Open Journal of Rheumatology and Autoimmune Diseases
Vol.1 No.2(2011), Article ID:8794,5 pages DOI:10.4236/ojra.2011.12002
A Fatal Case of Treatment-Related Adverse Effects in Granulomatosis with Polyangiitis (Wegener’s Granulomatosis)
![]()
Department of Medicine, Division of Rheumatology, Case Western Reserve University/Metro Health Medical Center, Cleveland, USA.
Email: mantonelli@metrohealth.org
Received September 2nd, 2011; revised October 11th, 2011; accepted October 25th, 2011.
Keywords: Granulomatosis with Polyangiitis, Wegener’s Granulomatosis, Cerebrovascular Accident, Infection, CNS Manifestation
ABSTRACT
Well-studied therapies have proven to be effective in treating Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis) (GPA). There has been considerable improvement in survival of patients with GPA but treatment related morbidity and mortality remains still high, particularly in patients with renal disease. We describe a case of 64-year old woman with recent onset GPA, who responds well to the initial cyclophosphamide based therapy but latter develops a fatal stroke. Infectious complications should be considered in patients with GPA who are on adequate immunosuppression but develop symptoms that may mimic a relapse. Aggressive diagnostic interventions should be undertaken to discriminate between an infection and a relapse of GPA.
1. Introduction
Granulomatosis with polyangiitis (Wegener’s granulomatosis) (GPA) is a necrotizing granulomatous vasculitis that predominantly affects airways and kidneys. The prognosis of GPA has improved greatly with the introduction of combined therapy with cyclophosphamide and prednisone. However, the therapy related morbidity and mortality still remains high.
2. Case Report
A 64-year-old female with recently diagnosed GPA on hemodialysis presented to the emergency department with progressive shortness of breath and hemoptysis. Five weeks earlier, she experienced joint pain, malaise, alveolar hemorrhage, acute hypoxic respiratory failure, and acute renal failure that required mechanical ventilation and hemodialysis. Serum creatinine was 5.15 mg/dl on that admission. Anti-neutrophillic cytoplasmic antibodies (ANCA) were present with a C (cytoplasmic) pattern and enzyme immunoassay results were positive for antibodies for proteinase 3 (PR3). Renal biopsy revealed crescentic glomerulonephritis with scant immune deposits. Treatment was initiated with intravenous methlyprednisone (1000 mg/d for 3 days), daily cyclophosphamide (CYC) 150 mg PO, and plasmapheresis. The patient improved and was discharged to a skilled nursing facility, and stayed there for 2 weeks until the current presentation.
The patient presented from the skilled nursing facility with hemoptysis and progressive dyspnea and was admitted. Treatment was initiated with broad-spectrum intravenous antibiotics and diuretics for presumed health care-associated pneumonia (HCAP) and volume overload. Her other past medical history was remarkable for breast cancer thirteen years prior, treated with lumpectomy, chemotherapy, which presumably led to cardiomyopathy, and radiation therapy. In addition, there were ongoing problems related to diabetes mellitus, type II, hypertension, fibromyalgia and hypothyroidism. She had a fiftypack year smoking history. Medications on admission included CYC (75 mg/day, adjusted for prior leukopenia, lowest known WBC = 6.4 × 10/mm, ALC = 192), prednisone (60 mg/day), trimethoprim-sulfameth-oxazole DS (three times per week for Pneumocystis jirovecii prophylaxis), insulin, glyburide (5 mg/day), levothyroxine (50 mcg/day), carvedilol (25 mg twice per day), diltiazem (360 mg/day), rosuvastatin (5 mg/day), aspirin (81 mg/ day), albuterol inhaler, and atrovent inhaler.
