Open Journal of Rheumatology and Autoimmune Diseases
Vol. 2 No. 4 (2012) , Article ID: 24842 , 4 pages DOI:10.4236/ojra.2012.24015
Is Chemokine Receptor CCR9 Required for Synovitis in Rheumatoid Arthritis? Deficiency of CCR9 in a Murine Model of Antigen-Induced Arthritis
![]()
1Keele University at Robert Jones and Agnes Hunt Orthopaedic Hospital, Oswestry, UK; 2Faculty of Medicine and Dentistry, School of Oral and Dental Sciences, University of Bristol, Bristol, UK.
Email: Alison.Cartwright@rjah.nhs.uk
Received July 27th, 2012; revised September 10th, 2012; accepted September 19th, 2012
Keywords: Chemokine Receptor CCR9; Rheumatoid Arthritis; Inflammation; Antigen-Induced Arthritis; Mouse Model; Monocytes/Macrophages
ABSTRACT
Objectives: Monocytes/macrophages accumulate in the synovial membrane in rheumatoid arthritis and play a key role in disease pathogenesis, contributing to inflammation, cartilage destruction and bone erosion. Identification of molecules involved in monocyte/macrophage recruitment in inflammation is crucial for development of therapeutic interventions. Chemokine receptor CCR9 is up-regulated on these cells in peripheral blood and synovium of rheumatoid patients. This study investigated the course of antigen-induced arthritis in CCR9 deficient C57BL/6 mice in comparison to wild type animals to determine whether CCR9 is critical for disease severity and progression. Methods: Methylated bovine serum albumin was used for induction of uni-lateral arthritis by direct injection into the knee joints of preimmunized animals. Arthritis is confined to the injected joint allowing comparison with the normal opposing joint. Clinical severity of arthritis was assessed by measuring swelling in the arthritic joint in comparison to the normal joint. Histological analysis was performed to assess the extent of leukocyte infiltration and cartilage depletion. Results: Levels of swelling were not significantly different between wild type and CCR9 deficient mice. Similarly there was no significant difference in histological severity of arthritis when comparing CCR9-deficient mice to wild type mice. Conclusions: CCR9 was not required for development of synovial inflammation and cartilage destruction in the antigen-induced model of arthritis in C57BL/6 mice in this study. This may reflect a true lack of a pathogenic role of CCR9 on monocyte/macrophage function in vivo or it may reflect differences in the current antigen-induced arthritis model when compared to human RA.
1. Introduction
Rheumatoid Arthritis (RA) is a chronic inflammatory disease characterized by the accumulation of leukocytes in the synovial membrane and fluid of affected joints. Chemokine receptors on leukocytes mediate the persistent recruitment of these cells and identification of involved receptors offers potential for development of therapeutic interventions. Monocytes/macrophages play a key role in RA, contributing to inflammation, cartilage destruction and bone erosion. Monocytes migrate from the blood across the vascular endothelium into the synovium where they differentiate into macrophages. Activated macrophages produce large quantities of pro-inflammatory cytokines and chemokines including interleukin-1beta (IL-1β), tumour necrosis factor alpha (TNFα), IL-6, chemokine ligands CXCL8, CCL2 and CCL3, and also proteases such as matrix metalloproteinase MMP-9 and MMP-12 [1,2]. The degree of macrophage infiltration correlates to the radiological progression of joint destruction [3].
Chemokine receptor CCR9 is constitutively expressed on T lymphocytes in the small intestine, thymus, lymph node and spleen [4,5] with involvement in T cell recruitment to the small intestine and T cell development and migration within the thymus.
CCR9 mediates migration of malignant cluster of differentiation 4+ (CD4+) T lymphocytes into various organs such as the lymph nodes, liver, spleen, lungs and intestinal tract in T-cell lineage acute lymphocytic leukaemia (T-ALL) [6]. CCR9 and its ligand CCL25 were found to be highly over-expressed on T-ALL CD4+ T cells when compared to normal CD4+ T cells. Human cutaneous melanoma cells are suggested to metastasize to the small intestine via action of CCR9 [7]. CCR9 also has roles in the metastatic spread of tumour cells in prostate cancer [8] and ovarian cancer [9] and the CCR9/ CCL25 axis is also involved in breast cancer cell migration and invasion [10].
CCR9 is expressed in inflammation with involvement in T cell recruitment in inflammatory bowel disease. A potent human CCR9 small molecule antagonist: GSK- 1605786 (CCX-282; Traficet-EN) is being developed by GlaxoSmithKline plc under licence from ChemoCentryx Inc., for potential treatment of inflammatory bowel disease including Crohn’s disease [11,12]. It is currently being used to conduct clinical trials with the aim that it will inhibit lymphocyte migration to the small intestine and ameliorate inflammation occurring in Crohn’s disease.
