-K+-ATPase: Transport Properties of ATP1al1 Assembled with Different Beta Subunits. American Journal of Physiology—Cell Physiology, 283, 305-314. http://dx.doi.org/10.1152/ajpcell.00590.2001
- Swarts, H., Koenderink, J., Willems, P. and De Pont, J. (2009) The Non-Gastric H,K-ATPase as a Tool to Study the Ouabain-Binding Site in Na,K-ATPase. Pflügers Archiv—European Journal of Physiology, 457, 623-634. http://dx.doi.org/10.1007/s00424-008-0467-8
- Radkov, R., Kharoubi-Hess, S., Schaer, D., Modyanov, N., Geering, K. and Horisberger, J. (2007) Role of Homolog- ous ASP334 and GLU319 in Human Non-Gastric H,K- and Na,K-ATPases in Cardiac Glycoside Binding. Biochemical and Biophysical Research Communications, 356, 142-146. http://dx.doi.org/10.1016/j.bbrc.2007.02.119
- Pestov, N.B., Korneenko, T.V., Radkov, R., Zhao, H., Shakhparonov, M.I. and Modyanov, N.N. (2004) Identification of the Beta-Subunit for Nongastric H-K-ATPase in Rat Anterior Prostate. American Journal of Physiology—Cell Physiology, 286, C1229-C1237. http://dx.doi.org/10.1152/ajpcell.00393.2003
- Codina, J., Delmas-Mata, J. and Dubose, T. (1998) The Alpha-Subunit of the Colonic H+,K+-ATPase Assembles with Beta1-Na+, K+-ATPase in Kidney and Distal Colon. The Journal of Biological Chemistry, 273, 7894-7899. http://dx.doi.org/10.1074/jbc.273.14.7894
- Codina, J., Pressley, T. and Dubose, T. (1997) Effect of Chronic Hypokalemia on H+-K+-ATPase Expression in Rat Colon. American Journal of Physiology, 272, 22-30.
- Jaisser, F., Escoubet, B., Coutry, N., Eugene, E., Bonvalet, J. and Farman, N. (1996) Differential Regulation of Putative K+-ATPase by Low-K+ Diet and Corticosteroids in Rat Distal Colon and Kidney. American Journal of Physiology, 270, 679-687.
- Sangan, P., Rajendran, V., Mann, A., Kashgarian, M. and Binder, H. (1997) Regulation of Colonic H-K-ATPase in Large Intestine and Kidney by Dietary Na Depletion and Dietary K Depletion. American Journal of Physiology, 272, 685-696.
- Burnay, M., Crambert, G., Kharoubi-Hess, S., Geering, K. and Horisberger, J. (2003) Electrogenicity of Na,K- and H,K- ATPase Activity and Presence of a Positively Charged Amino Acid in the Fifth Transmembrane Segment. The Journal of Biological Chemistry, 278, 19237-19244. http://dx.doi.org/10.1074/jbc.M300946200
- Jaisser, F. (1996) Molecular and Functional Diversity of Na,K-ATPase and Renal H,K-ATPases. Nephrologie, 17, 401- 408.
- Nakamura, S., Amlal, H., Galla, J. and Soleimani, M. (1998) Colonic H+-K+-ATPase Is Induced and Mediates Increased
 Reabsorption in Inner Medullary Collecing Duct in Potassium Depletion. Kidney International, 54, 1233-1239.http://dx.doi.org/10.1046/j.1523-1755.1998.00105.x
Reabsorption in Inner Medullary Collecing Duct in Potassium Depletion. Kidney International, 54, 1233-1239.http://dx.doi.org/10.1046/j.1523-1755.1998.00105.x - Zhao, N., Sui, G., Li, N., Wu, X., Yang, Z., Lai, G., Yang, Y., Lin, Z. and Tang, X. (2011) Study on Activities and Protein and Gene Expression of Renal H+-K+-ATPase in Rats Subchronic Exposed to Trimethyltin Chloride. Chinese Journal of Industrial Hygiene and Occupational Diseases, 29, 299-302.
- Poulsen, H., Morth, P., Egebjerg, J. and Nissen, P. (2010) Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATP- ase. FEBS Letters, 584, 2589-2595. http://dx.doi.org/10.1016/j.febslet.2010.04.035
- Witzke, A., Lindner, K., Munson, K. and Apell, H. (2010) Inhibition of the Gastric H,K-ATPase by Clotrimazole. Biochemistry, 49, 4524-4532. http://dx.doi.org/10.1021/bi1004014