Upon admission, her blood pressure was 120/67 mm Hg, temperature of 36.4˚C, pulse of 110, and respirations 28 per minute with oxygen saturation of 84% on room air. The lungs had bilateral crackles with rhonchi. Cardiac rhythm was regular with normal heart sounds and no murmurs. The lower extremities had bilateral pitting edema. Neurological examination was normal on admission. All other exam was unremarkable. Initial pertinent laboratory tests included normal complete blood count (CBC) with normal differential except for mild anemia (Hgb 9.7 gm/dL, above baseline from recent discharge), creatinine of 3.6 mg/dL (on hemodialysis), sodium of 115 mmoles/L and albumin 3.2 gm/dL (up from baseline 2.7 on recent admission). Computed Tomography (CT) of the chest without contrast on admission revealed diffuse bilateral ground-glass opacities and effusions (Figure 1). Doppler ultrasonography of lower extremities did not show any evidence of deep venous thrombosis.
The patient was initially treated with intravenous vancomycin and meropenum for 10 days for presumed HCAP, aggressive diuresis (IV furosemide) and continued on hemodialysis. She was continued on CYC therapy. The shortness of breath improved but continued to have intermittent hemoptysis. Two weeks after admission, fever (101.1˚F) and right-sided respirophasic chest pain occurred. Hypoxia worsened and required BIPAP. A repeat CT scan of chest revealed persistent diffuse ground glass opacities with new right hilar opacity measuring 1.9 cms × 1.7 cms and multiple other soft tissue opacities in the right lower lobe; some were solid and some with cavitation; air-bronchograms were also noted (Figure 2). The pleural effusions had decreased in size. On hospital day 19, intravenous antibiotic therapy was restarted with ciproflox-
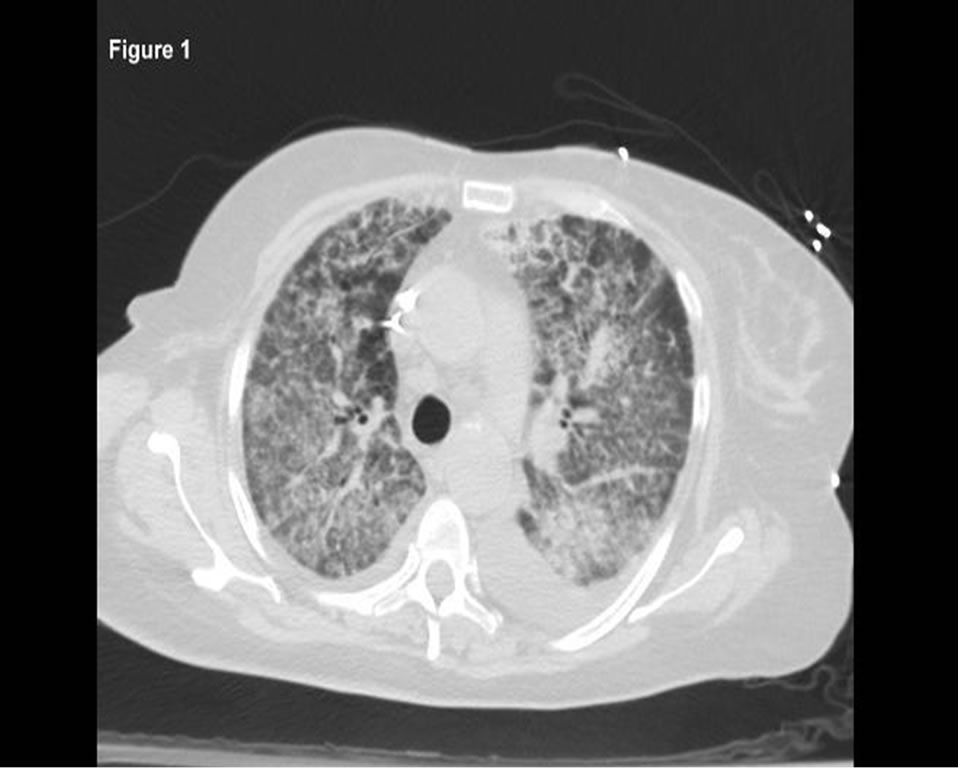
Figure 1. Chest CT on admission demonstrating diffusebilateral ground-glass opacities and effusions.