CCR9 expression has been observed on monocytes/macrophages but not on T or B cells in the synovium, with expression increasing in inflamed synovium in RA [13]. The number of CCR9+ CD14+ monocytes/ macrophages increases significantly in RA synovium compared to non-RA tissue. The differentiation of monocytes into macrophages within this tissue is suggested to involve CCR9 and its sole ligand CCL25, particularly under inflammatory conditions [13]. Expression of CCR9 also increases significantly on Peripheral Blood (PB) monocytes in RA suggesting a role for CCR9 and CCL25 in the pathogenesis of this disease.
This study investigated whether CCR9/CCL25 interactions are critical for the development of inflammation in a murine model of Antigen-Induced Arthritis (AIA). AIA is a mono-articular disease model caused by the direct injection of antigen into the knee joint of a preimmunized animal. Arthritis is confined to the injected joint allowing comparison with the normal opposing joint. The model was used to compare hyperplasia, leukocyte infiltration, and cartilage erosion in the synovial joint of CCR9-deficient (CCR9–/–) and wild-type (WT, CCR9+/+) mice, with the aim of investigating the role played by CCR9 in this experimental model which forms a close experimental analogue of human RA [14, 15]. The CCR9 deficient mice used in this study were C57BL/6 strain in which the model of methylated bovine serum albumin (mBSA) AIA shows good severity and chronicity [15]. The AIA model has the following characteristics:
1) preimmunisation induces humoral and cell-mediated immunity;
2) leukocytes: lymphocytes, plasma cells, macrophages and neutrophils, migrate into the injected joint;
3) uni-lateral arthritis with controlled onset;
4) pannus formation resulting in erosion of cartilage and bone;
5) antigen-specific local hyper-reactivity and antigen retention in cartilage;
6) chronicity by repeated flares.
2. Methods
2.1. Animals
Experiments were conducted in 7 to 8 week old inbred, male C57BL/6 wild-type (CCR9+/+) mice (Charles River UK, Margate, UK) and CCR9-deficient (CCR9–/–) C- 57BL/6 mice bred in-house from breeding pairs (MRC Harwell, Oxfordshire, UK). Animals were maintained under “conventional regime” and according to the institutional and national guide for care and use of laboratory animals. Procedures were conducted in accordance with Home Office Project License PPL-40/3047.
2.2. Induction of Antigen-Induced Arthritis (AIA)
Mice were immunized subcutaneously with 100 µl of an emulsion of 1 mg/ml·mBSA in Phosphate Buffered Saline (PBS) mixed with an equal volume of complete Freund’s adjuvant [16]. At the same time they were injected intraperitoneally with 160 ng heat-inactivated Bordetella pertussis toxin in 100 µl PBS (all reagents were from Sigma, Gillingham, UK). At one week the immune response was boosted by subcutaneous injection again with 100 µl of 1mg/ml mBSA in PBS mixed with an equal volume of complete Freund’s adjuvant. At three weeks AIA was induced by injecting 10 µl·10mg/ml mBSA in PBS intraarticularly into the right hind knee joint and 10 µl PBS into the left knee joint to act as control (day 0).
2.3. Clinical Assessment of Arthritis
Severity of arthritis was assessed by comparing the right hind limb joint in which AIA had been induced, with the left hind limb joint which acted as control. Both knee joint diameters were measured before and at 1, 2, 3, 5, 7, 14 and 21 days after arthritis induction using a digital micrometer (Kroeplin GmbH, Schlüchtern, Germany) to monitor swelling.
2.4. Histological Assessment of Arthritis
Animals were killed at day 3, 14 and 21 after induction of arthritis, with a minimum of 5 animals at each time point. For AIA in CCR9-deficient mice 18 animals were used over two experiments. 7 animals were sacrificed at day 3, 6 animals at day 14 and 5 animals at day 21. For AIA in WT mice 66 animals were used over 8 experiments. 20 animals were sacrificed at day 3, 23 animals at day 14, and 23 animals at day 21.
Knee joints were removed and fixed in neutral buffered formal saline. Decalcification in formic acid, embedding in paraffin and sectioning (of 4 µm thickness) was carried out by the histology department, RJAH Orthopaedic Hospital.
Serial sections were stained with haematoxylin and eosin (H&E) and safranin O-fast green. Sections were examined at ×100 and × 200 magnifications. Scoring of sections was carried out blind [16]. H&E sections were used to score synovial hyperplasia from 0 (normal: lining 1 cell thick) to 3 (severe: lining 4 or more cells thick); cellular exudate from 0 (no cells present in joint cavity) to 3 (20 or more cells present in joint cavity), and synovial leukocyte infiltration, from 0 (no infiltration) to 5 (extensive infiltration). Safranin O stains proteoglycans in cartilage orange/red in colour and stained sections were used to score cartilage depletion based on loss of red staining on femoral and tibial condyles, from 0 (no apparent loss of colour intensity) to 3 (extensive loss of cartilage staining). Parameters were then summed to give an arthritis index.