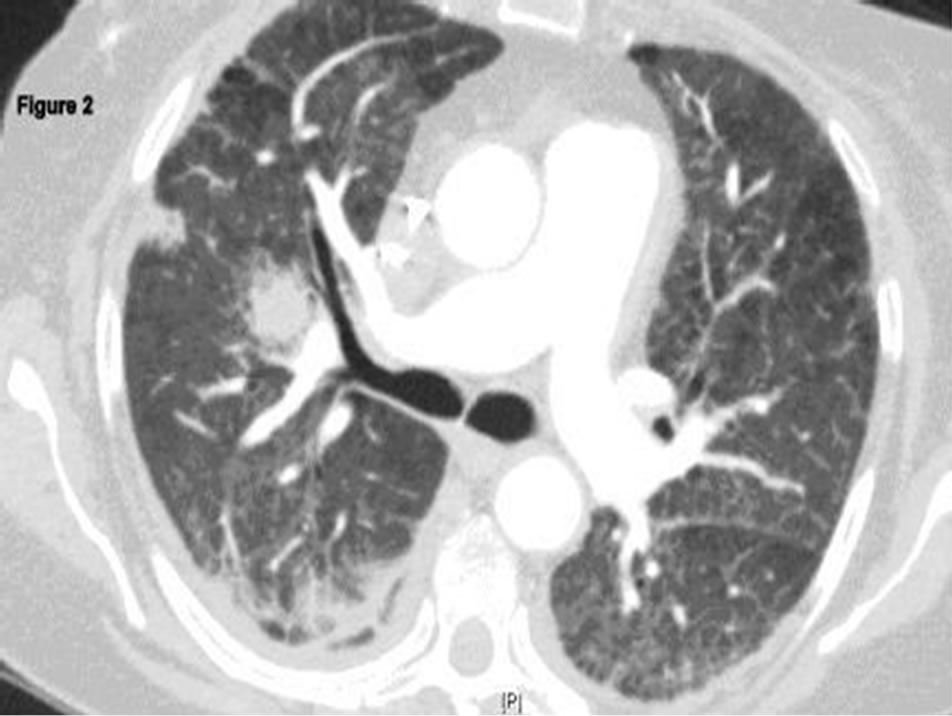
Figure 2. Repeat chest CT demonstrating new right hilar opacity, multiple other softtissue opacities, some with-cavitation and air-bronchograms.
acin, vancomycin and piperacillin/tazobactam. Blood and sputum cultures were negative. Despite therapy, lowgrade fevers continued. Caspofungin was added for empiric fungal coverage. Bronchoscopy was scheduled but hypoxia persisted and the patient developed atrial fibrillation with rapid ventricular response. Anti-coagulation was not initiated because of persistent hemoptysis. Patient became lethargic but arousable. Her speech was slurred with paresis of the right lower face and she had left arm weakness. The weakness progressed with leftsided paralysis. Motor examination revealed decreased strength of 3/5 on the left side. Sensory exam was normal. Brain CT revealed an ill-defined hypodensity involving the right basal ganglia, portions of the caudate head, putamen and globus pallidus (Figure 3).
Lumbar puncture revealed hazy fluid, protein 56 mg/dl, glucose 68 mg/dl (blood glucose 159), and 9 WBC (89% neutrophils, 4% lymphocytes and monocytes each). A Gram-stain of the CSF revealed rare polymorphonuclear cells with no organisms. A 2-D echocardiogram revealed no thrombus or vegetations. Carotid ultrasound was normal. Clinical deterioration followed. She became more somnolent. Repeat head CT showed large area of hypodensity in right basal ganglia, frontal lobe and corpus callosum with significant mass effect on third ventricle. Ventriculostomy was performed with no improvement in neurological status. The patient expired the following day.