2.5. Immunohistology
Sections were deparaffinized, rehydrated and antigen retrieval carried out overnight at 50˚C in 100 mM Tris/HCl, pH 9.0. Slides were rinsed in PBS and incubated with goat anti-mouse CCR9 (4 μg/ml; Genetex, Source BioScience, Nottingham, UK) and goat IgG control (R&D Systems, Abingdon, UK) for 60 minutes. The sections were rinsed in PBS and then further incubated with donkey anti-goat Alexa 594 (1:100; Molecular Probes, Invitrogen, Paisley, UK) in 10% mouse serum for 30 minutes. Sections were rinsed in PBS and nuclear staining was performed with 4’, 6-diamidino-2-phenyl indole dihydrochloride (DAPI; 2 μg/ml in PBS; Sigma) for 3 minutes before rinsing and mounting.
2.6. Statistical analysis
GraphPad Prism Version 5.01 was used for all statistical analysis. Joint swelling was compared between CCR9 deficient and WT mice by 2-way analysis of variance (ANOVA) followed by Bonferroni post tests to compare swelling at each time point. Comparisons were made between the arthritis index for CCR9 deficient and WT mice at days 3, 14 and 21 by Kruskal-Wallis test followed by Dunn’s post test. Individual parameters comprising the arthritis index were also compared by Kruskal-Wallis test followed by Dunn’s post test. Values of p < 0.05 were considered statistically significant.
3. Results
3.1. Clinical Assessment of Knee Joints
Joint swelling was measured following intra-articular injection with mBSA or PBS at day 0. The difference in knee joint diameter between the right limb in which arthritis had been induced, and the left limb (PBS injected) which acted as control, was recorded for each animal over a 21 day period (Figure 1). Animals were sacrificed at days 3, 14 and 21 for assessment of inflammatory parameters by histology. Joints were measured for remaining animals in each experiment. Both WT and CCR9 deficient mice demonstrated significant joint swelling (Figure 1; p < 0.0001) following injection of mBSA to induce arthritis, and swelling was most severe at 1 day post injection. Swelling then reduced by approaching 50% at day 2 in both strains of mice, and subsequently returned to approximately normal by day 21. However levels of swelling were not significantly different between WT and CCR9 deficient mice when analyzed by 2-way ANOVA with Bonferroni post tests to analyse the difference in swelling between strains at each time point (p > 0.05).
3.2. Histopathological Assessment of Knee Joints
Figures 2 and 3 show representative micrographs from CCR9+/+ and CCR9–/– mice. A pronounced arthritis was induced by intra-articular injection of mBSA which was not observed in contra-lateral control joints from the same animals. In both CCR9–/– (Figure 2a) and WT mice (Figure 2c) arthritis was characterized by the hyperplasia (thickening) of the synovial lining layer and infiltration of leukocytes in the sub-lining of the joint. Contra-lateral control joints from the same animals showed that they did not become inflamed (Figures 2b and 2d). Synovial exudate was present in the joints with AIA and consisted of neutrophils within the joint space.
The AIA model is characterized by cartilage depletion
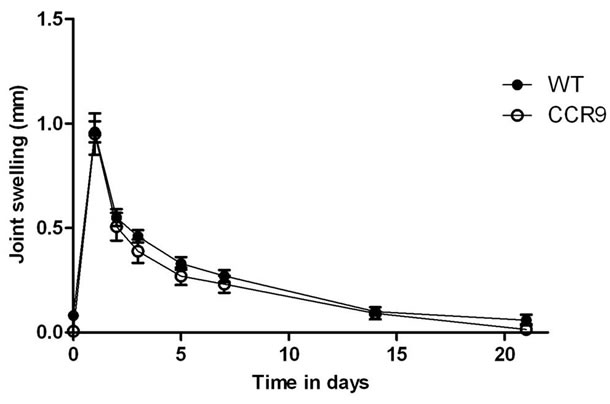
Figure 1. Joint swelling following antigen-induced arthritis in wild type (CCR9+/+) and CCR9 deficient (CCR9–/–) mice. AIA was induced in preimmunized CCR9–/– and CCR9+/+ mice by intraarticular injection of mBSA in PBS. Data represent mean ± standard error for swelling measurements for all mice remaining in the experiment before sacrifice for histology. For measurements on CCR9 deficient animals at days 0 - 3: n = 18; days 5 - 14: n = 13, and day 21, n = 7. For WT animals n = 66 at days 0 - 3; n = 47 at days 5 - 14 and n = 27 at day 21.