Pathologic Findings
On autopsy, right lung revealed diffuse necrosis and inflammation. There was cavitation of the lower lobe associated with thrombotic occlusion of the pulmonary artery and vein branches supplying the area. Non-septated, irregularly wide (6 mm to 50 mm) fungal hyphae
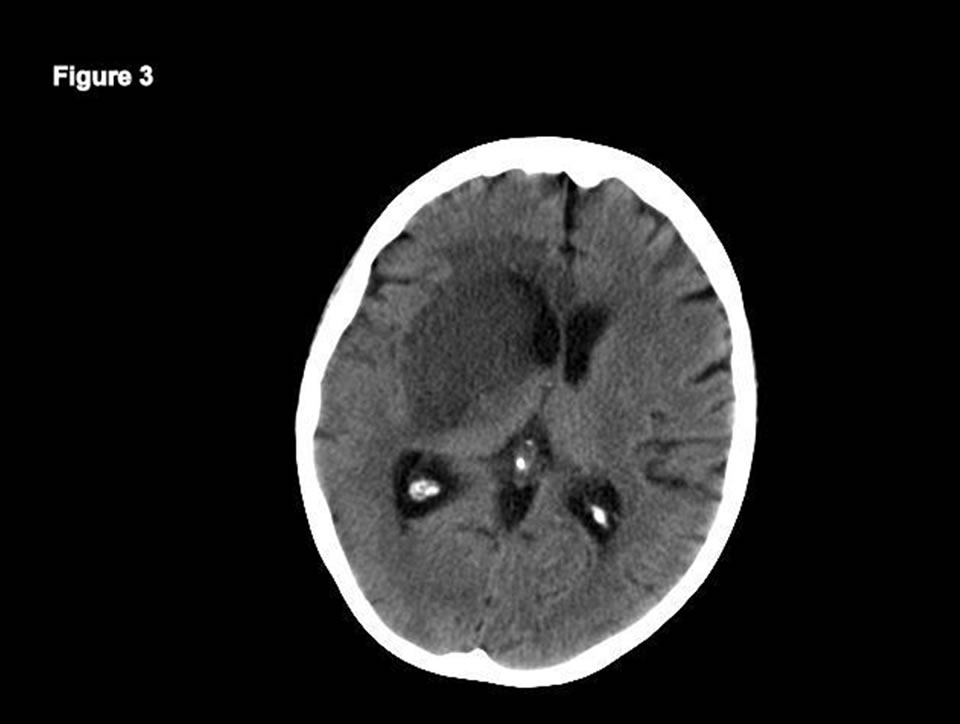
Figure 3. BrainCT demonstrating ill-defined hypodensity of right basal ganglia, portions of the caudate head, putamen and globus pallidus.
were identified in areas of inflammation and necrosis. Neuropathology showed subacute infarct involving the right basal ganglia and right temporal lobe. Special stains of brain tissue revealed fungal hyphae with morphology consistent with Mucor (Figure 4). Right kidney showed infarction with mucor infection as well as fibrotic crescents consistent with treated GPA. Heart pathology demonstrated cardiomegaly with atherosclerosis and unremarkable heart valves.
3. Discussion
We described a case of GPA, who following initiation of potent immunosuppressive therapies, improved, only later to develop disseminated mucormycosis, a fatal fungal infection. Prior to autopsy, it was unclear if the patient was suffering from CNS manifestations of GPA, cerebrovascular accident or infection.
This patient had multiple risk factors for stroke including diabetes, hypertension, atrial fibrillation and GPA itself. Thrombotic strokes may occur in patients with GPA [1]. Possible explanations include release of proinflammatory substances from damaged tissue, increased platelet aggregation [2], enhanced endothelial dysfunction and thrombogenicity [3,4]. High-dose steroids may also be thrombogenic, inducing factor VIII and causing hypofibrinolytic states.