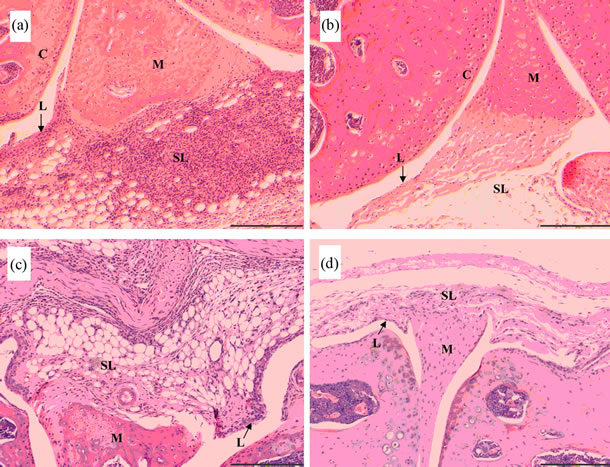
Figure 2. Antigen-induced arthritis in CCR9-deficient (CCR9–/–) and wild type (CCR9+/+) mice. Representative haematoxylin and eosin stained sections are shown for CCR9–/– (a) and (b) and CCR9+/+ (c) and (d) mice sacrificed on day 3, showing antigen-induced arthritis (a) and (c) and contralateral controls (b) and (d). (c), cartilage; L, synovial lining; SL, synovial sub-lining; M, meniscus; scale bar represents 200 µm.
which occurs due to proteoglycan loss, and this was visualized as loss of red staining (Figure 3a and 3c) when stained with Safranin O-fast green. Depletion of cartilage can be seen on the femoral condyle (Figure 3a) of a CCR9–/– mouse at day 3 of AIA. The marked area (*) shows a region of proteoglycan depletion. Likewise, cartilage depletion marked by (*) can be observed on the tibial condyle in a wild type mouse at day 3 (Figure 3c) of AIA. The contra-lateral controls for the same animals show that the cartilage did not become depleted (Figures 3b and 3d).
The severity of arthritis was quantified by histopatho logical assessment of joint sections from CCR9+/+ and
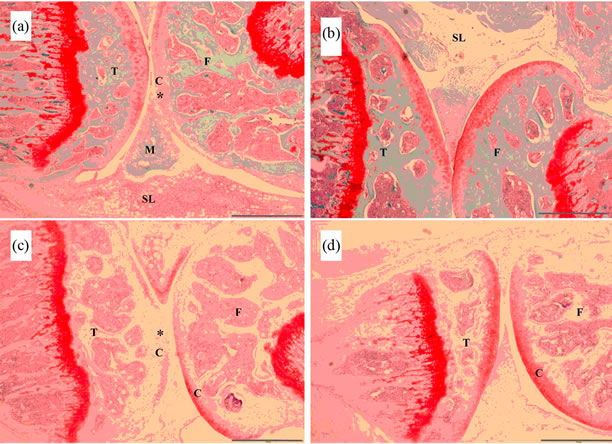
Figure 3.Antigen-induced arthritis in CCR9–/– and CCR9+/+ mice. Representative haematoxylin and safranin O-fast green stained sections are shown for CCR9–/– (a) and (b) and CCR9+/+ (c) and (d) mice sacrificed on day 3, showing antigen-induced arthritis (a) and (c) and contra-lateral controls (b) and (d). (c), cartilage; SL, sub-lining: M, meniscus; T, tibia; F, femur; (*) = region of proteoglycan depletion; scale bar represents 500 µm.
CCR9–/– mice sacrificed at 3, 14 and 21 days following induction of arthritis. 4 parameters were scored: synovial hyperplasia (thickening of the lining layer), cellular exudate, cartilage depletion/bone erosion and synovial infiltration and scores were summed to give an arthritis index (Table 1) [16]. Although arthritis developed following intraarticular injection of mBSA, joints from CCR9 deficient mice showed limited pathological changes.
Hyperplasia did not alter significantly with time in either the wild type or knockout mice or indeed between the 2 strains (p > 0.05). Infiltration of leukocytes in the sub-lining was highest at day 3 in both mouse strains and declined over the course of the model as did synovial exudate, with no significant difference observed between
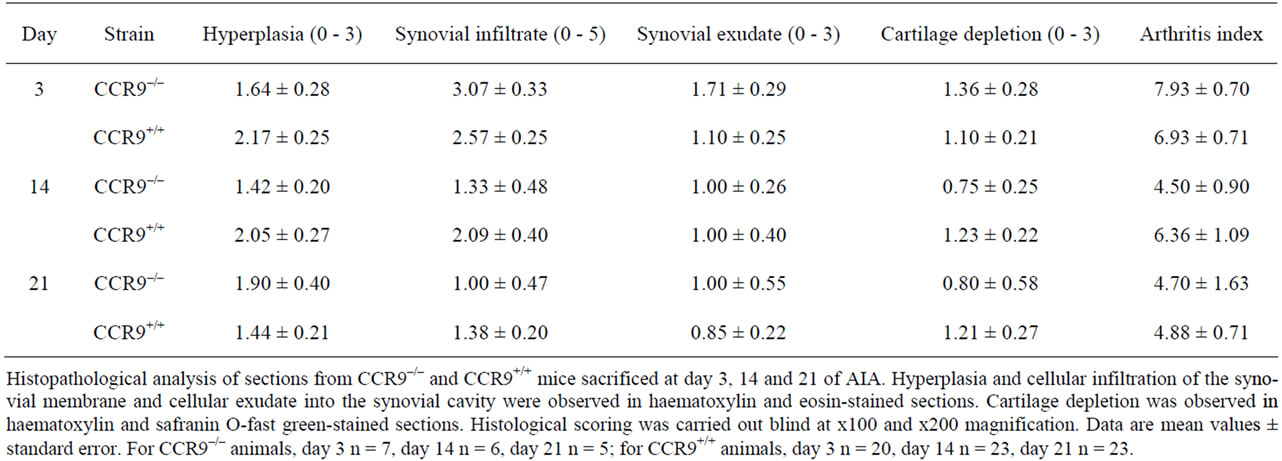
Table 1. Joint inflammation and cartilage damage on day 3, 14 and 21 of antigen-induced arthritis.