Cerebral involvement is uncommon in GPA (2% - 8%) [5-7]. Three patterns of CNS involvement in GPA have been described: (a) cerebral vasculitis; (b) meningeal involvement due to extension from adjacent disease in the sinuses, nose or orbit; and (c) isolated meningocerebral lesions [6]. The most common form of involvement is the granulomatous invasion by contiguity. The main clinical manifestations are headache, seizure, or loss of function owing to focal lesion and stroke. Stroke is usually
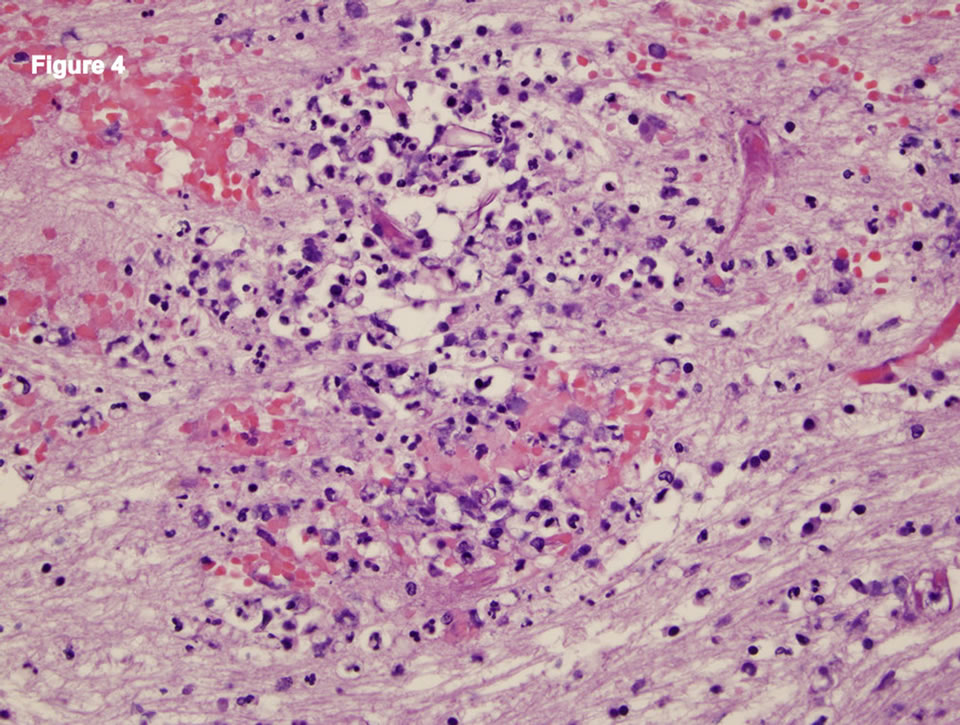
Figure 4. Neuropathology showing fungal hyphae consistent with Mucor.
caused by cerebral vasculitis resulting in hemorrhage (intracerebral or subarachnoid) or ischemic infarction. The diagnosis is usually obvious since most patients have established disease [6]. CSF analysis often shows only slight elevation in protein with mild lymphocytic pleocytosis. The diagnosis is made by clinical findings, MRI of the CNS, and stereotypic brain biopsy [7,8]. There are no classic findings of cerebral vasculitis in GPA on MRI, except non-specific white matter lesions [9,10,11].
Infections are common complications of the treatment of GPA. With the treatment of GPA, immunity is compromised that increases risk of ordinary and opportunistic infections (especially intracellular organisms e.g. Listeria, Nocardia, Cryptococcus neoformans, cytomegalovirus, brucellosis, Aspergillus, Mucor, Pneumocystis jirovecci). CNS infections in an immunocompromised host can manifest as meningitis, encephalopathy, seizures, or a stroke [11]. CNS Aspergillus infections present either as mass lesions (e.g., brain abscess), or as cerebral infarcts, but rarely as meningitis. Cryptococcus neoformans, in contrast, usually presents as meningitis but not as a cerebral mass lesion even when cryptococcal elements are present. Excluding stroke-like presentations, CNS mass lesions tend to present subacutely or chronically. Meningitis and encephalitis tend to present more acutely. Our patient, being on CYC was at increased risk for opportunistic infection. With the treatment of GPA, immunity is compromised and there is increased risk of ordinary and opportunistic infections (especially intracellular organisms). Infectious complications should be considered in all patients who are on adequate immunosuppression, not just CYC.
Although previously a fatal disease, treatment with CYC and immunosuppressive agents has greatly improved survival in GPA [12]. Despite a remarkable improvement in the outcome of patients with GPA, treatment-related mortality is still high in most series [13]. Early deaths are up to 3.5 times more often related to infection than active GPA [14-19]. A recent study demonstrated that one-year mortality in patients with systemic vasculitis was much higher due to therapyassociated adverse events (59%) as compared to active vasculitis (14%) [19].
This case illustrates that the most common complications of treatment in patients with GPA are infections. Various traditional risk factors for infection in GPA are leukopenia, severe lymphopenia, increasing age, and deteriorating renal function [17]. Severity of renal impairment and advancing age has been shown to pose the greatest threat during the first year of treatment [19]. Our patient, being a diabetic with renal failure and on high dose immunosuppressive therapy was at high risk of developing an infection. The greatest challenge facing clinicians treating these patients is to recognize those that are at a highest risk of death.
Opportunistic infections in GPA are caused by a wide variety of organisms. Mucormycosis is a rare but potentially devastating fungal infection. The mucoraceae are a ubiquitous saprophytic group of fungi found in soil, dust, decaying fruit and bread; as well as, in the nasal mucosa, oral cavity, and stools of health individuals. Despite the ubiquitous nature of the fungi, the virulence is low; hence, infection from these organisms is essentially limited to patients with poorly controlled diabetes mellitus, hematologic malignancy, organ transplant, chronic renal insufficiency, malnutrition and on deferoxamine therapy [19,20]. Our patient continued to have elevated blood sugars during the course of the hospitalization despite being on insulin. A high-glucose and acidic environment favors the growth of Mucor by attenuating neutrophil adhesion and chemotaxis to the hypha. The anti-micro-bial effecttiveness of pulmonary alveolar macrophage against the fungal spores and hypha is inhibited in hyperglycemic states [21]. Acidosis reduces the iron-binding capacity of serum ferritin, which in turn, results in increased free serum iron ions that promote the proliferation of the fungal pathogen. Disseminated mucormycosis is often fatal, since the fungus is very resistant to treatment. Lipid formulations of Amphotericin B have emerged as the first line therapy for mucormycosis. Posaconazole may be useful as salvage therapy.
This case also illustrates that symptoms caused by invasive fungal infections can mirror disease progression or relapse in GPA [22,23]. The patient presented with hemoptysis and dyspnea suggestive of relapse of GPA; however, pulmonary mucormycosis or other infections may also have a similar presentation. The fungi invade major blood vessels, leading to extensive necrosis, and in pulmonary disease, bleeding into the lungs may occur.
There are no specific blood tests or imaging studies that can aid in differentiating between the relapse and an infection. A rise in ANCA titer may be associated with relapses, but not always. Elevated ANCA titer should not be used as a sole parameter for preemptive therapy [24, 25]. Chest radiological findings of mucormycosis are nonspecific and include consolidation, nodules, masses, cavities, lymphadenopathy, and pleural effusion [26].
In patients who initially respond to appropriate therapy for GPA and then deteriorate during the first few months of therapy, worsening is most likely due to a comorbidity and not the primary disease. This sequence of events obligates aggressive diagnostic intervention, including biopsy if a definitive diagnosis cannot be made within hours to days. Bronchoalveolar lavage via a bronchoscope and microbiologic evaluation are the most efficient methods for detecting endobronchial infections, and should be undertaken early in the course of the illness.
4. Conclusions
This case illustrates the challenges faced by clinicians in treating patients with GPA particularly recognizing the complications of therapy. Infections complications should be considered in patients who are on adequate immuneosuppression.
Exploration of newer and safer treatments for GPA is needed. Rituximab has emerged as a new potential drug for treatment of GPA [27]. Whether it or other developing therapies will help to reduce side effects associated with conventional treatments in GPA will be determined over time.
5. Acknowledgements
Gary Hoffman for his helpful suggestions
REFERENCES
- B. V. Maramattom, C. Giannini, E. M. Manno and E. F. Wijdicks, “Wegener’s,” 2005. http://www.ncibi.nlm.nih.gov/pubmed/15980626granulo-matosis http://www.ncibi.nlm.nih.gov/pubmed/15980626
- H. A. Rula, R. Silverstein, G. Hoffman, L. Zhang, P. Imrey and C. Langford, “The Correlation between Circulating Microparticles and Platelet Aggregation and Disease Activity in Wegener Granulomatosis,” Arthritis and Rheumatism, Vol. 62, Supplement 10, 2010, p. S856.