strains (p > 0.05). Cartilage depletion was greatest at day 3 in CCR9 deficient mice, and declined by day 14, whereas it did not change much in the wild type animals. The overall arthritis index was greatest at day 3 with a higher score in CCR9 deficient mice. It declined by day 14 in both mouse strains, although the reduction was greater in the knockout mice. By day 21 it was the same for both strains. Kruskal-Wallis tests with Dunn’s post tests were carried out to compare the arthritis index and each individual parameter and showed that there was no significant difference between the CCR9-deficient and wild type mice.
3.3. CCR9 Expression in Synovium
CCR9 expression was examined by immunohistochemistry on paraffin embedded synovial sections obtained from WT mice sacrificed at day 3 of AIA. The receptor was detected in all animals observed on infiltrated leukocytes in the synovium (Figure 4). Investigation of CCR9 expression on CCR9–/– mice sacrificed at day 3 of AIA showed that infiltrated leukocytes and also stromal cells were CCR9 negative (data not shown).
4. Discussion
To investigate the requirement for chemokine receptor

Figure 4. Expression of CCR9 by infiltrating leukocytes in CCR9+/+ mice sacrificed on day 3 of antigen-induced arthritis. Sections were stained with antibody to CCR9 (a) and DAPI nuclear stain (b). (c) represents a merge of images (a) and (b). (d) is the isotype control for CCR9 and (e) is the same field of view stained with DAPI. The scale bar represents 100 µm.
CCR9 in synovitis in RA, a mouse AIA model was used which forms a close experimental analogue of human RA [14]. AIA was induced in WT and CCR9–/– mice. The severity of the inflammatory response was determined firstly by monitoring joint swelling over the 21 day time course of the arthritis model which showed that there was no significant difference in joint swelling between the two mouse strains. Secondly, histological assessment of joint sections from mice sacrificed at days 3, 14 and 21 showed that again, there was no significant difference in severity of arthritis when comparing CCR9-deficient mice to WT mice.
We then examined expression of CCR9 in WT arthritic joints at the peak of synovitis (day 3) observing strong expression of CCR9 on infiltrating leukocytes in the synovium. This observation agrees with our finding that CCR9 is expressed intensely on a subset of infiltrating leukocytes in human rheumatoid synovium [13]. The examination of corresponding sections from CCR9–/– mice showed joints were indeed CCR9 negative although synovia were equally infiltrated by leukocytes. Therefore although leukocytes recruited to the inflamed synovium in WT animals were populations positive for CCR9, expression of this receptor does not appear critical for leukocyte recruitment and development of arthritis in the AIA model in C57BL/6 mice.
The AIA model includes the initial adaptive immune response during which mBSA presentation to T cells stimulates T cell proliferation and down stream activation of B cells and production of antibodies to mBSA. This model also includes the inflammatory effector phase of disease in which neutrophils and mononuclear cells migrate into the joint producing swelling, cartilage depletion and bone erosions. Disease severity may be reduced in CCR9 deficient mice if the receptor is critical to either stage. In humans expression of CCR9 increased on rheumatoid monocytes/macrophages in blood and synovium but the receptor was not detected on synovial lymphocytes [13]. Furthermore, CCR9 expression was low on neutrophils (personal communication from Dr Caroline Schmutz) so CCR9 absence may not directly affect lymphocyte or neutrophil recruitment.
CCR9 is suggested to be involved in the differentiation of monocytes into macrophages once they have been recruited to the synovium [13] so CCR9 deficiency may impede the effector phase of the disease, inhibiting differentiation and activation of monocytes/macrophages so restricting production of proinflammatory cytokines such as TNFα and IL-6. It is possible however, that in the current study CCR9 may not be involved in differentiation of monocytes to macrophages. There may be differrences between human RA and the AIA mouse model, or CCR9 may be expressed on a non-pathogenic phenotypic subset of monocytes/macrophages in the mouse [17].