- M. DeBandt, V. Ollivier, O. Meyer, C. Babin-Chevaye, F. Khechai, D. de Prost, J. Hakim and C. Pasquier, “Induction of Interleukin-1 and Subsequent Tissue Factor Expression by Anti-Proteinase 3 Antibodies in Human Umbilical Vein Endothelial Cells,” Arthritis Rheum, Vol. 40, No. 11, 1997, pp. 2030-2038. doi:10.1002/art.1780401116
- P. A. Merkel, G. H. Lo, J. T. Holbrook, et al., “Brief Communication: High Incidence of Venous Thrombotic Events among Patients with Wegener Granulomatosis: The Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study,” Annals of Internal Medicine, Vol. 142, No. 8, 2005, pp. 620-626.
- G. S. Hoffman, G. S. Kerr, R. Y. Leavitt, C. W. Hallahan, R. S. Lebovics, W. D. Travis, et al., “Wegener Granulomatosis: An Analysis of 158 Patients,” Annals of Internal Medicine, Vol. 116, No. 6, 1992, pp. 488-498
- R. Seror, A. Mahr, J. Ramanoelina, C. Pagnoux, P. Cohen and L. Guillevin, “Central Nervous System Involvement in Wegener’s Granulomatosis,” Medicine, Vol. 85, No. 1, 2006, pp. 54-65.
- H. Nishino, F. A. Rubino and J. E. Parisi, “The Spectrum of Neurologic Involvement in Wegener’s Granulomatosis,” Neurology, Vol. 43, No. 7, 1993, pp. 1334-1337.
- W. Zhang, G. Zhou, Q. Shi, X. Zhang, X. F. Zeng and F. C. Zhang, “Clinical Analysis of Nervous System Involvement in ANCA-Associated Systemic Vasculitides,” Clinical and Experimental Rheumatology, Vol. 27, Supplement 52, 2009, pp. S65-S69.
- J. U. Holle and W. L Gross, “Neurological Involvement in Wegener’s Granulomatosis,” Current Opinion Rheumatology, Vol. 23, No. 1, 2011, pp. 7-11. doi:10.1097/BOR.0b013e32834115f9
- J. M. Provenzale and N. B. Allen, “Wegener Granulomatosis: CT and MR Findings,” American Journal of Neuroradiology, Vol. 17, 1996, pp. 785-792.
- B. A. Cunha, “Nervous System Infections in the Compromised Host: A Diagnostic Approach,” Infectious Disease Clinics of North America Central, Vol. 15, No. 1, 2001, pp. 567-590.
- J. U. Holle, W. L. Gross, U. Latza, B. Noelle, P. Ambrosch, M. Heller, R. Fertmann and E. Reinhold-Keller, “Improved Outcome of 445 Wegener’s Granulomatosis Patients in a German Vasculitis Center over Four Decades,” Arthritis & Rheumatism, Vol. 63, No. 1, 2010, pp. 257-266.
- R. Luqmani, R. Suppiah, C. J. Edwards, R. Phillip, J. Maskell, D. Culliford, D. Jayne, K. Morishita and N. Arden, “Mortality in Wegener’s Granulomatosis: A Bimodal Pattern,” Rheumatology, Vol. 50, No. 4, 2010, pp. 597-702.
- G. S. Hoffman, Y. Drucker, M. F. Cotch, et al., “Wegener’s Granulomatosis: Patient-Reported Effects of Disease on Health, Function, and Income,” Arthritis & Rheumatism, Vol. 41, No. 12, 1998, pp. 2257-2262. doi:10.1002/1529-0131(199812)41:12<2257::AID-ART22>3.0.CO;2-K
- E. Reinhold-Keller, N. Beuge, U. Latza, et al., “An Interdisciplinary Approach to the Care of Patients with Wegener’s Granulomatosis: Long-Term Outcome in 155 Patients,” Arthritis & Rheumatism, Vol. 43, No. 5, 2000, pp. 1021-1032. doi:10.1002/1529-0131(200005)43:5<1021::AID-ANR10>3.0.CO;2-J
- K. De Groot, L. Harper, D. R. Jayne, L. F. Flores Suarez, G. Gregorinin, W. L. Gross, et al., “Pulse versus Daily Oral Cyclophopshamide for Induction of Remission in Antineu-Trophil Cytoplasmic Antibody-Associated Vasculitis: A Randomized Trial,” Annals of Internal Medicine, Vol. 150, 2009, pp. 670-680.
- A. D. Booth, M. K. Almond, A. Burns, et al., “PanThames Renal Research Group. Outcome of ANCA-Associated Renal Vasculitis: A Five-Year Retrospective Study,” American Journal of Kidney Diseases, Vol. 41, No. 4, 2003, pp. 776-784. doi:10.1016/S0272-6386(03)00025-8
- C. Charlier, C. Henegar, O. Launay, C. Pagnoux, A. Berezné, B. Bienvenu, P. Cohen, L. Mouthon and L. Guillevin, “Risk Factors for Major Infections in Wegener Granulomatosis: Analysis of 113 Patients,” Annals of the Rheumatic Disease, Vol. 68, No. 5, 2009, pp. 658-663. doi:10.1136/ard.2008.088302
- M. Little, P. Nightingale, C. A. Verburgh, et al., “Early Mortality in Systemic Vasculitis: Relative Contribution of Adverse Events and Active Vasculitis,” Annals of the Rheumatic Disease, Vol. 69, No. 6, 2010, pp. 1036-1043. doi:10.1136/ard.2009.109389
- A. Ibrahim, B. Spellberg and J. Edwards, “Iron Acquisition: A Novel Prospective on Mucormycosis Pathogenesis and Treatment,” Current Opinion in Infectious Disease, Vol. 21, No. 6, 2008, pp. 620-625. doi:10.1097/QCO.0b013e3283165fd1
- W.-F. Li, H. E. Chao, X.-F. Liu, S.-Y. Wang, J.-L. Qu and Z.-F. Lin, “A Diagnosis Neglected for 6 Years: Report of a Misdiagnosed Case of Pulmonary Mucormycosis and Review of the Literature,” Chinese Medical Journal, Vol. 123, No. 17, 2010, pp. 2480-2482
- M. F. Liu, F. F. Chen, T. R. Hsiue and C. C. Liu, “Disseminated Zygomycosis Simulating Cerebrovascular Disease and Pulmonary Alveolar Haemorrhage in a Patient with Systemic Lupus Erythematosus,” Clinical Rheumatology, Vol. 19, No. 4, 2000, pp. 311-314. doi:10.1007/s100670070052
- E. Nogueira, P. W. Ind, J. Friedland and A. D. Salama, “Mucormycosis May Mimic Disease Relapse in Wegener’s Granulomatosis,” The Journal of Rheumatology, Vol. 37, No. 6, 2010, pp. 1364-1365. doi:10.3899/jrheum.091423
- J. D. Finkielman, P. A. Merkel, D. Schroeder, et al., “Antiproteinase 3 Antineutrophil Cytoplasmic Antibodies and Disease Activity in Wegener Granulomatosis,” Annals of Internal Medicine, Vol. 147, No. 9, 2007, pp. 611-619.
- R. J. Falk and G. S. Hoffman, “Controversies in Small Vessel Vasculitis,” Current Opinion in Rheumatology, Vol. 19, No. 1, 2007, pp. 1-9.
- H. P. McAdams, M. R. de Christenson, D. C. Strollo and E. F. Patz Jr., “Pulmonary Mucormycosis: Radiologic Findings in 32 Cases,” American Journal of Neuroradiology, Vol. 168, No. 6, 1997, pp. 1541-1548.
- J. H. Stone, P. A. Merkel, R. Spiera, P. Seo, C. A. Langford, G. S. Hoffman, U. Specks, et al., “RAVE-ITN Research Group,” The New England Journal of Medicine, Vol. 5, No. 363, 2010, pp. 221-232. doi:10.1056/NEJMoa0909905