K/BxN mice spontaneously develop an arthritis that is similar to human RA [18]. K/BxN mice produce arthritogenic autoantibodies which induce arthritis when transferred to recipient mouse strains. Jacobs et al., (2010) used the K/BxN serum-transfer model to investigate involvement of various chemokine receptors in the effector phase of autoantibody-mediated arthritis. K/BxN serum was transferred into CCR9–/– mice but ensuing arthritis assessed over a 20 day period was not assessed as significantly different from heterozygote controls demonstrating that CCR9 was not critical for development of synovitis in this model. CXCR2 was found to be necessary for development of arthritis and recruitment of neutrophils to the joints [19]. However, other receptors such as CCR1-7, CXCR3 and CXCR5 were not required in the model although some have been shown to function in arthritis using adaptive immune response models. It would therefore be interesting to examine function of CCR9 in other murine arthritis models.
CCR9 involvement in inflammation has been demonstrated in mouse models of hepatitis where CCR9+ macrophages were required for the induction of acute liver inflammation [20]. CCR9+ macrophages produced TNFα, stimulating naïve CD4+ T cells to become Th1 cells producing interferon-gamma. Additionally, PB from acute hepatitis patients had increased numbers of CCR9+ monocytes compared to controls. Therefore the importance of macrophage CCR9 may vary between different inflammatory diseases CCR9 and its ligand CCL25 mediate T cell migration to the small intestine under both homeostatic [4,21] and inflammatory conditions [22,23]. CCR9 is implicated in pathogenesis of small intestinal Crohn’s disease, a chronic inflammatory bowel disease characterised by an influx of T cells. CCR9+ T cells are elevated in the blood and CCL25 is increased in the intestine in areas close to lymphocytic infiltrates [22]. A mouse model of Crohn’s disease is provided by TNF-ΔARE mice which have raised levels of TNFα leading to spontaneous development of severe small intestinal inflammation with similarities to Crohn’s disease [12]. The CCR9 small molecule antagonist CCX-282-B, which has been developed for potential treatment of inflammatory bowel disease including Crohn’s disease [11,12], provided complete protection from severe intestinal inflammation in this mouse model. In contrast, Apostolaki et al. (2008) determined that development of inflammation in TNF-ΔARE mice occurred in the genetic absence of CCR9 and also CCL25 demonstrating a difference between pharmacological inhibition and genetic manipulation of CCR9 [24]. This suggests that gene deficient mice may use alternate genes to compensate for the function of the missing gene. It would therefore be interesting to use the CCR9 antagonist CCX-282-B in our AIA model in WT mice to investigate whether synovial inflammation could be similarly prevented.
CCR9 is not indispensable for lymphocyte recruitment into the small intestine or for T cell development in the thymus. Wurbel et al (2001) generated a CCR9–/– mouse strain in which the absence of CCR9 had no major effect on thymocyte development as a time lag of only one day was observed in the appearance of CD4+CD8+ cells [25]. When they looked at the ratio of intraepithelial lymphocytes (IELs) to epithelial cells in small intestine it was decreased 2-fold in the CCR9 knockout compared to wild type mouse. Further analysis of the isolated IEL subsets showed that the reduction was due to low numbers of the TCRγδ subset. This subset had actually decreased 5-fold in CCR9 knockout compared to wild type mice. Although the absence of CCR9 was obviously effecting IEL populations, the overall effect was not greater probably as a result of the action of other chemokine receptors expressed on intestinal lymphocytes. Receptors such as CXCR4, CCR6, CXCR3 and CCR5 may be able to recruit and retain lymphocytes so compensating for absence of CCR9.
5. Conclusion
CCR9 deficient mice developed arthritis not significantly different in severity to wild type animals. The data from the AIA model demonstrate that recruitment of inflammatory leukocytes into the arthritic joint does not critically require the function of CCR9 in C57BL/6 mice in this arthritis model.
6. Acknowledgements
We thank Pat Evans, Martin Pritchard and Nigel Harness for their histological expertise, staff at the Life Science Support Unit of Liverpool John Moores University for breeding and keeping of the mice, and Anwen Williams for help in setting up the animal model.
This work was supported by Keele University and the Medical Research Council [grant number G0401634].
Disclosure statement: The authors have declared no conflicts of interest.
REFERENCES
- Y. Ma and R. M. Pope, “The Role of Macrophages in Rheumatoid Arthritis,” Current Pharmaceutical Design, Vol. 11, No. 5, 2005, pp. 569-580. doi:10.2174/1381612053381927
- R. W. Kinne, B. Stuhlmüller and G. R. Burmester, “Cells of the Synovium in Rheumatoid Arthritis: Macrophages,” Arthritis Research and Therapy, Vol. 9, 2007, p. 224. doi:10.1186/ar2333
- D. Mulherin, O. Fitzgerald and B. Bresnihan, “Synovial Tissue Macrophage Populations and Articular Damage in Rheumatoid Arthritis,” Arthritis and Rheumatism, Vol. 39, No. 1, 1996, pp. 115-124. doi:10.1002/art.1780390116
- B. A. Zabel, W. W. Agace, J. J. Campbell, H. M. Heath, D. Parent, A. I. Roberts, E. C. Ebert, N. Kassam, S. Qin, M. Zovko, G. J. LaRosa, L. L. Yang, D. Soler, E. C. Butcher, P. D. Ponath, C. M. Parker and D. P. Andrew, “Human G Protein-Coupled Receptor GPR-9-6/CC Chemokine Receptor 9 Is Selectively Expressed on Intestinal homing T Lymphocytes, Mucosal Lymphocytes, and Thymocytes and Is Required for Thymus-Expressed Chemokine-Mediated Chemotaxis, Journal of Experimental Medicine, Vol. 190, No. 9, 1999, pp. 1241-1256. doi:10.1084/jem.190.9.1241
- E. J. Kunkel, J. J. Campbell, G. Haraldsen, J. Pan, J. Boisvert, A. I. Roberts, E. C. Ebert, M. A. Vierra, S. B. Goodman, M. C. Genovese, A. J. Wardlaw, H. B. Greenberg, C. M. Parker, E. C. Butcher, D. P. Andrew and W. W. Agace, “Lymphocyte CC Chemokine Receptor 9 and Epithelial Thymus-Expressed Chemokine (TECK) expression Distinguish the Small Intestinal Immune Compartment: Epithelial Expression of Tissue-Specific Chemokines as an Organizing Principle in Regional Immunity,” Journal of Experimental Medicine, Vol. 192, No. 5, 2000, pp. 761-768. doi:10.1084/jem.192.5.761
- Z. Qiuping, L. Qun, H. Chunsong, Z. Xiaolian, H. Baojun, Y. Mingzhen, L. Chengming, H. Jinshen, G. Qingping, Z. Kejian, S. Zhimin, Z. Xuejun, L. Junyan and T. Jinquan, “Selectively Increased Expression and Functions of Chemokine Receptor CCR9 on CD4+ T cells from Patients with T-Cell Lineage Acute Lymphocytic Leukaemia,” Cancer Research, Vol. 63, No. 19, 2003, pp. 6469-6477.
- F. F. Amersi, A. M. Terando, Y. Goto, R. A. Scolyer, J. F. Thompson, A. N. Tran, M. B. Faries, D. L. Morton and D. S. Hoon, “Activation of CCR9/CCL25 in Cutaneous Melanoma Mediates Preferential Metastasis to the Small Intestine,” Clinical Cancer Research, Vol. 14, 2008, pp. 638-645. doi:10.1158/1078-0432.CCR-07-2025
- S. Singh, U. P. Singh, J. K. Stiles, W. E. Grizzle and J. W. Lillard Jr., “Expression and Functional Role of CCR9 in Prostate Cancer Cell Migration and Invasion,” Clinical Cancer Research, Vol. 18, No. 21, 2004, pp. 8743-8750. doi:10.1158/1078-0432.CCR-04-0266
- E. L. Johnson, R. Singh, S. Singh, C. M. Johnson-Holiday, W. E. Grizzle, E. E. Partridge and J. W. Lillard Jr., “CCL25-CCR9 Interaction Modulates Ovarian Cancer Cell Migration, Metalloproteinase Expression, and Invasion,” World Journal of Surgical Oncology, Vol. 8, 2010, p. 62. doi:10.1186/1477-7819-8-62
- C. Johnson-Holiday, R. Singh, E. Johnson, S. Singh, C. R. Stockard, W. E. Grizzle and J. W. Lillard Jr., “CCL25 Mediates Migration, Invasion and Matrix Metalloproteinase Expression by Breast Cancer Cells in a CCR9- Dependent Fashion,” International Journal of Oncology, Vol. 38, No. 5, 2011, pp. 1279-1285.
- B. Eksteen and D. H. Adams, “GSK-1605786, a Selective Small-Molecule Antagonist of the CCR9 Chemokine Receptor for the Treatment of Crohn’s Disease,” IDrugs, Vol. 13, No. 7, 2010, pp.472-781.
- M. J. Walters, Y. Wang, N. Lai, T. Baumgart, B. N. Zhao, D. J. Dairaghi, P. Bekker, L. S. Ertl, M. E. Penfold, J. C. Jaen, S. Keshav, E. Wendt, A. Pennell, S. Ungashe, Z. Wei, J. J. Wright and T. J. Schall, “Characterization of CCX282-B, an Orally Bioavailable Antagonist of the CCR9 Chemokine Receptor, for Treatment of Inflammatory Bowel Disease,” Journal of Pharmacology and Experimental Therapeutics, Vol. 335, No. 1, 2010, pp. 61-69. doi:10.1124/jpet.110.169714
- C. Schmutz, A. Cartwright, H. Williams, O. Haworth, J. H. Williams, A. Filer, M. Salmon, C. D. Buckley and J. Middleton, “Monocytes/Macrophages Express CCR9 in Rheumatoid Arthritis and CCL25 Stimulates Their Differentiation,” Arthritis Research and Therapy, Vol. 12, No. 4, 2010, pp. R161. doi:10.1186/ar3120
- D. Brackertz, G. F. Mitchell and I. R. Mackay, “Antigen-Induced Arthritis in Mice,” Arthritis and Rheumatism, Vol. 20, No. 3, 1977, pp. 841-850. doi:10.1002/art.1780200314
- W. B. van den Berg, L. A. Joosten and P. L. van Lent, “Murine Antigen-Induced Arthritis,” Methods in Molecular Medicine, Vol. 136, No. 2, 2007, pp.243-253. doi:10.1007/978-1-59745-402-5_18
- M. A. Nowell, P. J. Richards, S. Horiuchi, N. Yamamoto, S. Rose-John, N. Topley, A. S. Williams and S. A. Jones, “Soluble IL-6 Receptor Governs IL-6 Activity in Experimental Arthritis: Blockade of Arthritis Severity by Soluble Glycoprotein,” Journal of Immunology, Vol. 171, 2003, pp. 3202-3209.
- A. Mantovani, A. Sica and M. Locati, “New Vistas on Macrophage Differentiation and Activation,” European Journal of Immunology, Vol. 37, No. 1, 2007, pp.14-16. doi:10.1002/eji.200636910
- M. Corr and B. Crain, “The Role of FcgammaR Signaling in the K/B × N Serum Transfer Model of Arthritis,” Journal of Immunology, Vol. 169, No. 11, 2002, pp. 6604-6609.
- J. P. Jacobs, A. Ortiz-Lopez, J. J. Campbell, C. J. Gerard, D. Mathis and C. Benoist, “Deficiency of CXCR2, but Not Other Chemokine Receptors, Attenuates a Murine Model of Autoantibody-Mediated Arthritis,” Arthritis and Rheumatism, Vol. 62, No. 7, 2010, pp.1921-1932.
- N. Nakamoto, H. Ebinuma H, T. Kanai, P. S. Chu, Y. Ono, Y. Mikami, K. Ojiro, M. Lipp, P. E. Love, H. Saito and T. Hibi, “CCR9(+) Macrophages Are Required for Acute Liver Inflammation in Mouse Models of Hepatitis,” Gastroenterology, Vol. 142, No. 2, 2012, pp.366-376. doi:10.1053/j.gastro.2011.10.039
- B. Johansson-Lindbom and W. W. Agace, “Generation of Gut-Homing T Cells and Their Localization to the Small Intestinal Mucosa,” Immunological Reviews, Vol. 215, 2007, pp. 226-242. doi:10.1111/j.1600-065X.2006.00482.x
- K. A. Papadakis, J. Prehn, S. T. Moreno, L. Cheng, E. A. Kouroumalis, R. Deem, T. Breaverman, P. D. Ponath, D. P. Andrew, P. H. Green, M. R. Hodge, S. W. Binder and S. R. Targan, “CCR9-Positive Lymphocytes and ThymusExpressed Chemokine Distinguish Small Bowel from Colonic Crohn’s Disease,” Gastroenterology, Vol. 121, No. 2, 2001, pp.246-254. doi:10.1053/gast.2001.27154
- C. Koenecke and R. Förster, “CCR9 and Inflammatory Bowel Disease,” Expert Opinion on Therapeutic Targets, Vol. 13, No. 3, 2009, pp.297-306. doi:10.1517/14728220902762928
- M. Apostolaki, M. Manoloukos, M. Roulis, M. A. Wurbel, and W. Müller, “Role of β7 Integrin and the Chemokine/Chemokine Receptor Pair CCL25/CCR9 in Modelled TNF-Dependent Crohn’s Disease,” Gastroenterology, Vol. 134, No. 7, 2008, pp. 2025-2035. doi:10.1053/j.gastro.2008.02.085
- M. A. Wurbel, M. Malissen, D. Guy-Grand, E. Meffre, M. C. Nussenzweig, M. Richelme, A. Carrier and B. Malissen, “Mice Lacking the CCR9 CC-Chemokine Receptor Show a Mild Impairment of Early Tand B-Cell Development and a Reduction in T-Cell Receptor Gammadelta(+) Gut Intraepithelial Lymphocytes,” Blood, Vol. 98, No. 9, 2001, pp. 2626-2632. doi:10.1182/blood.V98.9.2626